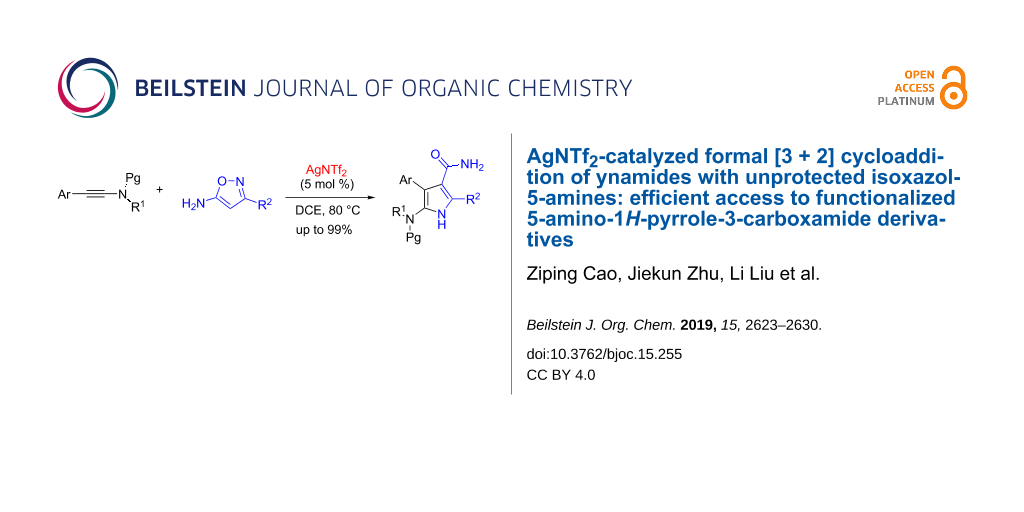
Abstract
A formal [3 + 2] cycloaddition between ynamides and unprotected isoxazol-5-amines has been developed in the presence of catalytic AgNTf2 in an open flask. By the protocol, a variety of functionalized 5-amino-1H-pyrrole-3-carboxamide derivatives can be obtained in up to 99% yield. The reaction mechanism might involve the generation of an unusual α-imino silver carbene intermediate (or a silver-stabilized carbocation) and subsequent cyclization/isomerization to build the significant pyrrole-3-carboxamide motif. The reaction features the use of an inexpensive catalyst, simple reaction conditions, simple work-up without column chromatographic purification for most of products and high yields.

Graphical Abstract
Introduction
Silver-catalyzed transformations of alkynes have attracted much attention over the past decade [1,2]. As a powerful π-activator, silver can promote various reactions of alkynes in high efficiencies [3-8]. Generally speaking, the proceeding of these reactions involves the activation of alkyne bond by coordination of [Ag] and then the attacking of nucleophilic partners, followed by a protodemetalation step to form the alkene motif (Scheme 1, path a) [9-11]. However, the formation of a silver carbene intermediate, which could be generated usually from relevant diazo precursors [12-14], appears to be an unusual event in silver-mediated reactions of alkynes (Scheme 1, path b). In 2007, Echavarren and co-workers have developed an intramolecular cyclopropanation reaction of 1,6-enynes by silver catalysis, involving probably the generation of a silver-carbene species [15]. Wang and co-workers reported a range of propargylic esters tethered to cyclohexadienones that can be converted into complex polycycles by Ag-carbenoid-initiated cascades [16]. Recently, Zhu’s group developed a tandem 1,3‑dipolar cycloaddition/cyclopropanation silver-catalyzed reaction of enynals with alkenes [17]. In our previous studies, this silver carbene species could be also involved mechanistically in the Ag-mediated reaction of enynals with conjugated dienes [18] and the homodimerization of enynals [19,20]. So, the studies on the reactions of alkynes involving the generation of silver carbene species from non-diazo precursors are of great value for getting some insight into silver-carbene chemistry.
![[1860-5397-15-255-i1]](/bjoc/content/inline/1860-5397-15-255-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Two modes of reactions of alkynes by silver catalysis.
Scheme 1: Two modes of reactions of alkynes by silver catalysis.
The generation of α-imino gold carbene intermediates in gold-catalyzed reactions of alkynes has been widely studied in recent years [21-30]. In 2015, Ye and co-workers, as pioneers, developed this chemistry with the employment of isoxazole nucleophiles in gold-catalyzed formal [3 + 2] cycloaddition reaction of ynamides [31,32], and zinc-catalyzed the reaction of ynol ethers [33], giving the respective multi-substituted pyrrole derivatives efficiently (Scheme 2a) [34,35]. The reaction proceeds via an α-imino gold carbene pathway presumed by mechanistic studies and theoretical calculations. Following our ongoing interest in the alkyne chemistry [36-38], we recently envisaged that the reaction of ynamides with isoxazoles could proceed under silver catalysis conditions, involving the generation of α-imino silver carbene and subsequent cyclization to pyrroles (Scheme 2b). Herein we want to provide some detailed results on the reaction (Scheme 2b), leading to the synthesis of a variety of functionalized 5-amino-1H-pyrrole-3-carboxamide derivatives in high yields. The reaction features the use of an inexpensive catalyst, mild reaction conditions, simple operation and product purification. Notably, the core skeleton of these products is the substructure of many biologically active molecules. For example (Figure 1), compound 1 has significant activities as DNA-cleaving agent [39] and sangivamycin 2 has been in clinical trials against colon cancer, gall bladder cancer and acute myelogenous leukemia in humans [40] and its 2-aza analogue 3 is also active against human cytomegalovirus (HCMV) and herpes simplex virus type 1 (HSV-1) [41]. To the best of our knowledge, this case of α-imino silver-carbene is not yet reported [42].
