Abstract
One new aromadendrane-type sesquiterpenoid, namely ximaocavernosin P [(+)-1], and three new cadinane-type sesquiterpenoids, namely (+)-maninsigin D [(+)-4], (+)- and (−)-ximaocavernosin Q [(+)- and (−)-5], together with five related known ones [2, 3, (−)-4, 6, and 7], were isolated from the Hainan sponge Acanthella cavernosa. Compounds 4 and 5 were isolated as racemic forms, which were further separated to the corresponding enantiomers [(+)-4/(−)-4 and (+)-5/(−)-5], respectively, by using chiral-phase HPLC. The structures of new compounds were elucidated by extensive spectroscopic analysis and comparison with the reported data. In addition, the absolute configuration of optically pure (+)-1 and 2 were determined by time-dependent density functional theory/electronic circular dichroism (TDDFT-ECD) calculations or X-ray diffraction analysis. A plausible biosynthetic pathway of these sesquiterpenoids and their internal correlation were proposed and discussed. In an in vitro bioassay, (+)-aristolone (3) exhibited promising anti-inflammatory activity by the inhibition of LPS-induced TNF-α and CCL2 release in RAW 264.7 macrophages.

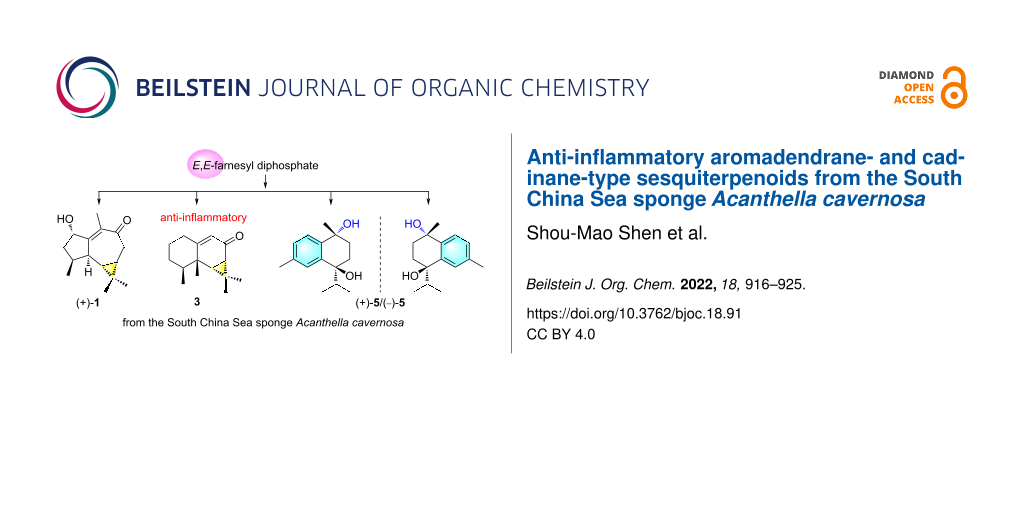
Graphical Abstract
Introduction
Marine sponges of the genus Acanthella (class Demospongiae, order Halichondrida, family Axinellidae) are one of the most common marine invertebrates natively distributed throughout tropical and subtropical regions of the Indo-Pacific Ocean, in particular, the South China Sea [1]. They are well-known producers of various nitrogenous sesquiterpenes and diterpenes with characteristic isocyano, isothiocyano, and formamido functionalities [2-5]. Many of these secondary metabolites merit further investigation due to their intriguing structural diversity and wide spectra of biological activities ranging from antifeedant, antifouling, and cytotoxic to antibiotic effects [3,5,6]. Acanthella sponges have thus attracted much attention from marine natural products chemists and pharmacologists. The title animal is the most chemically studied species among the Acanthella sponges. Till now, more than 100 secondary metabolites belonging to sesquiterpenoids and diterpenes [7,8], alkaloids [9], and steroids [10], have been isolated and characterized.
In connection with our continuing studies of Chinese marine invertebrates to search for novel and bioactive secondary metabolites, we have recently studied the sponge A. cavernosa collected from Ximao Island of Hainan Province, China, resulting in the isolation of fifteen new nitrogenous sesquiterpenoids, exemplified by (+)-ximaocavernosin A (8) (Figure 1) [11]. To accumulate more amounts of these sesquiterpenoids to perform more in-depth pharmacological screening, we carried out the chemical investigation of the same sample on a large scale manner (310 g, dry weight). As a result, besides the already reported nitrogenous sesquiterpenoids, nine non-nitrogenous sesquiterpenoids, including a new aromadendrane-type sesquiterpenoid [(+)-1] and three new cadinane-type sesquiterpenoids [(+)-4, (+)-5, and (−)-5], together with five related known ones [2, 3, (−)-4, 6, and 7] (Figure 1), were obtained. Herein, we report the isolation, chiral separation of racemic mixtures of 4 and 5, structural elucidation, plausible biosynthetic pathway, and biological evaluation of these isolated compounds.
![[1860-5397-18-91-1]](/bjoc/content/figures/1860-5397-18-91-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of compounds 1–8.
Figure 1: Chemical structures of compounds 1–8.
Results and Discussion
By the similar workup [11] of the Et2O-soluble portion of the dichloromethane/methanol 1:1 extract of the title animal, five optically pure sesquiterpenoids [(+)-1 (1.2 mg), 2 (4.1 mg), 3 (3.6 mg), 6 (1.3 mg), and 7 (0.5 mg)], as well as two non-optically pure compounds 4 (2.7 mg) and 5 (4.6 mg) were isolated.
Among them, known compounds 2, 3, 6, and 7 were readily identified as ent-4β,10α-dihydroxyaromadendrane (2) [12-14], (+)-aristolone (3) [15,16], cadalene (6) [17], and trans-4,5-dihydroxycorocalane (7) [18], respectively, by comparing their spectroscopic data and optical rotation values with those reported in the literature. In addition, the full structure of 2, which was previously isolated from the soft coral Sinularia mayi [12], was unambiguously confirmed by X-ray diffraction analysis using Cu Kα radiation (λ = 1.54178 Å) [Flack parameter: 0.00 (11)] (Figure 2), since it was crystallized from MeCN at 4 °C in the present study.
![[1860-5397-18-91-2]](/bjoc/content/figures/1860-5397-18-91-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP drawing of 2 (displacement ellipsoids are drawn at the 50% probability level).
Figure 2: ORTEP drawing of 2 (displacement ellipsoids are drawn at the 50% probability level).
Compound (+)-1 was obtained as an optically active colorless oil. Its molecular formula was deduced to be C15H22O2 on the basis of HRESIMS [quasi-molecular ion peak at m/z 235.1700 ([M + H]+, calcd for 235.1693)]. The IR spectrum displayed an absorption band of an α,β-unsaturated ketone group (1644 cm−1), as additionally supported by the UV absorption at λmax 245 nm (log ε 3.8). Careful analysis of the NMR spectra of (+)-1 (Table 1 and Figures S8–S13 in Supporting Information File 1) showed a close similarity with those of co-occurring 2 [12-14], indicating compound (+)-1 also being an aromadendrane-type sesquiterpenoid containing a gem-dimethylcyclopropyl unit [δH 0.80 (dd, J = 10.6, 9.6 Hz, H-6), 0.68 (m, H-7), 1.07 (s, 12-Me), 1.19 (s, 13-Me)]. Further literature survey revealed that the overall 1D and 2D NMR data of compound (+)-1 (Table 1), except the optical rotation signal (![[Graphic 1]](/bjoc/content/inline/1860-5397-18-91-i2.svg?max-width=637&scale=1.18182) +169.6 (c 0.12, CHCl3) for (+)-1,
+169.6 (c 0.12, CHCl3) for (+)-1, ![[Graphic 2]](/bjoc/content/inline/1860-5397-18-91-i3.svg?max-width=637&scale=1.18182) −186.0 (c 6.30, CHCl3) for (−)-1), were almost identical to those of 2β-hydroxyaromadendr-1(10)-en-9-one [(−)-1] [19], namely (1aR,5R,7R,7aS,7bR)-1,1a,2,5,6,7,7a,7b-octahydro-5-hydroxy-1,1,4,7-tetramethyl-3H-cycloprop[e]azulen-3-one, a biotransformation product of squamulosone [aromadendr-1(10)-en-9-one] by the fungus Curvularia lunata ATCC 12017 [19], indicating (+)-1 and (−)-1 were enantiomeric each other.
