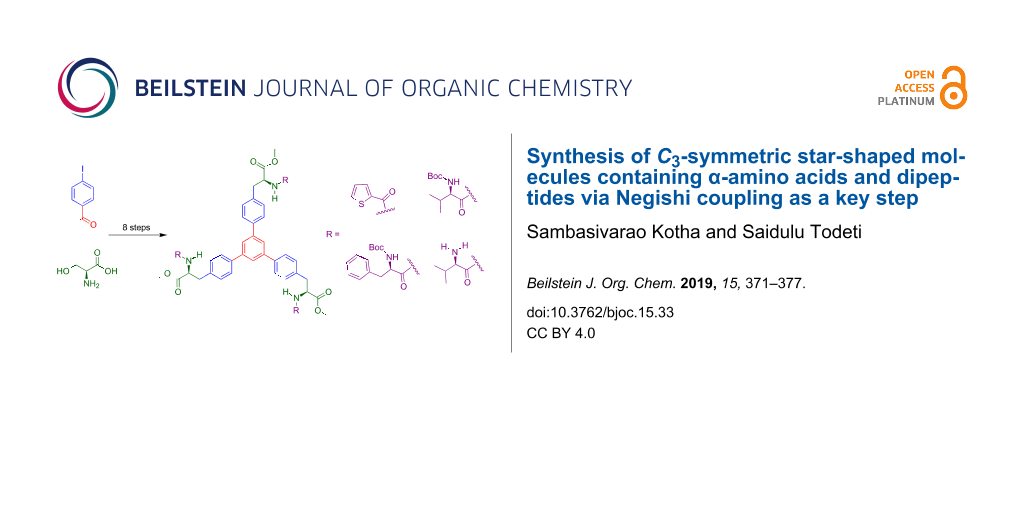
Abstract
We demonstrate a new synthetic strategy toward star-shaped C3-symmetric molecules containing α-amino acid (AAA) derivatives and dipeptides. In this regard, trimerization and Negishi cross-coupling reactions are used as the key steps starting from readily available 4’-iodoacetophenone and L-serine. These C3-symmetric molecules containing AAA moieties are useful to design new ligands suitable for asymmetric synthesis and peptide dendrimers.

Graphical Abstract
Introduction
Optically active C3-symmetric molecules are valuable synthons to design dendrimers, chiral ligands, polymers, and supramolecules [1-4]. In this regard, 1,3,5-triarylbenzene derivatives are helpful to design star-shaped α-amino acids (AAAs) and they play an important role in biological systems. The tumor necrosis factor (TNF) superfamily belongs to trimeric ligands that form in the shape of C3-symmetric molecules [5]. Trimeric proteins containing star-shaped compounds are also involved in the complex interactions between cells and pathogens, e.g., the human immunodeficiency virus (HIV-1) [6]. The HIV-1 envelope protein is present as a C3-symmetric trimer on the viruses’ surface [7], and the virus entry into the cell is mediated by its interactions with cellular receptors. To explore the structural and chemical nature of protein–protein interactions, synthetic peptides and unnatural AAAs [8-15] can be useful as molecular tools. Moreover, C3-symmetric peptides are valuable in studying the molecular interactions involving proteins that are derived from trimers and synthetic access to such amino acids is vital. In this regard, new star-shaped C3-symmetric molecules [16-26] have been used in photovoltaics [27,28], organic light-emitting diodes (OLEDs) [29,30], organic field-effect transistors (OFETs) [31,32] and electroluminescent devices [33]. To address these challenges, we [34] and others [35,36] have synthesized functionalized C3-symmetric molecules containing amino acids and peptides.
The Negishi cross coupling [37,38] is a reliable synthetic method, which involves palladium or nickel-catalyzed coupling of organozinc reagents [39,40] with various halo derivatives (e.g., aryl, vinyl, benzyl, or allyl) and has a broad scope to assemble diverse targets. This reaction was first reported in 1977, and it is an elegant and versatile method that allows the preparation of biaryls and olefins in good yields. To the best of our knowledge only a limited number of reports is available for the synthesis of C3-symmetric peptides (Figure 1) [8,41]. To fill this gap, we have explored a new synthetic strategy to star-shaped C3-symmetric AAA derivatives and peptides by using trimerization and the Negishi cross coupling as key steps.
