Abstract
The total chemical synthesis of the pentasaccharide repeating unit of the O-polysaccharide from E. coli O132 is accomplished in the form of its 2-aminoethyl glycoside. The 2-aminoethyl glycoside is particularly important as it allows further glycoconjugate formation utilizing the terminal amine without affecting the stereochemistry of the reducing end. The target was achieved through a [3 + 2] strategy where the required monosaccharide building blocks are prepared from commercially available sugars through rational protecting group manipulation. The NIS-mediated activation of thioglycosides was used extensively for the glycosylation reactions throughout.

Graphical Abstract
Introduction
O-Polysaccharides are the most complex molecular systems present in the bacterial cell wall owing to the presence of various sugar residues connected through diverse glycosidic linkages. These complex structures play a key role in determining and regulating the biology of these organisms [1] and act as the elicitor of the innate immune response [2]. As these polysaccharides can protect the bacteria concerned by killing the serum complements of the host system and can stop phagocytosis, they are extremely important for the pathogenicity of the microorganisms [2]. Their antigenic character has made them attractive targets for designing potential vaccine candidates. Arguably these complex structures can be isolated from the biological sources, however, often this isolation is cumbersome and becomes too expensive to get reasonable quantities of material with adequate purity. Therefore, the chemical syntheses of these complex structures become the only option to afford the material for detailed biological studies leading to the exploration of the vaccine potential.
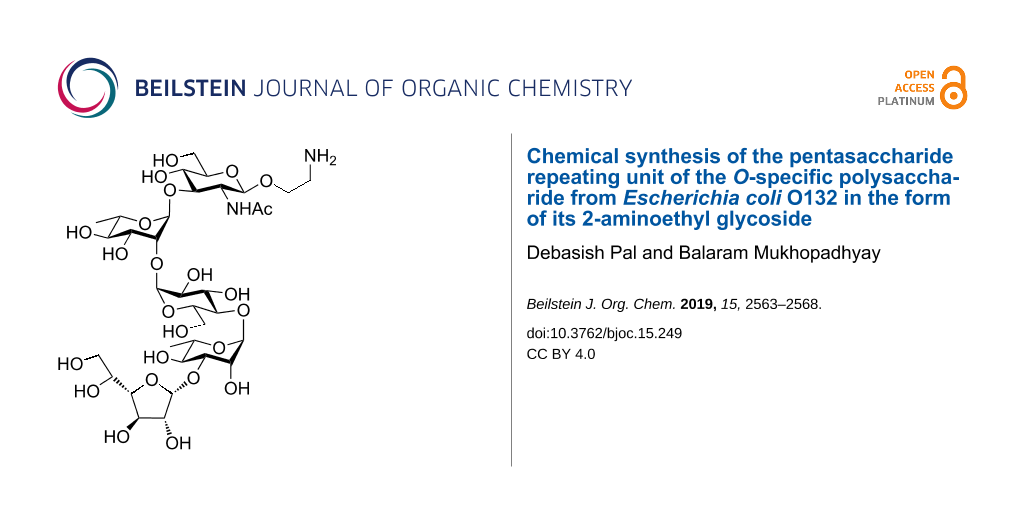
Escherichia coli is a Gram-negative bacteria present in the guts of human and other warm-blooded animals. It is usually harmless and beneficial to the host’s body, however, there are other variants of E. coli having virulence factors and causing diseases like diarrhea, urinary tract infection, septicaemia etc. E. coli O132 is reported to be the causative agent for septicemia in chicken [3], diarrhea in children under three years of age [4], diarrhea in calves and rabbits [5,6]. Recently, Knirel et al. [7] reported the structure of the O-polysaccharide of E. coli O132 as a pentasaccharide repeating unit consisting of GlcNAcp, Glcp, Rhap and Galf. In this communication we report on the total synthesis of this pentasaccharide repeating unit of the O-specific polysaccharide in the form of its 2-aminoethyl glycoside (Figure 1) through a [3 + 2] converging strategy. The building blocks were prepared by suitable protecting group manipulations on the commercially available monosaccharides and stereoselective chemical glycosylations. The 2-aminoethyl glycoside at the reducing end will facilitate further glycoconjugate formation without hampering the stereochemistry of the anomeric center. We have used similar glycosides in case of other chemically synthesized oligosaccharides before [8-10]. The corresponding aminopropyl linker has also been used by others [11]. The chemically synthesized oligosaccharide structure will help to elucidate further biological implications of the O-antigen concerned and possible vaccine potential.