−186.0 (c 6.30, CHCl3) for (−)-1), were almost identical to those of 2β-hydroxyaromadendr-1(10)-en-9-one [(−)-1] [19], namely (1aR,5R,7R,7aS,7bR)-1,1a,2,5,6,7,7a,7b-octahydro-5-hydroxy-1,1,4,7-tetramethyl-3H-cycloprop[e]azulen-3-one, a biotransformation product of squamulosone [aromadendr-1(10)-en-9-one] by the fungus Curvularia lunata ATCC 12017 [19], indicating (+)-1 and (−)-1 were enantiomeric each other.
Table 1: 1H (J in Hz) and 13C NMR data of compounds (+)-1, 4, and 5.
| no. | (+)-1 | 4 | 5 | |||||
| δHa,b | δCa,c | δHb,d | δCc,d | δCa,e | δHb,d | δCc,d | δCa,e | |
| 1 | – | 163.7 s | – | 142.8 s | 141.7 | – | 140.4 s | 139.0 |
| 2 | 4.92 d (4.8) | 74.5 d | 7.47 d (8.4) | 126.6 d | 125.6 | 7.44 d (7.8) | 127.2 d | 125.8 |
| 3a | 1.92 overlap | 41.7 t | 7.08 dd (8.4, 1.8) | 129.2 d | 129.0 | 7.08 dd (7.8, 1.8) | 129.0 d | 128.9 |
| 3b | 1.56 dd (13.2, 4.8) | – | – | – | – | – | – | – |
| 4 | 2.68 m | 33.8 d | – | 137.6 s | 137.5 | – | 137.8 s | 137.9 |
| 5 | 2.73 dd (9.6, 9.6) | 43.7 d | 7.28 brs | 127.4 d | 126.0 | 7.33 brs | 127.9 d | 126.7 |
| 6 | 0.80 dd (10.6, 9.6) | 32.1 d | – | 140.9 s | 139.9 | – | 142.1 s | 140.6 |
| 7 | 0.68 m | 22.6 d | – | 75.0 s | 74.3 | – | 75.2 s | 74.5 |
| 8a | 2.38 dd (14.4, 11.4) | – | 1.92 ddd (15.6, 10.8, 3.0) | 28.4 t | 27.9 | 2.26 ddd (13.8, 10.2, 3.0) | 28.3 t | 26.9 |
| 8b | 2.84 dd (14.4, 4.8) | 42.1 t | 1.84 m | – | – | 1.70 ddd (13.8, 8.0, 3.0) | – | – |
| 9a | – | – | 2.09 ddd (15.6, 10.8, 3.0) | 35.6 t | 35.6 | 2.00 ddd (13.8, 10.2, 3.0) | 36.2 t | 35.0 |
| 9b | – | 201.9 s | 1.81 m | – | – | 1.92 ddd (13.8, 8.0, 1.2) | – | – |
| 10 | – | 134.1 s | – | 71.5 s | 71.0 | – | 70.3 s | 69.5 |
| 11 | – | 25.9 s | 2.41 m | 38.4 d | 37.2 | 2.35 m | 38.5 d | 37.6 |
| 12 | 1.07 s | 28.3 q | 0.62 d (6.6) | 16.5 q | 16.2 | 0.71 d (6.6) | 17.0 q | 16.5 |
| 13 | 1.19 s | 16.1 q | 1.08 d (6.6) | 18.9 q | 18.7 | 1.08 d (6.6) | 19.1 q | 18.9 |
| 14 | 1.94 d (1.8) | 14.8 q | 1.40 s | 30.2 q | 29.7 | 1.54 s | 31.2 q | 30.6 |
| 15 | 1.05 d (6.6) | 15.3 q | 2.31 s | 21.4 q | 21.4 | 2.32 s | 21.3 q | 21.5 |
aRecorded in CDCl3, chemical shifts refer to CHCl3 (δH 7.26, δC 77.2); brecorded at 600 MHz; crecorded at 125 MHz; drecorded in CD3OD, chemical shifts refer to CD3OD (δH 3.31, δC 49.0); erecorded at 150 MHz.
To secure the absolute configuration of optically pure (+)-1, a TDDFT-ECD calculation, which has proven to be a reliable tool for the absolute configuration determination of natural products with stereogenic centers near the chromophore groups [20], was applied, since there is an α,β-unsaturated ketone chromophore nearby C-5 and C-2 in compound (+)-1. Thus, the theoretical ECD spectrum of (+)-1 was calculated by the DFT calculation method at the b3lyp/6-311G** level of theory (see Supporting Information File 1 for details). A detailed comparison of the Boltzmann-averaged ECD spectrum with that of the experimental one (Figure 3) confirmed the absolute configuration of (+)-1 as 2S, 4S, 5R, 6S, 7S. Consequently, the structure of (+)-1 was assigned as depicted in Figure 1, and named (+)-ximaocavernosin P.
![[1860-5397-18-91-3]](/bjoc/content/figures/1860-5397-18-91-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Experimental and calculated ECD curves of (+)-1.
Figure 3: Experimental and calculated ECD curves of (+)-1.
Compounds 4 and 5 showed NMR data diagnostic of tetrahydronaphthalene-bearing cadinane-type sesquiterpenoids, in line with the co-occurring compounds 6 and 7. In addition, 4 and 5 as optically inactive white powder implied the possibility that both of them were present as racemic mixtures.
Compound 4 possessed a molecular formula of C15H22O2. Its structural elucidation was straightforward. The planar structure and relative configuration of 4 were immediately identified to be the same as those of (−)-maninsigin D [(−)-4] (Figure 1), a cadinane-type sesquiterpenoid of plant origin (leaves and stems of Manglietia insignis) [21], based on their identical 1H and 13C NMR data (Table 1 and Figures S16–S20 in Supporting Information File 1). Since the relative configuration of 4 (7S*, 10R*) was assigned, its absolute configuration was worth to be determined. However, the optical value at zero of 4 differed from that of (−)-maninsigin D [(−)-4: lit. ![[Graphic 3]](/bjoc/content/inline/1860-5397-18-91-i4.svg?max-width=637&scale=1.18182) −9.4 (c 0.70, MeOH)] [21], suggesting that 4 should be present as a racemic mixture.
−9.4 (c 0.70, MeOH)] [21], suggesting that 4 should be present as a racemic mixture.
Ximaocavernosin Q (5) had the same molecular formula (C15H22O2) as 4, implying compound 5 to be isomeric with 4. The IR absorption band at 3386 cm−1 suggested the presence of hydroxy groups. A careful analysis of its 1D and 2D NMR spectra revealed that the NMR spectroscopic features of compound 5 (Table 1 and Figures S24–S31 in Supporting Information File 1) extremely resembled those of 4. The main difference between them was obvious at C-10 and its neighboring carbons (e.g., C-1 and C-14). Considering the insignificant distinction between these chemical shift values (∆δ ≤ 2.4), compounds 4 and 5 were further analyzed using the same RP-HPLC conditions to examine whether they were the same compound (see Supporting Information File 1 for details). As a result, their distinct retention time [MeCN/H2O (35:65), 3.0 mL/min, 210 nm, 5: tR = 10.6 min, 4: tR = 17.6 min] (Figure S1, Supporting Information File 1) confirmed their different identities. Detailed analysis of their NMR spectroscopic data suggested an epimeric relationship between 5 and 4. Epimerization at C-10 caused upfield shifts for C-10 and C-1 (δC 71.5 and 142.8 in 4; δC 70.3 and 140.4 in 5), whereas the resonance of C-14 was shifted downfield from δC 30.2 in 4 to δC 31.2 in 5). Further, the diagnostic NOESY correlations between 14-Me (δH 1.54) and H-8b (δH 1.70)/H-9a (δH 2.00), between H-5 (δH 7.33) and H-11 (δH 2.35), as well as between H-8a (δH 2.26) and 12-Me (δH 0.71)/13-Me (δH 1.08), as shown in a computer-generated 3D drawing (Figure 4), determined the 7,10-trans relationship.