![[1860-5397-15-33-1]](/bjoc/content/figures/1860-5397-15-33-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Exemplar C3-symmetric peptide scaffolds reported in the literature.
Figure 1: Exemplar C3-symmetric peptide scaffolds reported in the literature.
Results and Discussion
The required zinc insertion compound 7 was prepared from L-serine (3). Thus, commercially available L-serine (3) was treated with acetyl chloride in methanol to give methyl ester 4, which was subjected to N-Boc protection with di-tert-butyl dicarbonate (Boc2O) and triethylamine in tetrahydrofuran (THF) to obtain the N-Boc-serine methyl ester (5) in 93% yield [42]. Afterwards, the protected methyl ester 5 was subjected to iodination in the presence of iodine (I2), triphenylphosphane (PPh3) and imidazole in CH2Cl2 at 0 °C to deliver the iodo derivative 6 in 63% yield [43,44]. Finally, the iodo compound 6 was treated with freshly activated Zn in DMF at room temperature to afford the zinc insertion product 7 (Scheme 1) [43].
![[1860-5397-15-33-i1]](/bjoc/content/inline/1860-5397-15-33-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of compound 7 from L-serine (3).
Scheme 1: Preparation of compound 7 from L-serine (3).
With the organozinc compound 7 at hand we turned to the synthesis of the halide component for the attempted Negishi coupling. For this 4-iodoacetophenone (8) was treated with silicon tetrachloride and ethanol (SiCl4/EtOH) at room temperature for 6 h to produce the iodonated trimerized product 9 in 71% yield (Scheme 2) [45,46].
![[1860-5397-15-33-i2]](/bjoc/content/inline/1860-5397-15-33-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of the trimerized product 9.
Scheme 2: Preparation of the trimerized product 9.
Then, the organozinc reagent 7 was coupled with triiodo derivative 9 in the presence of tetrakis(triphenylphosphane)palladium(0) (Pd(PPh3)4) as catalyst to provide the Negishi coupling product 10 (68%). Having the trimeric AAA derivative 10 in hand, it was treated with trifluoroacetic acid (TFA) in CH2Cl2 (1:1) at room temperature for 1 h to deliver the Boc-deprotected compound. Then, without further purification the deprotected product was directly treated with thiophene-2-carboxylic acid in the presence of 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIPEA) in CH2Cl2 at room temperature for 5 h to give trimer 11 in 86% yield (Scheme 3).
![[1860-5397-15-33-i3]](/bjoc/content/inline/1860-5397-15-33-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of compound 11 via Negishi cross-coupling reaction.
Scheme 3: Synthesis of compound 11 via Negishi cross-coupling reaction.
In addition, different amino acids were incorporated in the star-shaped molecule. In this regard, the Negishi cross-coupling product 10 was treated with TFA in CH2Cl2 at room temperature for 1 h to give the Boc-deprotection product, which was directly treated with Boc-Val-OH or Boc-Phe-OH in the presence of HBTU and DIPEA in CH2Cl2 at room temperature for 5 h to give trimeric derivatives 12 (73%) and 13 (81%), respectively. Further, trimer 12 was subjected to another Boc-deprotection to give the tris-amine 14 in 95% yield (Scheme 4).
![[1860-5397-15-33-i4]](/bjoc/content/inline/1860-5397-15-33-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of C3-symmetric trimers 12, 13 and 14.
Scheme 4: Synthesis of C3-symmetric trimers 12, 13 and 14.
Conclusion
We have demonstrated a simple synthetic strategy toward star-shaped molecules containing unusual AAA units through cyclotrimerization and Negishi cross-coupling reaction as key steps under operationally simple reaction conditions. Here, we have used the readily available starting materials 4-iodoacetophenone (8) and L-serine (3). The C3-symmetric building blocks prepared were coupled with different AAAs to produce the C3-symmetric dipeptide trimers.