![[1860-5397-15-249-1]](/bjoc/content/figures/1860-5397-15-249-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the target pentasaccharide repeating unit in the form of its 2-aminoethyl glycoside.
Figure 1: Structure of the target pentasaccharide repeating unit in the form of its 2-aminoethyl glycoside.
Results and Discussion
It is important to select a suitable glycoside at the reducing end of the target oligosaccharide keeping in mind that the glycoside should allow further glycoconjugate formation without disturbing the stereochemistry of the anomeric center. Therefore, the 2-aminoethyl glycoside was selected at the reducing end of the target pentasaccharide that will allow further conjugation using the terminal amine without affecting the glycosidic stereochemistry. Further retrosynthetic analysis of the target pentasaccharide 1 revealed that a [3 + 2] strategy will be the most suitable one for the total synthesis of the aforementioned target. Therefore, the first disconnection gave the trisaccharide acceptor 11 and the disaccharide donor 16. Further disconnection on the trisaccharide 11 gave disaccharide 5 and the monosaccharide 9. Disaccharide 5 is accessible from monosaccharide acceptor 2 and donor 3. Similarly, the disaccharide donor 16 may be prepared from monosaccharide acceptor 15 and donor 14. The monosaccharide building blocks can be prepared from commercially available ᴅ-glucose, ᴅ-galactose, ᴅ-glucosamine hydrochloride and ʟ-rhamnose through rational protecting group manipulations (Figure 2).
![[1860-5397-15-249-2]](/bjoc/content/figures/1860-5397-15-249-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis for the synthesis of the target pentasaccharide 1.
Figure 2: Retrosynthetic analysis for the synthesis of the target pentasaccharide 1.
Glycosylation of the known GlcNPhth acceptor 2 [12] and the rhamnosyl donor 3 [13] through the activation of thioglycoside using N-iodosuccinimide (NIS) in the presence of TMSOTf gave the Rha-(1→3)-GlcNPhth disaccharide 4 in 84% yield. The presence of a participating acetate group at the 2-position of the rhamnosyl donor 3 ensured the formation of the desired 1,2-trans glycoside. Peaks at 4.52 ppm (s, 1H, H-1′) in the 1H NMR and at 98.1 (C-1′) in the 13C NMR spectra confirmed the stereochemistry of the newly formed glycosidic linkage. Further, Zemplén de-O-acetylation [14] gave the disaccharide acceptor 5 in 91% isolated yield (Scheme 1).
![[1860-5397-15-249-i1]](/bjoc/content/inline/1860-5397-15-249-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the disaccharide acceptor 5.
Scheme 1: Synthesis of the disaccharide acceptor 5.
In a separate experiment, the 4-O-hydroxy group of the known compound 6 [15] was alkylated using naphthyl bromide in the presence of NaH [16] to afford the corresponding naphthyl derivative 7 in 82% yield. Next, the TBDPS group was removed using tetrabutylammonium fluoride in THF [17] to give the 6-hydroxy compound 8 in 90% yield. It was then benzoylated using BzCl in pyridine [18] with a catalytic amount of DMAP to furnish the completely protected donor 9 in 90% yield. Glycosylation of donor 9 with disaccharide acceptor 5 through activation of the thioglycoside using NIS and TMSOTf afforded the trisaccharide 10 in 78% yield. The newly formed 1,2-cis glycosidic linkage was confirmed by the peaks at 4.27 ppm (d, J1′′,2′′ = 1.5 Hz, 1H, H-1′′) in the 1H NMR and at 93.8 ppm (C-1′′) in the 13C NMR spectra. The presence of the participating O-benzoyl group at the 6-O-position of the donor led exclusively to the 1,2-cis glycoside [19]. Finally, the oxidative removal of the naphthyl group using DDQ [20] afforded the trisaccharide acceptor 11 in 83% yield (Scheme 2).