![[1860-5397-18-91-4]](/bjoc/content/figures/1860-5397-18-91-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: 1H-1H COSY, key HMBC correlations, and NOESY correlations of compound 5.
Figure 4: 1H-1H COSY, key HMBC correlations, and NOESY correlations of compound 5.
To complete the full structure, the next step was to determine the stereochemistry at C-7 and C-10 of compounds 4 and 5. Since both 4 and 5, as mentioned above, are respective racemic mixtures, chiral-phase HPLC was then applied to separate each of them (see Figures S2 and S5 in Supporting Information File 1). As expected, new (+)-maninsigin D [(+)-4, 0.8 mg] and known (−)-maninsigin D [(−)-4, 0.8 mg] were afforded from 4 with opposite optical values [(+)-4: ![[Graphic 4]](/bjoc/content/inline/1860-5397-18-91-i5.svg?max-width=637&scale=1.18182) +26.0 (c 0.08, MeOH); (−)-4:
+26.0 (c 0.08, MeOH); (−)-4: ![[Graphic 5]](/bjoc/content/inline/1860-5397-18-91-i6.svg?max-width=637&scale=1.18182) −24.2 (c 0.08, MeOH)] (Figures S3 and S4 in Supporting Information File 1) and the mirror image-like ECD curves (Figure 5). (+)-Ximaocavernosin Q [(+)-5, 0.8 mg] and (−)-ximaocavernosin Q [(−)-5, 0.7 mg] were obtained in the same way from 5 with
−24.2 (c 0.08, MeOH)] (Figures S3 and S4 in Supporting Information File 1) and the mirror image-like ECD curves (Figure 5). (+)-Ximaocavernosin Q [(+)-5, 0.8 mg] and (−)-ximaocavernosin Q [(−)-5, 0.7 mg] were obtained in the same way from 5 with ![[Graphic 6]](/bjoc/content/inline/1860-5397-18-91-i7.svg?max-width=637&scale=1.18182) values of +8.8 (c 0.08, MeOH) and −8.3 (c 0.07, MeOH) (Figures S6 and S7 in Supporting Information File 1), and mirrored ECD curves (Figure 5). Having obtained two pairs of optically pure enantiomers [(+)-4/(−)-4 and (+)-5/(−)-5], it is worth determining their absolute configuration. Unfortunately, our efforts to obtain suitable crystals for X-ray diffraction analysis were unsuccessful. The lack of secondary hydroxy groups in these molecules prevented the use of chemical approaches. In addition, their weak Cotton effects (Figure 5) also restricted the application of computational methods. Therefore, the absolute configuration for (+)-4/(−)-4 and (+)-5/(−)-5 remain undefined.
values of +8.8 (c 0.08, MeOH) and −8.3 (c 0.07, MeOH) (Figures S6 and S7 in Supporting Information File 1), and mirrored ECD curves (Figure 5). Having obtained two pairs of optically pure enantiomers [(+)-4/(−)-4 and (+)-5/(−)-5], it is worth determining their absolute configuration. Unfortunately, our efforts to obtain suitable crystals for X-ray diffraction analysis were unsuccessful. The lack of secondary hydroxy groups in these molecules prevented the use of chemical approaches. In addition, their weak Cotton effects (Figure 5) also restricted the application of computational methods. Therefore, the absolute configuration for (+)-4/(−)-4 and (+)-5/(−)-5 remain undefined.
![[1860-5397-18-91-5]](/bjoc/content/figures/1860-5397-18-91-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Experimental ECD curves of compounds (+)-4, (−)-4 (top), (+)-5, and (−)-5 (bottom).
Figure 5: Experimental ECD curves of compounds (+)-4, (−)-4 (top), (+)-5, and (−)-5 (bottom).
As mentioned in a review by William’s group [22], the enantiomeric natural products can arise from a single species or different genera and/or species. Several examples of enantiomers formation catalyzed by different terpene synthases were also reported. Jiang et al. characterized two new fungal bifunctional terpene synthases, FoFS and AtAS (identity 27.8%), that catalyzed the formation of a pair of enantiomeric sesterterpenes [23]. Two groups independently reported that the quiannulatene synthases EvQS [24] and AtTPS25 [25] (identity < 10%) from fungus and plant produced (+)- and (−)-quiannulatene, respectively. Interestingly, the cadinane-type sesquiterpenoids obtained from A. cavernosa generally exist in enantiomeric forms, such as our recently reported ximaocavernosins A–C, E, and G [11], as well as compounds 4 and 5 obtained in this present study. However, aromadendrane-type sesquiterpenoids from A. cavernosa are generally produced in optically pure forms, occasionally enantiomeric with others from a different origin, exemplified by (+)-ximaocavernosin P [(+)-1]. Further, these isothiocyano-containing enantiomers were usually isolated as scalable mixture (where one enantiomer predominates) with an enantiomeric excess of ca. 80% [11]. In contrast, the two neutral cadinane-type enantiomers (4 and 5) were produced as racemic mixture (1:1 ratio). Different enantiomeric ratios could explain the properties of the active sites in the corresponding terpene synthases, which remain unclear for further investigations [22].
The diversified structures of terpenes were constructed by terpene synthase [26] along with the post-modification enzymes, such as P450 enzymes and other oxygenases [27]. The plausible biosynthetic pathways of the isolated sesquiterpenoids 1–7 were proposed, as shown in Scheme 1. Aromadendrane- [(+)-1 and 2], aristolane- (3) and candinane-type sesquiterpenes [(+)-4/(−)-4, (+)-5/(−)-5, 6 and 7)] were all originated from E,E-farnesyl diphosphate (E,E-FPP) as their linear precursor (Scheme 1).
![[1860-5397-18-91-i1]](/bjoc/content/inline/1860-5397-18-91-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed cyclization pathway of terpene intermediates and plausible post-modifications of compounds 1–7.
Scheme 1: Proposed cyclization pathway of terpene intermediates and plausible post-modifications of compounds ...
The 6,7-bond formation was triggered by eliminating the pyrophosphate group of E,E-FPP yielding a monocyclic carbocation intermediate A, followed by the 6,11-closure via deprotonation to afford bicyclogermacrene (B) containing a gem-dimethylcyclopropyl unit [28]. Bicyclogermacrene (B) served as the branch point of aromadendrane cyclization and aristolane cyclization routes (Scheme 1, I). On one hand, the protonation on the double bond Δ1,10 of B initialized the aromadendrane cyclization, followed by 1,5-bond formation to yield carbocation intermediate C. Firstly, deprotonation occurred to form the double bond Δ1,10 and led to the formation of (−)-ledene (D) [29], on which the multiple-step oxidation happened at C-2 and C-9 to generate (+)-1. Secondly, the reaction is quenched by an H2O attach at carbocation C-10 to form (+)-globulol (E) [30], which was further oxidized to afford 2. On the other hand, the aristolane cyclization route is started from protonation on Δ4,5 of B and followed by 5,10-cyclization to give maaliane-type carbocation intermediate F. Then, a successive transformation involving the concerted 1,2-hydride and 1,2-methyl shifts led to aristolane-type carbocation intermediate G, which was further deprotonated to afford 9-aristolene (H) [29]. Multiple-step oxidation on H furnished the structure of 3.