![[1860-5397-15-255-i2]](/bjoc/content/inline/1860-5397-15-255-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reactions of ynamides or ynol ethers with isoxazoles by transition metal catalysis.
Scheme 2: Reactions of ynamides or ynol ethers with isoxazoles by transition metal catalysis.
![[1860-5397-15-255-1]](/bjoc/content/figures/1860-5397-15-255-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected bioactive molecules containing the 5-amino-1H-pyrrole-3-carboxamide motif.
Figure 1: Selected bioactive molecules containing the 5-amino-1H-pyrrole-3-carboxamide motif.
Results and Discussion
An initial experiment was carried out with ynamide 4a and isoxazole 5 as the selected substrates based on the known procedure developed by Ye’s group [31]. With pyrrole 6, a 42% yield was obtained using AgNTf2 (5 mol %) catalyst and DCE as the solvent at 80 °C for 2 h (Scheme 3). The reaction conditions were then further optimized but without obvious improvement of the yield. We presumed the reason might be due to the low nucleophilic reactivity of isoxazole 5 to the ynamide motif by silver activation. Therefore, we considered that the introduction of an amine motif can enhance the nucleophilic ability of nitrogen on the isoxazole ring. However, this change could raise at least an issue, involving the direct addition of an amino motif to the ynamide substrate [43,44]. To answer this question, two isoxazoles 7 and 8a with an amino group at the distinct position were utilized for the current reaction. The results exhibited that the use of 5-methylisoxazol-3-amine (7) gave a complex mixture. In contrast, 3-methylisoxazol-5-amine (8a) led to the formation of the desired pyrrole 10aa in 99% yield. Notably, the hydroamination product cannot be found in the reaction of 4a and 8a.
![[1860-5397-15-255-i3]](/bjoc/content/inline/1860-5397-15-255-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of ynamide 4a with different isoxazoles 5, 7 and 8a.
Scheme 3: Reactions of ynamide 4a with different isoxazoles 5, 7 and 8a.
In order to study the effect of reaction conditions on the yield of 10aa, various silver salts and solvents were screened and the results were listed in Table 1. Among the silver salts screened, AgNTf2 led to the formation of 10aa in the best yield (Table 1, entry 1). Both AgSbF6 and AgOTF (silver trifluoroacetate) salts can catalyze the proceeding of the reaction at 80 °C, affording the desired pyrrole 10aa in 42% and 28% yields (Table 1, entries 2 and 3), respectively. The hydrolytic product 11 was also obtained in 55% yield when AgOTF was used as the catalyst. Other silver catalysts such as AgNO3, AgBF4 and AgOTf were not suitable for the current reaction, thus no desired product was observed (Table 1, entries 4–6). Furthermore, the solvent studies showed that 1,4-dioxane, THF and toluene are good alternatives, giving the pyrrole 10aa in similar yields (Table 1, entries 7–9), while the use of acetonitrile led to a lowered yield and the formation of a small amount of side product 11 (Table 1, entry 10). By considering the boiling points of the solvents and reaction efficiencies, DCE is believed to be the optimal choice. In addition, the reaction temperature can be lowered to 60 ˚C but a prolonged time was needed (Table 1, entry 11). Incomplete conversion was observed at 40 °C, and even no reaction happened at 20 °C (Table 1, entries 12 and 13). The yield of 10aa did not decrease obviously when the isoxazole 8a were reduced to 1.1 equiv (Table 1, entries 14-16). A control experiment showed the reaction could not proceed in the absence of Ag catalyst (Table 1, entry 17). Finally, the optimal reaction conditions were established with a slight excess of isoxazole 8a (1.1 equiv) and catalytic AgNTf2 (5 mol %) in DCE at 80 °C (Table 1, entry 15).