![[1860-5397-15-249-i2]](/bjoc/content/inline/1860-5397-15-249-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the trisaccharide acceptor 11.
Scheme 2: Synthesis of the trisaccharide acceptor 11.
The known galactofuranosyl derivative 12 [21] was prepared by following a literature procedure. It was subjected to Zemplén de-O-benzoylation using NaOMe in MeOH to give compound 13 in 93% yield. Further, per-O-benzylation using BnBr in the presence of NaH [22] afforded the galactofuranosyl donor 14 in 89% yield. Glycosylation of the donor 14 with known rhamnose acceptor 15 [23] through activation of the thioglycoside using NIS in the presence of TMSOTf at low temperature gave the disaccharide 16a (α) [1H NMR: 5.37 ppm (bs, 1H, H-1′), 13C NMR: 108.2 ppm (C-1′)] and 16b (β) [1H NMR: 5.36 ppm (d, J1′,2′ = 4.0 Hz, 1H, H-1′), 13C NMR: 95.6 ppm (C-1′)] in 89% yield (α:β = 2:1) (Scheme 3). Although the acceptor is also a thioglycoside, only the donor thioglycoside was activated in the NIS-catalyzed activation because of the higher reactivity of the furanosyl donor having benzyl protections [24,25].
![[1860-5397-15-249-i3]](/bjoc/content/inline/1860-5397-15-249-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the non-reducing end disaccharides (16a and 16b).
Scheme 3: Synthesis of the non-reducing end disaccharides (16a and 16b).
The mixture of α-linked and β-linked disaccharides was separated by flash chromatography to yield 59% pure α-linked disaccharide (16a) along with 30% β-linked disaccharide (16b). Compound 16a was used for further glycosylation with the trisaccharide acceptor 11 to afford the protected pentasaccharide 17. Once the protected pentasaccharide was at hand, the next challenge was the global deprotection to achieve the target molecule. First, the phthalimido group was removed using ethylenediamine [26] followed by acetylation with Ac2O in the presence of pyridine [27] to furnish the desired acetamido functionality. Next, the benzylidene group was hydrolyzed using 80% AcOH at 80 ºC [28]. Further, Zemplén de-O-acetylation using NaOMe in MeOH followed by hydrogenolysis in a ThalesNano continuous flow hydrogenation assembly using a 10% Pd-C cartridge [29]. After three cycles of hydrogenation, formation of the target pentasaccharide 1 was evident from the mass spectrum (Scheme 4).
![[1860-5397-15-249-i4]](/bjoc/content/inline/1860-5397-15-249-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the target pentasaccharide 1.
Scheme 4: Synthesis of the target pentasaccharide 1.
Conclusion
In conclusion, the pentasaccharide repeating unit of the O-specific polysaccharide of Escherichia coli O132 was accomplished through a convergent [3 + 2] strategy. The target pentasaccharide was obtained as its 2-aminoethyl glycoside that offers further possibilities for conjugation with suitable aglycons without hampering the stereochemistry of the reducing end sugar. The challenging stereoselective glycosylation with the galactofuranosyl unit was achieved through a chemoselective glycosylation approach depending on the higher reactivity of the furanose derivative over the pyranose system.
References
-
Larue, K.; Kimber, M. S.; Ford, R.; Whitfield, C. J. Biol. Chem. 2009, 284, 7395–7403. doi:10.1074/jbc.m809068200
Return to citation in text: [1] -
Beutin, L.; Wang, Q.; Naumann, D.; Han, W.; Krause, G.; Leomil, L.; Wang, L.; Feng, L. J. Med. Microbiol. 2007, 56, 177–184. doi:10.1099/jmm.0.46775-0
Return to citation in text: [1] [2] -
Blanco, J. E.; Blanco, M.; Mora, A.; Jansen, W. H.; Garcı́a, V.; Vázquez, M. L.; Blanco, J. Vet. Microbiol. 1998, 61, 229–235. doi:10.1016/s0378-1135(98)00182-5
Return to citation in text: [1] -
Peruski, L. F., Jr.; Kay, B. A.; El-Yazeed, R. A.; El-Etr, S. H.; Cravioto, A.; Wierzba, T. F.; Rao, M.; ElGhorab, N.; Shaheen, H.; Khalil, S. B.; Kamal, K.; Wasfy, M. O.; Svennerholm, A.-M.; Clemens, J. D.; Savarino, S. J. J. Clin. Microbiol. 1999, 37, 2974–2978.