The terpene cyclase catalyzed the cyclization of cadinene-type sesquiterpenes using FPP as the substrate, which is first isomerized to nerolidyl diphosphate (NPP), followed by the 6,7-bond formation to generate carbocation intermediate I (Scheme 1, II) [31]. Sequential 1,3-hydride shift and 1,6-cyclization occurred to afford cadinyl cation (J). Further 1,3-hydride shift and deprotonation on J resulted in cadina-1(6),4-diene (L) [32], the terpene precursor of compounds 4–6. Meanwhile, direct deprotonation of cadinyl cation (J) generates the double bond Δ9,10 of 1,5-cadinadiene (N) [32], the precursor of compound 7. Compounds 4–7 belong to the phenolic sesquiterpenes family, and the biosynthesis of the phenolic group has not yet been discovered. Phenolic sesquiterpenoids, for instance, illudacetalic acid and illudinine from Omphalotus olearius, were also discovered. Based on bioinformatics analysis, their aromatic rings were proposed to be constructed by putative P450 enzymes or oxidoreductase [33]. The Huang group characterized the function of a P450 enzyme CYP76AH1 which was responsible for the formation of the aromatic ring of ferruginol in the biosynthesis pathway of tanshinones [34]. Hence, we proposed that the oxidation occurred on L to furnish the aromatic ring of calamenene (M) [29], followed by the hydroxylation at C-7 and C-10 to give a pair of dihydroxy epimers 4 and 5. Compound 6 could be generated by the dehydration of 4/5 or by the multiple-step oxidation of M. Similarly, the plausible biosynthetic pathway of compound 7 could be proposed below. Multiple-step oxidation of N gave a phenolic sesquiterpene α-corocalene (O) [35]. An epoxidation at C-4/C-5 of O resulted in the formation of α-corocalene epoxide (P) [35], which was further hydrolyzed to generate the final product 7 [36].
In the anti-inflammatory assay, the transcriptional expression level of the representative inflammatory genes such as tumor necrosis factor-α (TNF-α) and C–C motif chemokine ligand 2 (CCL2) were investigated in lipopolysaccharide (LPS)-stimulated RAW 264.7 macrophages, using NF-κB inhibitor BAY-11-7082 as the positive control. Compound 3 displayed promising dose-dependent anti-inflammatory activity with the inhibition ratios of 74.1% in TNF-α and 64.1% in CCL2 at a concentration of 1 μM (Figure 6).
![[1860-5397-18-91-6]](/bjoc/content/figures/1860-5397-18-91-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Compound 3 reduced the mRNA levels of TNF-α (left) and CCL2 (right) in LPS-stimulated RAW264.7 macrophages. Data were normalized by DMSO group and are presented as means ± standard errors of the mean (n = 3). ###p < 0.001 vs the control group; ***p < 0.001 vs the DMSO group.
Figure 6: Compound 3 reduced the mRNA levels of TNF-α (left) and CCL2 (right) in LPS-stimulated RAW264.7 macr...
Conclusion
The continuous chemical investigation of the sponge A. cavernosa has resulted in the isolation of nine non-nitrogenous sesquiterpenoids, of which four optically pure compounds [(+)-1, (+)-4, (+)-5, and (−)-5] are new. Compounds (+)-1, 2, and 3 share a common gem-dimethylcyclopropyl unit, belonging to aromadendrane- and aristolane-type sesquiterpenoids, respectively. Whereas compounds 4–7 are a small group of cadinane-type sesquiterpenoids bearing a tetrahydronaphthalene system, which are produced in nature from isoprene units rather than by the much more common routes to aromatics involving acetate or shikimate [37]. In addition, the absolute stereochemistry of (+)-1 was unambiguously elucidated by TDDFT-ECD calculations, and the complete structure of 2 was confirmed for the first time by X-ray diffraction analysis using Cu Kα radiation. Two pairs of racemic cadinane-type sesquiterpenoids (4 and 5) were successfully separated, by chiral-phase HPLC, to their corresponding enantiomers [(+)-4/(−)-4 and (+)-5/(−)-5]. However, the determination of the absolute configuration of (+)-4/(−)-4 and (+)-5/(−)-5 requires further studies.
Notably, to our best knowledge, although a series of nitrogenous sesquiterpenoids from the genus Acanthella have been well documented in literature, less than six non-nitrogenous sesquiterpenoids have been reported from the title sponge [38-40]. The discovery of these sesquiterpenoids not only extended the members of the sesquiterpenoid family but also enriched the chemical diversity of the Acanthella sponges. The plausible biosynthetic pathway of isolated compounds 1–7 was also proposed. Up to date, no sesquiterpene cyclase has been characterized for aromadendrane and aristolane cyclization. The ubiquitous co-existence of enantiomeric cadinane-type sesquiterpenoids in the sponge revealed the unique catalytic properties of sponge-derived terpene synthases, which attracts our attention for further investigation of their structure–function relationship. Further elucidation of the cyclization mechanism and characterization of post-modification enzymes for the biosynthetic pathway of these sesquiterpenoids will provide insights into expanding the chemical space from marine sponges.
Experimental
General experimental procedure. The melting point was recorded using an SGW X-4 micro-melting point apparatus (Shanghai Precision Scientific Apparatuses Co., Ltd, Shanghai, China). Optical rotations were measured on a Perkin-Elmer 241MC polarimeter (PerkinElmer, Fremont, CA, USA). IR spectra were recorded on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA). ECD and UV spectra were measured with a JASCO J-815 instrument (JASCO, Japan). NMR spectra were measured on Bruker Avance III 500 and Bruker Avance III 600 instruments (Bruker Biospin AG, Rheinstetten, Germany) using TMS as an internal standard. Chemical shifts (δ) were reported with reference to the solvent signals, and coupling constants (J) were in Hz. HRESIMS spectra were recorded on an Agilent G6520 Q-TOF mass spectrometer, while HREIMS spectra were recorded on a Finnigan-MAT-95 mass spectrometer (Thermo Fisher Scientific, Waltham, USA). Commercial silica gel (Qingdao Haiyang Chemical Group Co., Ltd., Qingdao, China, 200–300 and 300–400 mesh), Sephadex LH-20 gel (Amersham Biosciences), and MCI gel (CHP 20P, 75–150 μm, Mitsubishi Chemical Co., Ltd., Tokyo, Japan) were used for column chromatography (CC). Reversed-phase (RP) HPLC was performed on an Agilent 1260 series liquid chromatography equipped with a DAD G1315D detector and an Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm). In contrast, chiral HPLC separation was operated on the chromatography equipped with CHIRALPAK IB N-3 column (5 μm, 4.6 × 250 mm). All solvents used for CC and HPLC were analytical grade (Shanghai Chemical Reagents Co., Ltd., Shanghai, China) and chromatographic grade (DiKMA Technologies Inc., CA, USA), respectively.
Animal materials. See ref [11].
Extraction and isolation. For the extraction and preliminary fractionation of the extract, see ref [11]. The extract was separated by MCI column chromatography (CC) eluted with a MeOH/H2O gradient solvent system (10% to 100%) to yield six fractions (A–F). The fraction B (570 mg) was then fractionated into subfractions B1–B6 by Sephadex LH-20 CC eluted with CH2Cl2. The subfraction B4 (96.2 mg) was further separated by Sephadex LH-20 CC (petroleum ether (PE)/CH2Cl2/MeOH 2:1:1) to give subfractions B4A and B4B. The subfraction B4B (81.0 mg) was chromatographed by RP-HPLC (35% to 50% MeCN in H2O, 3.0 mL/min), yielding compounds 5 (4.6 mg, tR = 9.9 min), 7 (0.5 mg, tR = 13.8 min), (+)-1 (1.2 mg, tR = 18.9 min) and 2 (4.1 mg, tR = 10.3 min). The subfraction B5 (80.1 mg) was purified by RP-HPLC (65% MeOH in H2O, 3.0 mL/min) to give 4 (2.7 mg, tR = 16.2 min). The subfraction C2 (66.5 mg) was successively separated by silica gel CC (PE/Et2O 5:1 to 1:1) and RP-HPLC (75% MeCN in H2O, 3.0 mL/min) to give 3 (3.6 mg, tR = 9.8 min). The subfraction E (34.9 mg) was successively separated by Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1) and RP-HPLC (55% MeCN in H2O, 3.0 mL/min) to afford compound 6 (1.3 mg, tR = 12.4 min).