Experimental
General procedure
Commercially available starting materials were used without further purification. Analytical thin layer chromatography (TLC) was performed on 7.5 × 2.5 cm glass plates coated with Acme’s silica gel GF254 (containing 13% calcium sulfate as binder) by using a suitable mixture of ethyl acetate and petroleum ether for development. The Negishi coupling was performed in oven-dried glassware under argon or nitrogen atmosphere and the transfer of moisture-sensitive materials was carried out in a glovebox by using standard syringe–septum techniques. All purchased solvents (CH2Cl2, THF, acetonitrile, and DMF) were dried over calcium hydride (CaH2) or sodium. Column chromatography was performed by using Acme’s silica gel (100–200 mesh) with an appropriate mixture of ethyl acetate, petroleum ether methanol and dichloromethane. The coupling constants (J) are given in hertz (Hz) and chemical shifts are denoted in parts per million (ppm) downfield from internal standard, tetramethylsilane (TMS). The abbreviations, s, d, t, q, m, and dd refer to singlet, doublet, triplet, quartet, multiplet, and doublet of doublets, respectively. Infrared (IR) spectra were recorded on a Nicolet Impact-400 FTIR spectrometer. Specific rotation experiments were measured at 589 nm (Na) and 25 °C (HPLC, CHCl3 stabilized with 0.7–1.0% ethanol). Proton nuclear magnetic resonance (1H NMR, 400 MHz and 500 MHz) spectra and carbon nuclear magnetic resonance (13C NMR, 100 MHz and 125 MHz) spectra were recorded on a Bruker spectrometer. The high-resolution mass measurements were carried out by using electrospray ionization (ESI) spectrometer. Melting points were recorded on a Veego melting point apparatus.
Negishi coupling product 10
Zinc (Zn) dust was activated by using 3 M aq HCl, then filtered and washed with water (until neutral pH) followed by acetone. Large particles were crushed until a fine powder was formed and transferred into a round-bottomed flask and dried under vacuum with heating and the flask was filled with nitrogen. A portion of the activated Zn dust (500 mg, 7.65 mmol, 3 equiv) was cooled to room temperature. Then, iodo compound 6 (842 mg, 2.55 mmol) was dissolved in DMF (10 mL) and added dropwise to the freshly activated Zn powder under a nitrogen atmosphere and the suspension was stirred at room temperature for 3 h. After completion of Zn insertion reaction, stirring was stopped and the solid was allowed to settle down. The supernatant was carefully transferred to a suspension of triiodo derivative 9 (500 mg, 0.73 mmol) in DMF (10 mL) at room temperature. Five mol % tetrakis(triphenylphosphane)palladium (Pd(PPh3)4) was added to this mixture under inert atmosphere and the reaction mixture stirred at 80 °C for 12 h. The reaction mixture was cooled to room temperature and washed with water, brine (3 × 15 mL), 1 M aq Na2S2O3 solution and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over Na2SO4 and concentrated at reduced pressure. The crude was purified by silica gel column chromatography (30% ethyl acetate/petroleum ether) to afford the Negishi coupling product 10 (458 mg, 68%) as a colorless solid. Rf = 0.73 (3:7 ethyl acetate/petroleum ether), [α]D25 +7.78 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 3H), 7.64 (d, J = 8.0 Hz, 6H), 7.28 (d, J = 8.0 Hz, 6H), 5.14 (d, J = 8.0 Hz, 3H), 4.67 (d, J = 6.8 Hz, 3H), 3.77 (s, 9H), 3.24–3.11 (m, 6H), 1.45 (s, 27H) ppm; 13C NMR (100 MHz, CDCl3) δ 172.4, 155.2, 141.9, 139.8, 135.5, 129.9, 127.4, 124.9, 80.0, 54.5, 52.3, 38.0, 28.3 ppm; HRMS–ESI (Q-Tof, m/z): [M + Na]+ calcd for C51H63N3NaO12, 932.4304; found, 932.4302; IR (neat) ![[Graphic 1]](/bjoc/content/inline/1860-5397-15-33-i5.svg?max-width=637&scale=1.18182) : 3661, 2349, 1716, 1495, 1163, 1044, 755 cm−1.
: 3661, 2349, 1716, 1495, 1163, 1044, 755 cm−1.
General procedure for the mono- and dipeptide products 11, 12 and 13
Negishi coupling product 10 was dissolved in dichloromethane/trifluoroacetic acid (CH2Cl2/TFA 1:1) and the reaction mixture was stirred at room temperature for 1 h. Then, the mixture was concentrated at reduced pressure to remove the solvent and dried under vacuum. Later, without further purification the Negishi coupling deprotection product was reacted with 3 equiv of thiophene 2-carboxylic acid or amino acids (N-Boc-L-valine or Boc-Phe-OH) in the presence of N,N-diisopropylethylamine (DIPEA, 4 equiv), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 9 equiv) in CH2Cl2. Afterwards, the reaction mixture was stirred at room temperature for 5 h under an inert atmosphere. After completion of the reaction, the mixture was washed with water, brine (3 × 10 mL) and extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was dried over Na2SO4 and concentrated at reduced pressure. The crude product was purified by silica gel column chromatography (80% ethyl acetate/petroleum ether) to afford the C3-symmetric mono- and dipeptide derivatives 11, 12 and 13, respectively.