Table 1: Effect of different reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-255-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | Catalyst (5 mol %) | Solvent | Temp. (°C) | Time (h) | Yield (%)b |
| 1 | AgNTf2 | DCE | 80 | 2 | 99 |
| 2 | AgSbF6 | DCE | 80 | 4 | 42 |
| 3 | AgOTF | DCE | 80 | 4 | 28, 55c |
| 4 | AgNO3 | DCE | 80 | 4 | 0, 20c |
| 5 | AgBF4 | DCE | 80 | 4 | 0 |
| 6 | AgOTf | DCE | 80 | 4 | 0 |
| 7 | AgNTf2 | 1,4-dioxane | 80 | 2 | 99 |
| 8 | AgNTf2 | THF | 80 | 24 | 95 |
| 9 | AgNTf2 | toluene | 80 | 12 | 92 |
| 10 | AgNTf2 | CH3CN | 80 | 12 | 85, 10c |
| 11 | AgNTf2 | DCE | 60 | 4 | 98 |
| 12 | AgNTf2 | DCE | 40 | 8 | 50 |
| 13 | AgNTf2 | DCE | 20 | 24 | 0 |
| 14d | AgNTf2 | DCE | 80 | 2 | 99 |
| 15e | AgNTf2 | DCE | 80 | 2 | 99 |
| 16f | AgNTf2 | DCE | 80 | 2 | 96 |
| 17 | – | DCE | 80 | 24 | 0 |
aAll reactions were carried out with ynamide 4a (0.2 mmol), isoxazole 8a (0.4 mmol, 2 equiv) with the indicated catalyst (5 mol %) in solvent (2.0 mL), unless otherwise noted. bYield of isolated product 10aa. cYield of hydrolytic product 11. d1.5 equiv of 8a was used. e1.1 equiv of 8a was used. f1.0 equiv of 8a was used.
Having identified the optimized conditions, the scope of substrates was subsequently investigated. Firstly, various ynamides were utilized for the current reaction under the optimized conditions and the results were summarized in Figure 2. It could be found that a variety of 5-amino-1H-pyrrole-3-carboxamides were obtained in up to 99% yields. For examples, the aromatic motifs of ynamides possessing an electron-donating group such as MeO-, Me- and t-Bu- are well tolerated to afford the desired cycloadducts 10ba–da in 85–99% yields. The ynamide substrates 4e–g with an electron-deficient halogen substituent (F-, Cl- and Br-) on the aromatic ring could be also successfully applied to the reaction, giving the 5-amino-1H-pyrrole-3-carboxamides 10ea–ga in high efficiencies. The reactions of meta-substituted aromatic ynamides 4h and 4i with 8a could proceed smoothly, affording the corresponding products in 99% yield. However, the use of alkylated ynamides such as 4j gave a complex mixture under current conditions, and no desired product (10ja) was isolated. Subsequently, the N-substituents of ynamides were investigated. The results showed that other alkyl (iPr-, n-Bu- and Bn-) and phenyl are compatible for this reaction, and the pyrroles 10ka–na were obtained in 50–98% yields. In addition, the Ts protecting group could be changed for other sulfonyl groups such as Ms (10na, 98%), o-Ns (10oa, 99%) and p-Ns (10pa, 99%), while the cyclic carbamate-derived ynamides such as 4q are no good substrates, leading to the formation of a complex mixture.
![[1860-5397-15-255-2]](/bjoc/content/figures/1860-5397-15-255-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Scope with regard to ynamide 4. All reactions were carried out with ynamide 4 (0.2 mmol), isoxazole 8 (0.22 mmol, 1.1 equiv) with AgNTf2 (5 mol %) in DCE (2.0 mL) at 80 °C, unless otherwise noted. Isolated yields are provided.
Figure 2: Scope with regard to ynamide 4. All reactions were carried out with ynamide 4 (0.2 mmol), isoxazole ...
The scope with regard to the 5-aminoisoxazole was next evaluated. As seen from Figure 3, aryl-substituted 5-aminoisoxazoles 8b and 8c, and secondary alkyl-substituted 5-aminoisoxazole 8d were suitable reaction partners, thus expanding the applicability of the present reaction. By the transformations, several desired 5-amino-1H-pyrrole-3-carboxamide products were easily obtained in 96–98% yields (10ab–ad). The structure of 10ad was further confirmed unambiguously by single crystal X-ray analysis (Figure 4). The sterically demanding t-Bu group installed at 5-aminoisoxazole 8e has an obvious inferior effect on the efficiency, leading to the generation of cycloadduct 10ae in <10% yield under current conditions. Nevertheless, the yield of 10ae could be increased to 38% by changing the reaction conditions to 2.0 equiv of 8e and 100 °C. It should be also noted, however, that the reaction could not proceed efficiently with 3,4-dimethylisoxazol-5-amine (8f), thus a mixture of unidentifiable decomposition products was observed.
![[1860-5397-15-255-3]](/bjoc/content/figures/1860-5397-15-255-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Scope with regard to the 5-aminoisoxazole 8 (see Figure 2). aReaction conditions: 2.0 equiv of 8e, 100 °C.
Figure 3: Scope with regard to the 5-aminoisoxazole 8 (see Figure 2). aReaction conditions: 2.0 equiv of 8e, 100 °C.
![[1860-5397-15-255-4]](/bjoc/content/figures/1860-5397-15-255-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure in the solid state of compound 10ad.
Figure 4: Molecular structure in the solid state of compound 10ad.
In order to investigate the feasibility of the present reaction on a large scale, a gram-grade experiment was performed (Scheme 4). The results indicated a similar yield was obtained in the reaction of ynamide 4a with 8a. Notably, after the completion of the reaction, a white precipitation was observed and filtered straightforwardly to afford the desired product 10aa in mostly quantitative yield.