The separation of racemic mixtures (4 and 5) was performed on chiral HPLC equipped with a CHIRALPAK IB N-3 column eluted with MeCN/H2O [4: 25:75, 5: 15:85, respectively, 1.0 mL/min] at room temperature, yielding (+)-4 (0.8 mg, tR = 18.4 min) and (−)-4 (0.8 mg, tR = 16.7 min), (+)-5 (0.8 mg, tR = 30.4 min), (−)-5 (0.7 mg, tR = 31.9 min), respectively.
(+)-Ximaocavernosin P [(+)-1]: colorless oil; ![[Graphic 7]](/bjoc/content/inline/1860-5397-18-91-i8.svg?max-width=637&scale=1.18182) +169.6 (c 0.12, CHCl3); UV (MeCN) λmax, nm (log ε): 245 (3.8) nm; ECD (MeCN) λ max, nm (Δε): 222 (−9.7), 257 (+7.2), 340 (+1.3); IR (KBr) νmax: 3435, 2956, 2927, 2872, 1644, 1383, 1261, 1109, 1037 cm−1; 1H and 13C NMR data, see Table 1; HRMS–ESI (m/z): [M + H]+ calcd for C15H23O2, 235.1693; found, 235.1700.
+169.6 (c 0.12, CHCl3); UV (MeCN) λmax, nm (log ε): 245 (3.8) nm; ECD (MeCN) λ max, nm (Δε): 222 (−9.7), 257 (+7.2), 340 (+1.3); IR (KBr) νmax: 3435, 2956, 2927, 2872, 1644, 1383, 1261, 1109, 1037 cm−1; 1H and 13C NMR data, see Table 1; HRMS–ESI (m/z): [M + H]+ calcd for C15H23O2, 235.1693; found, 235.1700.
ent-4β,10α-Dihydroxyaromadendrane (2): colorless crystal; mp 117.0–118.0 °C; ![[Graphic 8]](/bjoc/content/inline/1860-5397-18-91-i9.svg?max-width=637&scale=1.18182) +23.8 (c 0.40, MeOH).
+23.8 (c 0.40, MeOH).
(+)-Maninsigin D [(+)-4]: white powder; ![[Graphic 9]](/bjoc/content/inline/1860-5397-18-91-i10.svg?max-width=637&scale=1.18182) +26.0 (c 0.08, MeOH); ECD (MeCN) λmax, nm (Δε): 203 (−0.5), 218 (+0.2); IR (KBr) νmax: 2925, 1699, 1632, 1435, 1384, 1259, 1243, 1116, 1069 cm−1; 1H and 13C NMR data, see Table 1; HRMS–ESI (m/z): [M + H]+ calcd for C15H23O2, 235.1693; found, 235.1689.
+26.0 (c 0.08, MeOH); ECD (MeCN) λmax, nm (Δε): 203 (−0.5), 218 (+0.2); IR (KBr) νmax: 2925, 1699, 1632, 1435, 1384, 1259, 1243, 1116, 1069 cm−1; 1H and 13C NMR data, see Table 1; HRMS–ESI (m/z): [M + H]+ calcd for C15H23O2, 235.1693; found, 235.1689.
(+)-Ximaocavernosin Q [(+)-5]: white powder; ![[Graphic 10]](/bjoc/content/inline/1860-5397-18-91-i11.svg?max-width=637&scale=1.18182) +8.8 (c 0.08, MeOH); ECD (MeCN) λ max, nm (Δε): 207 (−0.3), 216 (+0.1); IR (KBr) νmax: 3386, 2961, 2925, 2874, 1463, 1383, 1130, 993, 939, 820 cm−1; 1H and 13C NMR data, see Table 1; HRMS–EI (m/z): M+ calcd for C15H22O2, 234.1614; found, 234.1618.
+8.8 (c 0.08, MeOH); ECD (MeCN) λ max, nm (Δε): 207 (−0.3), 216 (+0.1); IR (KBr) νmax: 3386, 2961, 2925, 2874, 1463, 1383, 1130, 993, 939, 820 cm−1; 1H and 13C NMR data, see Table 1; HRMS–EI (m/z): M+ calcd for C15H22O2, 234.1614; found, 234.1618.
(−)-Ximaocavernosin Q [(−)-5]: white powder; ![[Graphic 11]](/bjoc/content/inline/1860-5397-18-91-i12.svg?max-width=637&scale=1.18182) −8.3 (c 0.07, MeOH); ECD (MeCN) λ max, nm (Δε) 208 (+0.2); IR (KBr) νmax: 3386, 2961, 2925, 2874, 1463, 1383, 1130, 993, 939, 820 cm−1; 1H and 13C NMR data, see Table 1; HRMS–EI (m/z): M+ calcd for C15H22O2, 234.1614; found, 234.1618.
−8.3 (c 0.07, MeOH); ECD (MeCN) λ max, nm (Δε) 208 (+0.2); IR (KBr) νmax: 3386, 2961, 2925, 2874, 1463, 1383, 1130, 993, 939, 820 cm−1; 1H and 13C NMR data, see Table 1; HRMS–EI (m/z): M+ calcd for C15H22O2, 234.1614; found, 234.1618.
X-ray crystallographic analysis. Compound 2 was crystallized from MeCN at 4 °C. The crystallographic data for compound 2 was collected on a Bruker D8 Venture diffractometer using Cu Kα radiation (λ = 1.54178 Å). The collected data integration and reduction were processed with SAINT V8.37A software, and multi-scan absorption correction was performed using the SADABS program. The structure was solved using ShelXTL and refined on F2 by the full-matrix least-squares technique using the SHELXL-2015 program package. The crystallographic data has been uploaded to the Cambridge Crystallographic Data Centre with CCDC number 2173439 (2). The data could be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif. Details of the crystallographic data were shown in Supporting Information File 1.
Computational section. TDDFT-ECD calculations. Conformational searches were conducted using the torsional sampling (MCMM) method and the OPLS_2005 force field. Conformers above 1% population were reoptimized at the B3LYP/6-311G** level of theory with IEFPCM (Polarizable Continuum Model using the Integral Equation Formalism variant) solvent model for acetonitrile. For the resulting geometries, ECD spectra were obtained by TDDFT calculations performed with the B3LYP/6-311G** level of theory with IEFPCM solvent model for acetonitrile. Finally, the Boltzmann-averaged ECD spectra were obtained with SpecDis1.62 [41].
In vitro anti-inflammatory assay. RAW264.7 cells, a murine macrophage cell line, was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). RAW264.7 cells were cultured in DMEM (Dulbecco’s modified Eagle medium) supplemented with 10% fetal bovine serum, antibiotics (100 U/mL penicillin and 100 U/mL streptomycin), and maintained at 37 °C in a humidified incubator of 5% CO2. RAW264.7 cells (1.6 × 105/well) were seeded in a 24-well plate overnight for cell adherence. Cells were treated with LPS alone (100 ng/mL, dissolved in DMSO) or with compounds at the indicated concentrations for 24 h. Total RNA was harvested from cells using TRIzolTM reagent (Invitrogen, USA) after being washed thrice with cold PBS. The total RNA (1 μg) was transcribed into cDNA in 20 μL reactions using the PrimeScript RT reagent kit (ABclonal, China) and then diluted to 200 μL. Real-time quantitative PCR (qPCR) was performed using SYBR GREEN QPCR KIT (Bimake, B21202) on Agilent Mx3000P following the manufacturer’s instructions. β-ACTIN was used as the normalization control. All reactions were performed in triplicate. The NF-κB inhibitor Bay 11-7082 (5 μM) was used as a positive control.