Peptide derivative 11
Colorless solid; yield 86% (89 mg, starting from 100 mg of 10); Rf = 0.46 (7:3 ethyl acetate/petroleum ether); mp 156–158 °C; [α]D25 +25.07 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 3H), 7.60 (d, J = 8.0 Hz, 6H), 7.48 (q, J = 3.5 Hz, 6H), 7.24 (d, J = 8.0 Hz, 6H), 7.04 (t, J = 4.5 Hz, 3H), 6.64 (d, J = 7.5 Hz, 3H), 5.10 (q, J = 5.5 Hz, 3H), 3.79 (s, 9H), 3.35–3.24 (m, 6H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.0, 161.5, 141.9, 139.9, 138.2, 135.3, 130.6, 129.9, 128.7, 127.8, 127.6, 125.0, 53.6, 52.6, 37.7 ppm; HRMS–ESI (Q-Tof, m/z): [M + H]+ calcd for C51H46N3O9S3, 940.2391; found, 940.2392; IR (neat) ![[Graphic 2]](/bjoc/content/inline/1860-5397-15-33-i6.svg?max-width=637&scale=1.18182) : 3769, 3327, 2932, 1664, 1169, 759 cm−1.
: 3769, 3327, 2932, 1664, 1169, 759 cm−1.
Dipeptide 12
Colorless solid; yield 73% (97 mg, starting from 100 mg of 10); Rf = 0.59 (6:4 ethyl acetate/petroleum ether); mp <230 °C (dec); [α]D25 +20.58 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 3H), 7.59 (d, J = 8.0 Hz, 6H), 7.21 (d, J = 7.6 Hz, 6H), 6.56 (d, J = 6.8 Hz, 3H), 5.12 (d, J = 7.2 Hz, 3H), 4.91 (d, J = 6.4 Hz, 3H), 3.95 (s, 3H), 3.72 (s, 9H), 3.16 (s, 6H), 2.09 (d, J = 6.0 Hz, 3H), 1.41 (s, 27 H), 0.92 (d, J = 6.8 Hz, 9H), 0.87 (d, J = 4.0 Hz, 9H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.8, 171.5, 155.9, 141.9, 139.9, 135.3, 129.8, 127.5, 124.9, 79.9, 60.0, 53.2, 52.4, 37.8, 31.0, 28.4, 19.3, 17.8 ppm; HRMS–ESI (Q-Tof, m/z): [M + Na]+ calcd for C66H90N6NaO15, 1229.6356; found, 1229.6359; IR (neat) ![[Graphic 3]](/bjoc/content/inline/1860-5397-15-33-i7.svg?max-width=637&scale=1.18182) : 3342, 2938, 2332, 1742, 1635, 1534, 1213, 754 cm−1.
: 3342, 2938, 2332, 1742, 1635, 1534, 1213, 754 cm−1.
Dipeptide 13
Colorless solid; yield 81% (96 mg, starting from 80 mg of 10); Rf = 0.73 (6:4 ethyl acetate/petroleum ether); mp 204–206 °C; 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 3H), 7.56 (d, J = 8.0 Hz, 6H), 7.27 (d, J = 7.6 Hz, 6H), 7.20 (t, J = 5.6 Hz, 9H), 7.10 (d, J = 8.0 Hz, 6H), 6.35 (d, J = 6.80 Hz, 3H), 4.98 (br, 3H), 4.83 (d, J = 6.0 Hz, 3H), 4.35 (d, J = 5.20 Hz, 3H), 3.70 (s, 9H), 3.10–3.00 (m, 12H), 1.34 (s, 27H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.5, 171.0, 155.4, 142.0, 140.0, 136.6, 135.2, 129.9, 129.5, 128.8, 127.6, 127.1, 125.0, 80.4, 55.9, 53.5, 52.5, 38.5, 37.8, 28.4 ppm; HRMS–ESI (Q-Tof, m/z): [M + Na]+ calcd for C78H90N6NaO15, 1373.6356; found, 1373.6359; IR (neat) ![[Graphic 4]](/bjoc/content/inline/1860-5397-15-33-i8.svg?max-width=637&scale=1.18182) : 3738, 3644, 2919, 2850, 2343, 1666, 1517, 814, 751 cm−1.