![[1860-5397-15-255-i4]](/bjoc/content/inline/1860-5397-15-255-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
A tentative mechanism for the formation of 5-amino-1H-pyrrole-3-carboxamide 10aa is provided in Scheme 5 [21,31,32]. Initial activation of ynamide 4a by silver catalyst can afford the Ag complex A, which can isomerize to the keteniminium ion intermediate B. A nucleophilic addition of 5-aminoisoxazole 8a to silver species B leads to the formation of alkenylsilver species C, followed by the fragmentation process to give an unusual α-imino silver carbene species D. The intermediate D should be able to isomerize to D’ by conformation rotation to facilitate the addition of activated methene to Ag-carbene, thus forming a new silver species E with a 5-membered ring. The leaving of silver catalyst from E along with the formation of enamide motif affords 3H-pyrrole F. A final aromatization step by isomerization provides the desire cyclic product 10aa. Notably, two possible cyclization routes from D’ (or D) to give 7-membered rings G and H cannot be achieved through the attack of the O- and N-nucleophilic sites, respectively.
![[1860-5397-15-255-i5]](/bjoc/content/inline/1860-5397-15-255-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanistic hypotheses for Ag-catalyzed reaction of ynamide 4a with aminoisoxazole 8a.
Scheme 5: Mechanistic hypotheses for Ag-catalyzed reaction of ynamide 4a with aminoisoxazole 8a.
In addition, silver-stabilized carbocation intermediate I generated from intermediate C might be another possible process to form E, although it was rarely mentioned due to weak Ag–C bond (Scheme 6). It should be also mentioned that a direct protodemetalation step of C was not existing, thus compound 12 could be not formed in current reaction.
![[1860-5397-15-255-i6]](/bjoc/content/inline/1860-5397-15-255-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible reaction routes of intermediate C.
Scheme 6: Possible reaction routes of intermediate C.
Conclusion
In conclusion, we have developed a reaction of ynamides with unprotected isoxazol-5-amines that can achieve the synthesis of a variety of functionalized 5-amino-1H-pyrrole-3-carboxamide derivatives in high efficiency. The reaction conditions involve the use of catalytic AgNTf2 with DCE as the solvent at 80 °C, without needing to exclude moisture or air. The presumed reaction mechanism might involve the generation of an unusual α-imino silver carbene species (or a silver-stabilized carbocation) and then cyclization/isomerization to complete the formal [3 + 2] cycloaddition process. This reaction highlights the use of an inexpensive catalyst, and simple work-up without column chromatographic purification for most of the products. Furthermore, these products contain the core structure of many bioactive molecules, thus providing a practical method for the construction of similar skeleton compounds. Our next investigation will focus on the studies of the detailed reaction mechanism and the applications of this methodology for the synthesis of interesting molecules.
Experimental
General information. Reactions were carried out in an open flask and monitored by thin-layer chromatography (TLC) on silica plates, visualized by irradiation with UV light. Commercially available reagents were used without further purification. 1H and 13C NMR spectra were recorded at 500 MHz for 1H nuclei, and 125.8 MHz for 13C nuclei. Chemical shifts (δ) are reported in units of parts per million (ppm); signals are referenced to TMS (0.00 ppm) or solvent residual peak (DMSO-d6, 2.5 ppm for 1H and 39.5 ppm for 13C) as an internal standard. Coupling constants (J) are given in Hz, and multiplicity is abbreviated as: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), and m (multiplet). All melting points are uncorrected and determined on an X-4 digital microscopic melting point apparatus. HRMS were measured using electrospray ionization (ESI).
Ynamide compounds 4a–q were prepared according to the known literature procedures [45]. The isoxazol-5-amines 8a–e are commercially available reagents.
Typical procedure for the AgNTf2-catalyzed reaction of ynamide 4a with 8a: To a stirred solution of ynamide 4a (57.0 mg, 0.2 mmol) in DCE (2.0 mL, 0.1 M) was added isoxazol-5-amine 8a (21.6 mg, 0.22 mmol, 1.1 equiv), followed by AgNTf2 (3.9 mg, 5 mol %). The resulting mixture was placed into an oil bath of 80 °C with stirring for 2 h generally, monitored by TLC. After completion, the reaction mixture was cooled and the desired product was precipitated. The solid was filtered and washed with DCM twice, then dried in a vacuum drying oven at 50 °C for 24 h to give the pure pyrrole product 10aa, 75.8 mg, 99% yield.
For products 10ab–ae, 10ka–la, the purification method was as follows: evaporation of volatiles under reduced pressure to give the residue, which was suffered from column chromatography on silica gel (petrol ether/ethyl acetate 1:1–1:2, v/v) to afford the pure pyrrole.
5-((N,4-Dimethylphenyl)sulfonamido)-2-methyl-4-phenyl-1H-pyrrole-3-carboxamide (10aa): white solid, mp 176.5–178.5 °C, yield 75.9 mg, 99%; Rf = 0.16 (hexanes/EtOAc 1:1); 1H NMR (DMSO-d6, 500 MHz) δ 11.22 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 7.23–7.13 (m, 3H), 6.99 (d, J = 6.4 Hz, 2H), 6.76 (s, 1H) 5.87 (s, 1H), 2.96 (s, 3H), 2.39 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6, 125.8 MHz) δ 166.7, 143.3, 135.1, 134.0, 129.57, 129.52, 128.8, 127.7, 127.3, 126.3, 122.3, 119.4, 114.4, 38.6, 21.0, 14.5; HRMS–ESI (m/z): [M + H]+ calcd for C20H22N3O3S, 384.1376; found, 384.1375.