Return to citation in text: [1] -
Aidar-Ugrinovich, L.; Blanco, J.; Blanco, M.; Blanco, J. E.; Leomil, L.; Dahbi, G.; Mora, A.; Onuma, D. L.; Silveira, W. D.; Pestana de Castro, A. F. Int. J. Food Microbiol. 2007, 115, 297–306. doi:10.1016/j.ijfoodmicro.2006.10.046
Return to citation in text: [1] -
Penteado, A. S.; Ugrinovich, L. A.; Blanco, J.; Blanco, M.; Blanco, J. E.; Mora, A.; Andrade, J. R. C.; Corrêa, S. S.; Pestana de Castro, A. F. Vet. Microbiol. 2002, 89, 41–51. doi:10.1016/s0378-1135(02)00148-7
Return to citation in text: [1] -
Shashkov, A. S.; Zhang, W.; Perepelov, A. V.; Weintraub, A.; Liu, B.; Widmalm, G.; Knirel, Y. A. Carbohydr. Res. 2016, 427, 44–47. doi:10.1016/j.carres.2016.03.016
Return to citation in text: [1] -
Mitra, A.; Mukhopadhyay, B. Eur. J. Org. Chem. 2019, 4869–4878. doi:10.1002/ejoc.201900815
Return to citation in text: [1] -
Adak, A.; Mukhopadhyay, B. Carbohydr. Res. 2019, 476, 1–7. doi:10.1016/j.carres.2019.02.009
Return to citation in text: [1] -
Sarkar, V.; Mukhopadhyay, B. Carbohydr. Res. 2018, 470, 13–18. doi:10.1016/j.carres.2018.09.006
Return to citation in text: [1] -
Mandal, P. K. Beilstein J. Org. Chem. 2014, 10, 2724–2728. doi:10.3762/bjoc.10.287
Return to citation in text: [1] -
Dhara, D.; Kar, R. K.; Bhunia, A.; Misra, A. K. Eur. J. Org. Chem. 2014, 4577–4584. doi:10.1002/ejoc.201402392
Return to citation in text: [1] -
Geng, X.; Wang, L.; Gu, G.; Guo, Z. Carbohydr. Res. 2016, 427, 13–20. doi:10.1016/j.carres.2016.03.023
Return to citation in text: [1] -
Zemplén, G. Ber. Dtsch. Chem. Ges. B 1926, 59, 1254–1266. doi:10.1002/cber.19260590626
Return to citation in text: [1] -
Lu, L.-D.; Shie, C.-R.; Kulkarni, S. S.; Pan, G.-R.; Lu, X.-A.; Hung, S.-C. Org. Lett. 2006, 8, 5995–5998. doi:10.1021/ol062464t
Return to citation in text: [1] -
Wang, L.; Overkleeft, H. S.; van der Marel, G. A.; Codée, J. D. C. J. Am. Chem. Soc. 2018, 140, 4632–4638. doi:10.1021/jacs.8b00669
Return to citation in text: [1] -
Limberg, G.; Thiem, J. Carbohydr. Res. 1995, 275, 107–115. doi:10.1016/0008-6215(95)00124-c
Return to citation in text: [1] -
Jacquinet, J.-C. Carbohydr. Res. 2004, 339, 349–359. doi:10.1016/j.carres.2003.10.012
Return to citation in text: [1] -
Sato, K.-i.; Akai, S.; Sakai, K.; Kojima, M.; Murakami, H.; Idoji, T. Tetrahedron Lett. 2005, 46, 7411–7414. doi:10.1016/j.tetlet.2005.08.109
Return to citation in text: [1] -
Tanaka, H.; Hamaya, Y.; Nishiwaki, N.; Ishida, H. Carbohydr. Res. 2018, 455, 23–31. doi:10.1016/j.carres.2017.11.005
Return to citation in text: [1] -
Completo, G. C.; Lowary, T. L. J. Org. Chem. 2008, 73, 4513–4525. doi:10.1021/jo800457j
Return to citation in text: [1] -