Supporting Information
| Supporting Information File 1: HPLC chromatograms of 4 and 5, chiral separation of 4 and 5, X-ray crystallographic data for 2, spectra of compounds (+)-1, 4 and 5, TDDFT-ECD calculation of compound (+)-1. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Huang, Z.-G. Species and distribution of Marine life in China; China Ocean Press Publishing: Beijing, 2008.
Return to citation in text: [1] -
Edenborough, M. S.; Herbert, R. B. Nat. Prod. Rep. 1988, 5, 229–245. doi:10.1039/np9880500229
Return to citation in text: [1] -
Garson, M. J.; Simpson, J. S. Nat. Prod. Rep. 2004, 21, 164–179. doi:10.1039/b302359c
Return to citation in text: [1] [2] -
Emsermann, J.; Kauhl, U.; Opatz, T. Mar. Drugs 2016, 14, 16. doi:10.3390/md14010016
Return to citation in text: [1] -
Massarotti, A.; Brunelli, F.; Aprile, S.; Giustiniano, M.; Tron, G. C. Chem. Rev. 2021, 121, 10742–10788. doi:10.1021/acs.chemrev.1c00143
Return to citation in text: [1] [2] -
Patra, A.; Chang, C. W. J.; Scheuer, P. J.; Van Duyne, G. D.; Matsumoto, G. K.; Clardy, J. J. Am. Chem. Soc. 1984, 106, 7981–7983. doi:10.1021/ja00337a061
Return to citation in text: [1] -
Omar, S.; Albert, C.; Fanni, T.; Crews, P. J. Org. Chem. 1988, 53, 5971–5972. doi:10.1021/jo00260a034
Return to citation in text: [1] -
Wu, Q.; Chen, W.-T.; Li, S.-W.; Ye, J.-Y.; Huan, X.-J.; Gavagnin, M.; Yao, L.-G.; Wang, H.; Miao, Z.-H.; Li, X.-W.; Guo, Y.-W. Mar. Drugs 2019, 17, 56. doi:10.3390/md17010056
Return to citation in text: [1] -
Grkovic, T.; Blees, J. S.; Bayer, M. M.; Colburn, N. H.; Thomas, C. L.; Henrich, C. J.; Peach, M. L.; McMahon, J. B.; Schmid, T.; Gustafson, K. R. Mar. Drugs 2014, 12, 4593–4601. doi:10.3390/md12084593
Return to citation in text: [1] -
Qiu, Y.; Deng, Z. W.; Xu, M.; Li, Q.; Lin, W. H. Steroids 2008, 73, 1500–1504. doi:10.1016/j.steroids.2008.08.006
Return to citation in text: [1] -
Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Beechan, C. M.; Djerassi, C.; Eggert, H. Tetrahedron 1978, 34, 2503–2508. doi:10.1016/0040-4020(78)88378-1
Return to citation in text: [1] [2] [3] -
Nagashima, F.; Tanaka, H.; Toyota, M.; Hashimoto, T.; Kan, Y.; Takaoka, S.; Tori, M.; Asakawa, Y. Phytochemistry 1994, 36, 1425–1430. doi:10.1016/s0031-9422(00)89735-6
Return to citation in text: [1] [2] -
Sun, Z.-H.; Zhang, S.; Shi, J.; Hu, C.-Q. Chin. J. Org. Chem. 2004, 24, 806–810.
Return to citation in text: [1] [2] -
Piers, E.; Britton, R. W.; De Waal, W. Can. J. Chem. 1969, 47, 831–840. doi:10.1139/v69-132
Return to citation in text: [1] -
Harrigan, G. G.; Ahmad, A.; Baj, N.; Glass, T. E.; Gunatilaka, A. A. L.; Kingston, D. G. I. J. Nat. Prod. 1993, 56, 921–925. doi:10.1021/np50096a016
Return to citation in text: [1] -
Kitagawa, I.; Cui, Z.; Son, B. W.; Kobayashi, M.; Kyogoku, Y. Chem. Pharm. Bull. 1987, 35, 124–135. doi:10.1248/cpb.35.124
Return to citation in text: [1] -
Kuo, Y.-H.; Yu, M.-T. Chem. Pharm. Bull. 1999, 47, 1017–1019. doi:10.1248/cpb.47.1017
Return to citation in text: [1] -
Collins, D. O.; Buchanan, G. O.; Reynolds, W. F.; Reese, P. B. Phytochemistry 2001, 57, 377–383. doi:10.1016/s0031-9422(01)00060-7
Return to citation in text: [1] [2] -
Mándi, A.; Kurtán, T. Nat. Prod. Rep. 2019, 36, 889–918. doi:10.1039/c9np00002j
Return to citation in text: [1] -
Shang, S.-Z.; Kong, L.-M.; Yang, L.-P.; Jiang, J.; Huang, J.; Zhang, H.-B.; Shi, Y.-M.; Zhao, W.; Li, H.-L.; Luo, H.-R.; Li, Y.; Xiao, W.-L.; Sun, H.-D. Fitoterapia 2013, 84, 58–63. doi:10.1016/j.fitote.2012.10.010
Return to citation in text: [1] [2] -
Finefield, J. M.; Sherman, D. H.; Kreitman, M.; Williams, R. M. Angew. Chem., Int. Ed. 2012, 51, 4802–4836. doi:10.1002/anie.201107204
Return to citation in text: [1] [2] -
Jiang, L.; Zhang, X.; Sato, Y.; Zhu, G.; Minami, A.; Zhang, W.; Ozaki, T.; Zhu, B.; Wang, Z.; Wang, X.; Lv, K.; Zhang, J.; Wang, Y.; Gao, S.; Liu, C.; Hsiang, T.; Zhang, L.; Oikawa, H.; Liu, X. Org. Lett. 2021, 23, 4645–4650. doi:10.1021/acs.orglett.1c01361
Return to citation in text: [1] -
Okada, M.; Matsuda, Y.; Mitsuhashi, T.; Hoshino, S.; Mori, T.; Nakagawa, K.; Quan, Z.; Qin, B.; Zhang, H.; Hayashi, F.; Kawaide, H.; Abe, I. J. Am. Chem. Soc. 2016, 138, 10011–10018. doi:10.1021/jacs.6b05799
Return to citation in text: [1] -
Huang, A. C.; Kautsar, S. A.; Hong, Y. J.; Medema, M. H.; Bond, A. D.; Tantillo, D. J.; Osbourn, A. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6005–E6014. doi:10.1073/pnas.1705567114
Return to citation in text: [1] -
Christianson, D. W. Chem. Rev. 2017, 117, 11570–11648. doi:10.1021/acs.chemrev.7b00287
Return to citation in text: [1] -
Narita, K.; Chiba, R.; Minami, A.; Kodama, M.; Fujii, I.; Gomi, K.; Oikawa, H. Org. Lett. 2016, 18, 1980–1983. doi:10.1021/acs.orglett.6b00552
Return to citation in text: [1] -
Durán-Peña, M. J.; Botubol Ares, J. M.; Hanson, J. R.; Collado, I. G.; Hernández-Galán, R. Nat. Prod. Rep. 2015, 32, 1236–1248. doi:10.1039/c5np00024f
Return to citation in text: [1] -
Warmers, U.; Wihstutz, K.; Bülow, N.; Fricke, C.; König, W. A. Phytochemistry 1998, 49, 1723–1731. doi:10.1016/s0031-9422(98)00283-0
Return to citation in text: [1] [2] [3] -
Stodůlková, E.; Šulc, M.; Císařová, I.; Novák, P.; Kolařík, M.; Flieger, M. Folia Microbiol. 2008, 53, 15–22. doi:10.1007/s12223-008-0002-5
Return to citation in text: [1] -
Rinkel, J.; Rabe, P.; Garbeva, P.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 13593–13596. doi:10.1002/anie.201608042
Return to citation in text: [1] -
Ngo, K.-S.; Wong, W.-T.; Brown, G. D. J. Nat. Prod. 1999, 62, 549–553. doi:10.1021/np980289m
Return to citation in text: [1] [2] -
Wawrzyn, G. T.; Quin, M. B.; Choudhary, S.; López-Gallego, F.; Schmidt-Dannert, C. Chem. Biol. 2012, 19, 772–783. doi:10.1016/j.chembiol.2012.05.012
Return to citation in text: [1] -
Guo, J.; Zhou, Y. J.; Hillwig, M. L.; Shen, Y.; Yang, L.; Wang, Y.; Zhang, X.; Liu, W.; Peters, R. J.; Chen, X.; Zhao, Z. K.; Huang, L. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12108–12113. doi:10.1073/pnas.1218061110
Return to citation in text: [1] -
Weyerstahl, P.; Marschall, H.; Thefeld, K.; Subba, G. C. Flavour Fragrance J. 1998, 13, 377–388. doi:10.1002/(sici)1099-1026(199811/12)13:6<377::aid-ffj755>3.0.co;2-f
Return to citation in text: [1] [2] -
Wu, Q.; Li, S.-W.; Xu, H.; Wang, H.; Hu, P.; Zhang, H.; Luo, C.; Chen, K.-X.; Nay, B.; Guo, Y.-W.; Li, X.-W. Angew. Chem., Int. Ed. 2020, 59, 12105–12112. doi:10.1002/anie.202003643
Return to citation in text: [1] -
Dewick, P. M. Medicinal natural products: a biosynthetic approach; John Wiley & Sons: Chichester, UK, 2002.