References
-
Fang, G.; Bi, X. Chem. Soc. Rev. 2015, 44, 8124–8173. doi:10.1039/c5cs00027k
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Han, G.; Xue, L.; Zhao, L.; Zhu, T.; Hou, J.; Song, Y.; Liu, Y. Adv. Synth. Catal. 2019, 361, 678–682. doi:10.1002/adsc.201801482
Return to citation in text: [1] -
Dabral, S.; Bayarmagnai, B.; Hermsen, M.; Schießl, J.; Mormul, V.; Hashmi, A. S. K.; Schaub, T. Org. Lett. 2019, 21, 1422–1425. doi:10.1021/acs.orglett.9b00156
Return to citation in text: [1] -
Yang, Y.; Antoni, P.; Zimmer, M.; Sekine, K.; Mulks, F. F.; Hu, L.; Zhang, L.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2019, 58, 5129–5133. doi:10.1002/anie.201812577
Return to citation in text: [1] -
Hashmi, A. S. K.; Schwarz, L.; Bats, J. W. J. Prakt. Chem. 2000, 342, 40–51. doi:10.1002/(sici)1521-3897(200001)342:1<40::aid-prac40>3.3.co;2-m
Return to citation in text: [1] -
Zheng, S.-C.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2019, 58, 1494–1498. doi:10.1002/anie.201812654
Return to citation in text: [1] -
Su, X.; Chen, B.; Wang, S.; Chen, H.; Chen, C. ACS Catal. 2018, 8, 7760–7765. doi:10.1021/acscatal.8b02448
Return to citation in text: [1] -
Fernández, P.; Valdés, C.; Fañanás, F. J.; Rodríguez, F. J. Org. Chem. 2019, 84, 3184–3191. doi:10.1021/acs.joc.8b03081
Return to citation in text: [1] -
Wang, Q.; Zhang, L.; Yao, J.; Qiu, G.; Li, X.; Zhou, H. J. Org. Chem. 2018, 83, 4092–4098. doi:10.1021/acs.joc.7b03257
Return to citation in text: [1] -
Clarke, A. K.; Lynam, J. M.; Taylor, R. J. K.; Unsworth, W. P. ACS Catal. 2018, 8, 6844–6850. doi:10.1021/acscatal.8b00745
Return to citation in text: [1] -
Luo, H.; Wu, G.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2015, 54, 14503–14507. doi:10.1002/anie.201507219
Return to citation in text: [1] -
Briones, J. F.; Davies, H. M. L. Org. Lett. 2011, 13, 3984–3987. doi:10.1021/ol201503j
Return to citation in text: [1] -
Caballero, A.; Despagnet-Ayoub, E.; Díaz-Requejo, M. M.; Díaz-Rodríguez, A.; González-Núñez, M. E.; Mello, R.; Muñoz, B. K.; Ojo, W.-S.; Asensio, G.; Etienne, M.; Pérez, P. J. Science 2011, 332, 835–838. doi:10.1126/science.1204131
Return to citation in text: [1] -
Porcel, S.; Echavarren, A. M. Angew. Chem., Int. Ed. 2007, 46, 2672–2676. doi:10.1002/anie.200605041
Return to citation in text: [1] -
Wang, L.; Cai, S.; Xing, X.; Gao, Y.; Wang, T.; Wang, D. Z. Org. Lett. 2013, 15, 2362–2365. doi:10.1021/ol4006954
Return to citation in text: [1] -
Liang, R.; Ma, T.; Zhu, S. Org. Lett. 2014, 16, 4412–4415. doi:10.1021/ol5017299
Return to citation in text: [1] -
Cao, Z.; Zhu, H.; Meng, X.; Tian, L.; Chen, G.; Sun, X.; You, J. J. Org. Chem. 2016, 81, 12401–12407. doi:10.1021/acs.joc.6b02529
Return to citation in text: [1] -
Guo, M.; Meng, X.; Zhao, Y.; Dong, Y.; Sun, X.; Tian, L.; Cao, Z. RSC Adv. 2019, 9, 2703–2707. doi:10.1039/c8ra09269a
Return to citation in text: [1] -
Beeler, A. B.; Su, S.; Singleton, C. A.; Porco, J. A., Jr. J. Am. Chem. Soc. 2007, 129, 1413–1419. doi:10.1021/ja0674744
Return to citation in text: [1] -
Aguilar, E.; Santamaría, J. Org. Chem. Front. 2019, 6, 1513–1540. doi:10.1039/c9qo00243j
Return to citation in text: [1] [2] -
Tian, X.; Song, L.; Han, C.; Zhang, C.; Wu, Y.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2019, 21, 2937–2940. doi:10.1021/acs.orglett.9b01011
Return to citation in text: [1] -
Hsu, Y.-C.; Hsieh, S.-A.; Liu, R.-S. Chem. – Eur. J. 2019, 25, 5288–5297. doi:10.1002/chem.201806083