Lee, Y. J.; Lee, B.-Y.; Jeon, H. B.; Kim, K. S. Org. Lett. 2006, 8, 3971–3974. doi:10.1021/ol061444o
Return to citation in text: [1] -
Mukhopadhyay, B.; Field, R. A. Carbohydr. Res. 2003, 338, 2149–2152. doi:10.1016/s0008-6215(03)00349-5
Return to citation in text: [1] -
Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988, 110, 5583–5584. doi:10.1021/ja00224a060
Return to citation in text: [1] -
Konradsson, P.; Mootoo, D. R.; McDevitt, R. E.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1990, 270–272. doi:10.1039/c39900000270
Return to citation in text: [1] -
Kanie, O.; Crawley, S. C.; Palcic, M. M.; Hindsgaul, O. Carbohydr. Res. 1993, 243, 139–164. doi:10.1016/0008-6215(93)84087-m
Return to citation in text: [1] -
Mitra, A.; Mukhopadhyay, B. Synthesis 2015, 47, 3061–3066. doi:10.1055/s-0034-1380922
Return to citation in text: [1] -
Medgyes, G.; Jerkovich, G.; Kuszmann, J.; Fügedi, P. Carbohydr. Res. 1989, 186, 225–239. doi:10.1016/0008-6215(89)84037-6
Return to citation in text: [1] -
Sarkar, V.; Mukhopadhyay, B. ChemistrySelect 2016, 1, 5948–5951. doi:10.1002/slct.201601386
Return to citation in text: [1]
| 1. | Larue, K.; Kimber, M. S.; Ford, R.; Whitfield, C. J. Biol. Chem. 2009, 284, 7395–7403. doi:10.1074/jbc.m809068200 |
| 4. | Peruski, L. F., Jr.; Kay, B. A.; El-Yazeed, R. A.; El-Etr, S. H.; Cravioto, A.; Wierzba, T. F.; Rao, M.; ElGhorab, N.; Shaheen, H.; Khalil, S. B.; Kamal, K.; Wasfy, M. O.; Svennerholm, A.-M.; Clemens, J. D.; Savarino, S. J. J. Clin. Microbiol. 1999, 37, 2974–2978. |
| 17. | Limberg, G.; Thiem, J. Carbohydr. Res. 1995, 275, 107–115. doi:10.1016/0008-6215(95)00124-c |
| 3. | Blanco, J. E.; Blanco, M.; Mora, A.; Jansen, W. H.; Garcı́a, V.; Vázquez, M. L.; Blanco, J. Vet. Microbiol. 1998, 61, 229–235. doi:10.1016/s0378-1135(98)00182-5 |
| 18. | Jacquinet, J.-C. Carbohydr. Res. 2004, 339, 349–359. doi:10.1016/j.carres.2003.10.012 |
| 2. | Beutin, L.; Wang, Q.; Naumann, D.; Han, W.; Krause, G.; Leomil, L.; Wang, L.; Feng, L. J. Med. Microbiol. 2007, 56, 177–184. doi:10.1099/jmm.0.46775-0 |
| 15. | Lu, L.-D.; Shie, C.-R.; Kulkarni, S. S.; Pan, G.-R.; Lu, X.-A.; Hung, S.-C. Org. Lett. 2006, 8, 5995–5998. doi:10.1021/ol062464t |
| 2. | Beutin, L.; Wang, Q.; Naumann, D.; Han, W.; Krause, G.; Leomil, L.; Wang, L.; Feng, L. J. Med. Microbiol. 2007, 56, 177–184. doi:10.1099/jmm.0.46775-0 |
| 16. | Wang, L.; Overkleeft, H. S.; van der Marel, G. A.; Codée, J. D. C. J. Am. Chem. Soc. 2018, 140, 4632–4638. doi:10.1021/jacs.8b00669 |
| 11. | Mandal, P. K. Beilstein J. Org. Chem. 2014, 10, 2724–2728. doi:10.3762/bjoc.10.287 |