Return to citation in text: [1] -
Okino, T.; Yoshimura, E.; Hirota, H.; Fusetani, N. J. Nat. Prod. 1996, 59, 1081–1083. doi:10.1021/np960496r
Return to citation in text: [1] -
Hirota, H.; Tomono, Y.; Fusetani, N. Tetrahedron 1996, 52, 2359–2368. doi:10.1016/0040-4020(95)01079-3
Return to citation in text: [1] -
Nogata, Y.; Yoshimura, E.; Shinshima, K.; Kitano, Y.; Sakaguchi, I. Biofouling 2003, 19 (Suppl. 1), 193–196. doi:10.1080/0892701031000065963
Return to citation in text: [1] -
Wu, Q.; Ye, F.; Li, X.-L.; Liang, L.-F.; Sun, J.; Sun, H.; Guo, Y.-W.; Wang, H. J. Org. Chem. 2019, 84, 3083–3092. doi:10.1021/acs.joc.8b02912
Return to citation in text: [1]
| 27. | Narita, K.; Chiba, R.; Minami, A.; Kodama, M.; Fujii, I.; Gomi, K.; Oikawa, H. Org. Lett. 2016, 18, 1980–1983. doi:10.1021/acs.orglett.6b00552 |
| 28. | Durán-Peña, M. J.; Botubol Ares, J. M.; Hanson, J. R.; Collado, I. G.; Hernández-Galán, R. Nat. Prod. Rep. 2015, 32, 1236–1248. doi:10.1039/c5np00024f |
| 29. | Warmers, U.; Wihstutz, K.; Bülow, N.; Fricke, C.; König, W. A. Phytochemistry 1998, 49, 1723–1731. doi:10.1016/s0031-9422(98)00283-0 |
| 1. | Huang, Z.-G. Species and distribution of Marine life in China; China Ocean Press Publishing: Beijing, 2008. |
| 9. | Grkovic, T.; Blees, J. S.; Bayer, M. M.; Colburn, N. H.; Thomas, C. L.; Henrich, C. J.; Peach, M. L.; McMahon, J. B.; Schmid, T.; Gustafson, K. R. Mar. Drugs 2014, 12, 4593–4601. doi:10.3390/md12084593 |
| 19. | Collins, D. O.; Buchanan, G. O.; Reynolds, W. F.; Reese, P. B. Phytochemistry 2001, 57, 377–383. doi:10.1016/s0031-9422(01)00060-7 |
| 34. | Guo, J.; Zhou, Y. J.; Hillwig, M. L.; Shen, Y.; Yang, L.; Wang, Y.; Zhang, X.; Liu, W.; Peters, R. J.; Chen, X.; Zhao, Z. K.; Huang, L. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12108–12113. doi:10.1073/pnas.1218061110 |
| 7. | Omar, S.; Albert, C.; Fanni, T.; Crews, P. J. Org. Chem. 1988, 53, 5971–5972. doi:10.1021/jo00260a034 |
| 8. | Wu, Q.; Chen, W.-T.; Li, S.-W.; Ye, J.-Y.; Huan, X.-J.; Gavagnin, M.; Yao, L.-G.; Wang, H.; Miao, Z.-H.; Li, X.-W.; Guo, Y.-W. Mar. Drugs 2019, 17, 56. doi:10.3390/md17010056 |
| 19. | Collins, D. O.; Buchanan, G. O.; Reynolds, W. F.; Reese, P. B. Phytochemistry 2001, 57, 377–383. doi:10.1016/s0031-9422(01)00060-7 |
| 29. | Warmers, U.; Wihstutz, K.; Bülow, N.; Fricke, C.; König, W. A. Phytochemistry 1998, 49, 1723–1731. doi:10.1016/s0031-9422(98)00283-0 |
| 3. | Garson, M. J.; Simpson, J. S. Nat. Prod. Rep. 2004, 21, 164–179. doi:10.1039/b302359c |
| 5. | Massarotti, A.; Brunelli, F.; Aprile, S.; Giustiniano, M.; Tron, G. C. Chem. Rev. 2021, 121, 10742–10788. doi:10.1021/acs.chemrev.1c00143 |
| 6. | Patra, A.; Chang, C. W. J.; Scheuer, P. J.; Van Duyne, G. D.; Matsumoto, G. K.; Clardy, J. J. Am. Chem. Soc. 1984, 106, 7981–7983. doi:10.1021/ja00337a061 |
| 12. | Beechan, C. M.; Djerassi, C.; Eggert, H. Tetrahedron 1978, 34, 2503–2508. doi:10.1016/0040-4020(78)88378-1 |
| 32. | Ngo, K.-S.; Wong, W.-T.; Brown, G. D. J. Nat. Prod. 1999, 62, 549–553. doi:10.1021/np980289m |
| 2. | Edenborough, M. S.; Herbert, R. B. Nat. Prod. Rep. 1988, 5, 229–245. doi:10.1039/np9880500229 |
| 3. | Garson, M. J.; Simpson, J. S. Nat. Prod. Rep. 2004, 21, 164–179. doi:10.1039/b302359c |
| 4. | Emsermann, J.; Kauhl, U.; Opatz, T. Mar. Drugs 2016, 14, 16. doi:10.3390/md14010016 |
| 5. | Massarotti, A.; Brunelli, F.; Aprile, S.; Giustiniano, M.; Tron, G. C. Chem. Rev. 2021, 121, 10742–10788. doi:10.1021/acs.chemrev.1c00143 |
| 12. | Beechan, C. M.; Djerassi, C.; Eggert, H. Tetrahedron 1978, 34, 2503–2508. doi:10.1016/0040-4020(78)88378-1 |
| 13. | Nagashima, F.; Tanaka, H.; Toyota, M.; Hashimoto, T.; Kan, Y.; Takaoka, S.; Tori, M.; Asakawa, Y. Phytochemistry 1994, 36, 1425–1430. doi:10.1016/s0031-9422(00)89735-6 |
| 14. | Sun, Z.-H.; Zhang, S.; Shi, J.; Hu, C.-Q. Chin. J. Org. Chem. 2004, 24, 806–810. |
| 33. | Wawrzyn, G. T.; Quin, M. B.; Choudhary, S.; López-Gallego, F.; Schmidt-Dannert, C. Chem. Biol. 2012, 19, 772–783. doi:10.1016/j.chembiol.2012.05.012 |
| 12. | Beechan, C. M.; Djerassi, C.; Eggert, H. Tetrahedron 1978, 34, 2503–2508. doi:10.1016/0040-4020(78)88378-1 |
| 13. | Nagashima, F.; Tanaka, H.; Toyota, M.; Hashimoto, T.; Kan, Y.; Takaoka, S.; Tori, M.; Asakawa, Y. Phytochemistry 1994, 36, 1425–1430. doi:10.1016/s0031-9422(00)89735-6 |
| 14. | Sun, Z.-H.; Zhang, S.; Shi, J.; Hu, C.-Q. Chin. J. Org. Chem. 2004, 24, 806–810. |