Return to citation in text: [1] -
Tian, X.; Song, L.; Rudolph, M.; Rominger, F.; Oeser, T.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2019, 58, 3589–3593. doi:10.1002/anie.201812002
Return to citation in text: [1] -
Allegue, D.; González, J.; Fernández, S.; Santamaría, J.; Ballesteros, A. Adv. Synth. Catal. 2019, 361, 758–768. doi:10.1002/adsc.201801484
Return to citation in text: [1] -
Song, L.; Tian, X.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Chem. Commun. 2019, 55, 9007–9010. doi:10.1039/c9cc04027g
Return to citation in text: [1] -
Tian, X.; Song, L.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2019, 21, 4327–4330. doi:10.1021/acs.orglett.9b01501
Return to citation in text: [1] -
Tian, X.; Song, L.; Rudolph, M.; Wang, Q.; Song, X.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2019, 21, 1598–1601. doi:10.1021/acs.orglett.9b00140
Return to citation in text: [1] -
Jin, H.; Huang, L.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem. 2016, 128, 804–808. doi:10.1002/ange.201508309
Return to citation in text: [1] -
Jin, H.; Tian, B.; Song, X.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem. 2016, 128, 12880–12884. doi:10.1002/ange.201606043
Return to citation in text: [1] -
Zhou, A.-H.; He, Q.; Shu, C.; Yu, Y.-F.; Liu, S.; Zhao, T.; Zhang, W.; Lu, X.; Ye, L.-W. Chem. Sci. 2015, 6, 1265–1271. doi:10.1039/c4sc02596b
Return to citation in text: [1] [2] [3] -
Xiao, X.-Y.; Zhou, A.-H.; Shu, C.; Pan, F.; Li, T.; Ye, L.-W. Chem. – Asian J. 2015, 10, 1854–1858. doi:10.1002/asia.201500447
Return to citation in text: [1] [2] -
Zhu, X.-Q.; Yuan, H.; Sun, Q.; Zhou, B.; Han, X.-Q.; Zhang, Z.-X.; Lu, X.; Ye, L.-W. Green Chem. 2018, 20, 4287–4291. doi:10.1039/c8gc02051e
Return to citation in text: [1] -
Zhu, X.-Q.; Sun, Q.; Zhang, Z.-X.; Zhou, B.; Xie, P.-X.; Shen, W.-B.; Lu, X.; Zhou, J.-M.; Ye, L.-W. Chem. Commun. 2018, 54, 7435–7438. doi:10.1039/c8cc03140a
Return to citation in text: [1] -
Giri, S. S.; Liu, R.-S. Chem. Sci. 2018, 9, 2991–2995. doi:10.1039/c8sc00232k
Return to citation in text: [1] -
Cao, Z.; Gagosz, F. Angew. Chem., Int. Ed. 2013, 52, 9014–9018. doi:10.1002/anie.201304497
Return to citation in text: [1] -
Meng, X.; Guo, M.; Zhu, J.; Zhu, H.; Sun, X.; Tian, L.; Cao, Z. Eur. J. Org. Chem. 2019, 1952–1956. doi:10.1002/ejoc.201900118
Return to citation in text: [1] -
Cao, Z.; Zhu, H.; Meng, X.; Tian, L.; Sun, X.; Chen, G.; You, J. Chem. – Eur. J. 2016, 22, 9125–9129. doi:10.1002/chem.201601430
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Bevilacqua, E.; Gandolfo, V.; Giannini, G.; Guanti, G.; Musso, L.; Paravidino, M.; Riva, R. Bioorg. Med. Chem. 2008, 16, 3501–3518. doi:10.1016/j.bmc.2008.02.022
Return to citation in text: [1] -
Robins, R. K.; Revankar, G. R. Med. Res. Rev. 1985, 5, 273–296. doi:10.1002/med.2610050302
Return to citation in text: [1] -
Migawa, M. T.; Drach, J. C.; Townsend, L. B. J. Med. Chem. 2005, 48, 3840–3851. doi:10.1021/jm0402014
Return to citation in text: [1] -
Su, H.; Bao, M.; Huang, J.; Qiu, L.; Xu, X. Adv. Synth. Catal. 2019, 361, 826–831. doi:10.1002/adsc.201801425
Return to citation in text: [1] -
Kramer, S.; Dooleweerdt, K.; Lindhardt, A. T.; Rottländer, M.; Skrydstrup, T. Org. Lett. 2009, 11, 4208–4211. doi:10.1021/ol901565p
Return to citation in text: [1] -
Chen, Z.; Huang, J.; Wang, Z. J. Org. Chem. 2016, 81, 9308–9314. doi:10.1021/acs.joc.6b01891