| 13. | Geng, X.; Wang, L.; Gu, G.; Guo, Z. Carbohydr. Res. 2016, 427, 13–20. doi:10.1016/j.carres.2016.03.023 |
| 8. | Mitra, A.; Mukhopadhyay, B. Eur. J. Org. Chem. 2019, 4869–4878. doi:10.1002/ejoc.201900815 |
| 9. | Adak, A.; Mukhopadhyay, B. Carbohydr. Res. 2019, 476, 1–7. doi:10.1016/j.carres.2019.02.009 |
| 10. | Sarkar, V.; Mukhopadhyay, B. Carbohydr. Res. 2018, 470, 13–18. doi:10.1016/j.carres.2018.09.006 |
| 14. | Zemplén, G. Ber. Dtsch. Chem. Ges. B 1926, 59, 1254–1266. doi:10.1002/cber.19260590626 |
| 7. | Shashkov, A. S.; Zhang, W.; Perepelov, A. V.; Weintraub, A.; Liu, B.; Widmalm, G.; Knirel, Y. A. Carbohydr. Res. 2016, 427, 44–47. doi:10.1016/j.carres.2016.03.016 |
| 5. | Aidar-Ugrinovich, L.; Blanco, J.; Blanco, M.; Blanco, J. E.; Leomil, L.; Dahbi, G.; Mora, A.; Onuma, D. L.; Silveira, W. D.; Pestana de Castro, A. F. Int. J. Food Microbiol. 2007, 115, 297–306. doi:10.1016/j.ijfoodmicro.2006.10.046 |
| 6. | Penteado, A. S.; Ugrinovich, L. A.; Blanco, J.; Blanco, M.; Blanco, J. E.; Mora, A.; Andrade, J. R. C.; Corrêa, S. S.; Pestana de Castro, A. F. Vet. Microbiol. 2002, 89, 41–51. doi:10.1016/s0378-1135(02)00148-7 |
| 12. | Dhara, D.; Kar, R. K.; Bhunia, A.; Misra, A. K. Eur. J. Org. Chem. 2014, 4577–4584. doi:10.1002/ejoc.201402392 |
| 21. | Completo, G. C.; Lowary, T. L. J. Org. Chem. 2008, 73, 4513–4525. doi:10.1021/jo800457j |
| 19. | Sato, K.-i.; Akai, S.; Sakai, K.; Kojima, M.; Murakami, H.; Idoji, T. Tetrahedron Lett. 2005, 46, 7411–7414. doi:10.1016/j.tetlet.2005.08.109 |
| 20. | Tanaka, H.; Hamaya, Y.; Nishiwaki, N.; Ishida, H. Carbohydr. Res. 2018, 455, 23–31. doi:10.1016/j.carres.2017.11.005 |
| 29. | Sarkar, V.; Mukhopadhyay, B. ChemistrySelect 2016, 1, 5948–5951. doi:10.1002/slct.201601386 |
| 27. | Mitra, A.; Mukhopadhyay, B. Synthesis 2015, 47, 3061–3066. doi:10.1055/s-0034-1380922 |
| 28. | Medgyes, G.; Jerkovich, G.; Kuszmann, J.; Fügedi, P. Carbohydr. Res. 1989, 186, 225–239. doi:10.1016/0008-6215(89)84037-6 |
| 24. | Mootoo, D. R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. J. Am. Chem. Soc. 1988, 110, 5583–5584. doi:10.1021/ja00224a060 |
| 25. | Konradsson, P.; Mootoo, D. R.; McDevitt, R. E.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1990, 270–272. doi:10.1039/c39900000270 |
| 26. | Kanie, O.; Crawley, S. C.; Palcic, M. M.; Hindsgaul, O. Carbohydr. Res. 1993, 243, 139–164. doi:10.1016/0008-6215(93)84087-m |
| 22. | Lee, Y. J.; Lee, B.-Y.; Jeon, H. B.; Kim, K. S. Org. Lett. 2006, 8, 3971–3974. doi:10.1021/ol061444o |
| 23. | Mukhopadhyay, B.; Field, R. A. Carbohydr. Res. 2003, 338, 2149–2152. doi:10.1016/s0008-6215(03)00349-5 |
© 2019 Pal and Mukhopadhyay; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)