| 17. | Kitagawa, I.; Cui, Z.; Son, B. W.; Kobayashi, M.; Kyogoku, Y. Chem. Pharm. Bull. 1987, 35, 124–135. doi:10.1248/cpb.35.124 |
| 31. | Rinkel, J.; Rabe, P.; Garbeva, P.; Dickschat, J. S. Angew. Chem., Int. Ed. 2016, 55, 13593–13596. doi:10.1002/anie.201608042 |
| 11. | Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676 |
| 18. | Kuo, Y.-H.; Yu, M.-T. Chem. Pharm. Bull. 1999, 47, 1017–1019. doi:10.1248/cpb.47.1017 |
| 32. | Ngo, K.-S.; Wong, W.-T.; Brown, G. D. J. Nat. Prod. 1999, 62, 549–553. doi:10.1021/np980289m |
| 11. | Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676 |
| 30. | Stodůlková, E.; Šulc, M.; Císařová, I.; Novák, P.; Kolařík, M.; Flieger, M. Folia Microbiol. 2008, 53, 15–22. doi:10.1007/s12223-008-0002-5 |
| 10. | Qiu, Y.; Deng, Z. W.; Xu, M.; Li, Q.; Lin, W. H. Steroids 2008, 73, 1500–1504. doi:10.1016/j.steroids.2008.08.006 |
| 15. | Piers, E.; Britton, R. W.; De Waal, W. Can. J. Chem. 1969, 47, 831–840. doi:10.1139/v69-132 |
| 16. | Harrigan, G. G.; Ahmad, A.; Baj, N.; Glass, T. E.; Gunatilaka, A. A. L.; Kingston, D. G. I. J. Nat. Prod. 1993, 56, 921–925. doi:10.1021/np50096a016 |
| 29. | Warmers, U.; Wihstutz, K.; Bülow, N.; Fricke, C.; König, W. A. Phytochemistry 1998, 49, 1723–1731. doi:10.1016/s0031-9422(98)00283-0 |
| 21. | Shang, S.-Z.; Kong, L.-M.; Yang, L.-P.; Jiang, J.; Huang, J.; Zhang, H.-B.; Shi, Y.-M.; Zhao, W.; Li, H.-L.; Luo, H.-R.; Li, Y.; Xiao, W.-L.; Sun, H.-D. Fitoterapia 2013, 84, 58–63. doi:10.1016/j.fitote.2012.10.010 |
| 20. | Mándi, A.; Kurtán, T. Nat. Prod. Rep. 2019, 36, 889–918. doi:10.1039/c9np00002j |
| 35. | Weyerstahl, P.; Marschall, H.; Thefeld, K.; Subba, G. C. Flavour Fragrance J. 1998, 13, 377–388. doi:10.1002/(sici)1099-1026(199811/12)13:6<377::aid-ffj755>3.0.co;2-f |
| 21. | Shang, S.-Z.; Kong, L.-M.; Yang, L.-P.; Jiang, J.; Huang, J.; Zhang, H.-B.; Shi, Y.-M.; Zhao, W.; Li, H.-L.; Luo, H.-R.; Li, Y.; Xiao, W.-L.; Sun, H.-D. Fitoterapia 2013, 84, 58–63. doi:10.1016/j.fitote.2012.10.010 |
| 35. | Weyerstahl, P.; Marschall, H.; Thefeld, K.; Subba, G. C. Flavour Fragrance J. 1998, 13, 377–388. doi:10.1002/(sici)1099-1026(199811/12)13:6<377::aid-ffj755>3.0.co;2-f |
| 36. | Wu, Q.; Li, S.-W.; Xu, H.; Wang, H.; Hu, P.; Zhang, H.; Luo, C.; Chen, K.-X.; Nay, B.; Guo, Y.-W.; Li, X.-W. Angew. Chem., Int. Ed. 2020, 59, 12105–12112. doi:10.1002/anie.202003643 |
| 22. | Finefield, J. M.; Sherman, D. H.; Kreitman, M.; Williams, R. M. Angew. Chem., Int. Ed. 2012, 51, 4802–4836. doi:10.1002/anie.201107204 |
| 26. | Christianson, D. W. Chem. Rev. 2017, 117, 11570–11648. doi:10.1021/acs.chemrev.7b00287 |
| 11. | Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676 |
| 41. | Wu, Q.; Ye, F.; Li, X.-L.; Liang, L.-F.; Sun, J.; Sun, H.; Guo, Y.-W.; Wang, H. J. Org. Chem. 2019, 84, 3083–3092. doi:10.1021/acs.joc.8b02912 |
| 11. | Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676 |
| 24. | Okada, M.; Matsuda, Y.; Mitsuhashi, T.; Hoshino, S.; Mori, T.; Nakagawa, K.; Quan, Z.; Qin, B.; Zhang, H.; Hayashi, F.; Kawaide, H.; Abe, I. J. Am. Chem. Soc. 2016, 138, 10011–10018. doi:10.1021/jacs.6b05799 |
| 11. | Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676 |
| 25. | Huang, A. C.; Kautsar, S. A.; Hong, Y. J.; Medema, M. H.; Bond, A. D.; Tantillo, D. J.; Osbourn, A. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6005–E6014. doi:10.1073/pnas.1705567114 |
| 11. | Shen, S.-M.; Zhang, Z.-Y.; Yao, L.-G.; Wang, J.-R.; Guo, Y.-W.; Li, X.-W. Chin. J. Chem. 2022, 40, 235–246. doi:10.1002/cjoc.202100676 |
| 22. | Finefield, J. M.; Sherman, D. H.; Kreitman, M.; Williams, R. M. Angew. Chem., Int. Ed. 2012, 51, 4802–4836. doi:10.1002/anie.201107204 |
| 37. | Dewick, P. M. Medicinal natural products: a biosynthetic approach; John Wiley & Sons: Chichester, UK, 2002. |
| 23. | Jiang, L.; Zhang, X.; Sato, Y.; Zhu, G.; Minami, A.; Zhang, W.; Ozaki, T.; Zhu, B.; Wang, Z.; Wang, X.; Lv, K.; Zhang, J.; Wang, Y.; Gao, S.; Liu, C.; Hsiang, T.; Zhang, L.; Oikawa, H.; Liu, X. Org. Lett. 2021, 23, 4645–4650. doi:10.1021/acs.orglett.1c01361 |
| 38. | Okino, T.; Yoshimura, E.; Hirota, H.; Fusetani, N. J. Nat. Prod. 1996, 59, 1081–1083. doi:10.1021/np960496r |
| 39. | Hirota, H.; Tomono, Y.; Fusetani, N. Tetrahedron 1996, 52, 2359–2368. doi:10.1016/0040-4020(95)01079-3 |
| 40. | Nogata, Y.; Yoshimura, E.; Shinshima, K.; Kitano, Y.; Sakaguchi, I. Biofouling 2003, 19 (Suppl. 1), 193–196. doi:10.1080/0892701031000065963 |
© 2022 Shen et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.