Return to citation in text: [1] -
Zhu, J.; Wang, Q.; Meng, X.; Zhao, C.; Sun, X.; Tian, L.; Cao, Z. Eur. J. Org. Chem. 2019, 4066–4070. doi:10.1002/ejoc.201900604
Return to citation in text: [1]
| 31. | Zhou, A.-H.; He, Q.; Shu, C.; Yu, Y.-F.; Liu, S.; Zhao, T.; Zhang, W.; Lu, X.; Ye, L.-W. Chem. Sci. 2015, 6, 1265–1271. doi:10.1039/c4sc02596b |
| 41. | Migawa, M. T.; Drach, J. C.; Townsend, L. B. J. Med. Chem. 2005, 48, 3840–3851. doi:10.1021/jm0402014 |
| 42. | Su, H.; Bao, M.; Huang, J.; Qiu, L.; Xu, X. Adv. Synth. Catal. 2019, 361, 826–831. doi:10.1002/adsc.201801425 |
| 1. | Fang, G.; Bi, X. Chem. Soc. Rev. 2015, 44, 8124–8173. doi:10.1039/c5cs00027k |
| 2. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 15. | Porcel, S.; Echavarren, A. M. Angew. Chem., Int. Ed. 2007, 46, 2672–2676. doi:10.1002/anie.200605041 |
| 39. | Banfi, L.; Basso, A.; Bevilacqua, E.; Gandolfo, V.; Giannini, G.; Guanti, G.; Musso, L.; Paravidino, M.; Riva, R. Bioorg. Med. Chem. 2008, 16, 3501–3518. doi:10.1016/j.bmc.2008.02.022 |
| 12. | Luo, H.; Wu, G.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2015, 54, 14503–14507. doi:10.1002/anie.201507219 |
| 13. | Briones, J. F.; Davies, H. M. L. Org. Lett. 2011, 13, 3984–3987. doi:10.1021/ol201503j |
| 14. | Caballero, A.; Despagnet-Ayoub, E.; Díaz-Requejo, M. M.; Díaz-Rodríguez, A.; González-Núñez, M. E.; Mello, R.; Muñoz, B. K.; Ojo, W.-S.; Asensio, G.; Etienne, M.; Pérez, P. J. Science 2011, 332, 835–838. doi:10.1126/science.1204131 |
| 40. | Robins, R. K.; Revankar, G. R. Med. Res. Rev. 1985, 5, 273–296. doi:10.1002/med.2610050302 |
| 9. | Fernández, P.; Valdés, C.; Fañanás, F. J.; Rodríguez, F. J. Org. Chem. 2019, 84, 3184–3191. doi:10.1021/acs.joc.8b03081 |
| 10. | Wang, Q.; Zhang, L.; Yao, J.; Qiu, G.; Li, X.; Zhou, H. J. Org. Chem. 2018, 83, 4092–4098. doi:10.1021/acs.joc.7b03257 |
| 11. | Clarke, A. K.; Lynam, J. M.; Taylor, R. J. K.; Unsworth, W. P. ACS Catal. 2018, 8, 6844–6850. doi:10.1021/acscatal.8b00745 |
| 34. | Zhu, X.-Q.; Sun, Q.; Zhang, Z.-X.; Zhou, B.; Xie, P.-X.; Shen, W.-B.; Lu, X.; Zhou, J.-M.; Ye, L.-W. Chem. Commun. 2018, 54, 7435–7438. doi:10.1039/c8cc03140a |
| 35. | Giri, S. S.; Liu, R.-S. Chem. Sci. 2018, 9, 2991–2995. doi:10.1039/c8sc00232k |
| 3. | Han, G.; Xue, L.; Zhao, L.; Zhu, T.; Hou, J.; Song, Y.; Liu, Y. Adv. Synth. Catal. 2019, 361, 678–682. doi:10.1002/adsc.201801482 |
| 4. | Dabral, S.; Bayarmagnai, B.; Hermsen, M.; Schießl, J.; Mormul, V.; Hashmi, A. S. K.; Schaub, T. Org. Lett. 2019, 21, 1422–1425. doi:10.1021/acs.orglett.9b00156 |
| 5. | Yang, Y.; Antoni, P.; Zimmer, M.; Sekine, K.; Mulks, F. F.; Hu, L.; Zhang, L.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2019, 58, 5129–5133. doi:10.1002/anie.201812577 |
| 6. | Hashmi, A. S. K.; Schwarz, L.; Bats, J. W. J. Prakt. Chem. 2000, 342, 40–51. doi:10.1002/(sici)1521-3897(200001)342:1<40::aid-prac40>3.3.co;2-m |
| 7. | Zheng, S.-C.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2019, 58, 1494–1498. doi:10.1002/anie.201812654 |
| 8. | Su, X.; Chen, B.; Wang, S.; Chen, H.; Chen, C. ACS Catal. 2018, 8, 7760–7765. doi:10.1021/acscatal.8b02448 |
| 36. | Cao, Z.; Gagosz, F. Angew. Chem., Int. Ed. 2013, 52, 9014–9018. doi:10.1002/anie.201304497 |
| 37. | Meng, X.; Guo, M.; Zhu, J.; Zhu, H.; Sun, X.; Tian, L.; Cao, Z. Eur. J. Org. Chem. 2019, 1952–1956. doi:10.1002/ejoc.201900118 |
| 38. | Cao, Z.; Zhu, H.; Meng, X.; Tian, L.; Sun, X.; Chen, G.; You, J. Chem. – Eur. J. 2016, 22, 9125–9129. doi:10.1002/chem.201601430 |
| 19. | Guo, M.; Meng, X.; Zhao, Y.; Dong, Y.; Sun, X.; Tian, L.; Cao, Z. RSC Adv. 2019, 9, 2703–2707. doi:10.1039/c8ra09269a |
| 20. | Beeler, A. B.; Su, S.; Singleton, C. A.; Porco, J. A., Jr. J. Am. Chem. Soc. 2007, 129, 1413–1419. doi:10.1021/ja0674744 |
| 31. | Zhou, A.-H.; He, Q.; Shu, C.; Yu, Y.-F.; Liu, S.; Zhao, T.; Zhang, W.; Lu, X.; Ye, L.-W. Chem. Sci. 2015, 6, 1265–1271. doi:10.1039/c4sc02596b |
| 32. | Xiao, X.-Y.; Zhou, A.-H.; Shu, C.; Pan, F.; Li, T.; Ye, L.-W. Chem. – Asian J. 2015, 10, 1854–1858. doi:10.1002/asia.201500447 |
| 45. | Zhu, J.; Wang, Q.; Meng, X.; Zhao, C.; Sun, X.; Tian, L.; Cao, Z. Eur. J. Org. Chem. 2019, 4066–4070. doi:10.1002/ejoc.201900604 |
| 18. | Cao, Z.; Zhu, H.; Meng, X.; Tian, L.; Chen, G.; Sun, X.; You, J. J. Org. Chem. 2016, 81, 12401–12407. doi:10.1021/acs.joc.6b02529 |
| 33. | Zhu, X.-Q.; Yuan, H.; Sun, Q.; Zhou, B.; Han, X.-Q.; Zhang, Z.-X.; Lu, X.; Ye, L.-W. Green Chem. 2018, 20, 4287–4291. doi:10.1039/c8gc02051e |
| 17. | Liang, R.; Ma, T.; Zhu, S. Org. Lett. 2014, 16, 4412–4415. doi:10.1021/ol5017299 |
| 43. | Kramer, S.; Dooleweerdt, K.; Lindhardt, A. T.; Rottländer, M.; Skrydstrup, T. Org. Lett. 2009, 11, 4208–4211. doi:10.1021/ol901565p |
| 44. | Chen, Z.; Huang, J.; Wang, Z. J. Org. Chem. 2016, 81, 9308–9314. doi:10.1021/acs.joc.6b01891 |
| 16. | Wang, L.; Cai, S.; Xing, X.; Gao, Y.; Wang, T.; Wang, D. Z. Org. Lett. 2013, 15, 2362–2365. doi:10.1021/ol4006954 |
| 21. | Aguilar, E.; Santamaría, J. Org. Chem. Front. 2019, 6, 1513–1540. doi:10.1039/c9qo00243j |
| 22. | Tian, X.; Song, L.; Han, C.; Zhang, C.; Wu, Y.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2019, 21, 2937–2940. doi:10.1021/acs.orglett.9b01011 |
| 23. | Hsu, Y.-C.; Hsieh, S.-A.; Liu, R.-S. Chem. – Eur. J. 2019, 25, 5288–5297. doi:10.1002/chem.201806083 |
| 24. | Tian, X.; Song, L.; Rudolph, M.; Rominger, F.; Oeser, T.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2019, 58, 3589–3593. doi:10.1002/anie.201812002 |
| 25. | Allegue, D.; González, J.; Fernández, S.; Santamaría, J.; Ballesteros, A. Adv. Synth. Catal. 2019, 361, 758–768. doi:10.1002/adsc.201801484 |
| 26. | Song, L.; Tian, X.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Chem. Commun. 2019, 55, 9007–9010. doi:10.1039/c9cc04027g |
| 27. | Tian, X.; Song, L.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2019, 21, 4327–4330. doi:10.1021/acs.orglett.9b01501 |
| 28. | Tian, X.; Song, L.; Rudolph, M.; Wang, Q.; Song, X.; Rominger, F.; Hashmi, A. S. K. Org. Lett. 2019, 21, 1598–1601. doi:10.1021/acs.orglett.9b00140 |
| 29. | Jin, H.; Huang, L.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem. 2016, 128, 804–808. doi:10.1002/ange.201508309 |
| 30. | Jin, H.; Tian, B.; Song, X.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A. S. K. Angew. Chem. 2016, 128, 12880–12884. doi:10.1002/ange.201606043 |
| 21. | Aguilar, E.; Santamaría, J. Org. Chem. Front. 2019, 6, 1513–1540. doi:10.1039/c9qo00243j |
| 31. | Zhou, A.-H.; He, Q.; Shu, C.; Yu, Y.-F.; Liu, S.; Zhao, T.; Zhang, W.; Lu, X.; Ye, L.-W. Chem. Sci. 2015, 6, 1265–1271. doi:10.1039/c4sc02596b |
| 32. | Xiao, X.-Y.; Zhou, A.-H.; Shu, C.; Pan, F.; Li, T.; Ye, L.-W. Chem. – Asian J. 2015, 10, 1854–1858. doi:10.1002/asia.201500447 |
© 2019 Cao et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)