: 3738, 3644, 2919, 2850, 2343, 1666, 1517, 814, 751 cm−1.
Trisamine derivative 14
Compound 12 (95 mg, 0.07 mmol) was dissolved in CH2Cl2/TFA 1:1 and this mixture was stirred at room temperature for 1 h. At the conclusion of the reaction (TLC monitoring), the reaction mixture was concentrated at reduced pressure and dried under vacuum. The crude product was purified by silica gel column chromatography (2% MeOH/CHCl3) to obtain the Boc-deprotection product 14 (68 mg, 95%) as a colorless solid. Rf = 0.46 (0.5:9.5 methanol/chloroform); mp: <250 °C (dec); [α]D24 +0.97 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.75 (s, 3H), 7.67 (d, J = 7.5 Hz, 6H), 7.39 (d, J = 8.0 Hz, 6H), 3.75 (d, J = 4.5 Hz, 3H), 3.71 (s, 9H), 3.28–3.24 (m, 3H), 3.13–3.09 (m, 3H), 2.25 (q, J = 6.0 Hz, 3H), 1.27 (s, 3H), 1.08 (d, J = 6.5 Hz, 9H), 1.04 (d, J = 7.0 Hz, 9H) ppm; 13C NMR (125 MHz, CDCl3) δ 173.5,170.4, 143.9, 141.5, 138.1, 131.4, 129.0, 126.1, 60.0, 56.1, 38.4, 32.2, 19.4, 18.1 ppm; HRMS–ESI (Q-Tof m/z): [M + H]+ calcd for C51H67N6O9, 907.4964; found, 907.4963; IR (neat) ![[Graphic 5]](/bjoc/content/inline/1860-5397-15-33-i9.svg?max-width=637&scale=1.18182) : 3779, 3240, 2928, 1606, 1596, 1434, 783, 505 cm−1.
: 3779, 3240, 2928, 1606, 1596, 1434, 783, 505 cm−1.
Supporting Information
| Supporting Information File 1: Copies of 1H, 13C NMR and HRMS spectra of new compounds. | ||
| Format: PDF | Size: 1.3 MB | Download |
Acknowledgements
We thank the Department of Science and Technology (DST), New Delhi, India, for financial support and IIT Bombay, for recording spectral data. S.K. thanks the Department of Science and Technology for the award of a J. C. Bose fellowship (SR/S2/JCB-33/2010), Praj industries, Pune, for Pramod Chaudhari, Chair Professor (Green Chemistry) and CSIR (02(0272)/16/EMR-II). S.T. thanks the IIT Bombay for the award of a research fellowship.
References
-
Dangel, B.; Clarke, M.; Haley, J.; Sames, D.; Polt, R. J. Am. Chem. Soc. 1997, 119, 10865–10866. doi:10.1021/ja972135j
Return to citation in text: [1] -
Etayo, P.; Ayats, C.; Pericàs, M. A. Chem. Commun. 2016, 52, 1997–2010. doi:10.1039/c5cc08961a
Return to citation in text: [1] -
Klajnert, B.; Bryszewska, M. Acta Biochim. Pol. 2001, 48, 199–208.
Return to citation in text: [1] -
Romagnoli, B.; Hayes, W. J. Mater. Chem. 2002, 12, 767–799. doi:10.1039/b110218b
Return to citation in text: [1] -
Tansey, M. G.; Szymkowski, D. E. Drug Discovery Today 2009, 14, 1082–1088. doi:10.1016/j.drudis.2009.10.002
Return to citation in text: [1] -
Liu, J.; Bartesaghi, A.; Borgnia, M. J.; Sapiro, G.; Subramaniam, S. Nature 2008, 455, 109–113. doi:10.1038/nature07159
Return to citation in text: [1] -
Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J. D.; Grisé, H.; Ofek, G. A.; Taylor, K. A.; Roux, K. H. Nature 2006, 441, 847–852. doi:10.1038/nature04817
Return to citation in text: [1] -
Berthelmann, A.; Lach, J.; Gräwert, M. A.; Groll, M.; Eichler, J. Org. Biomol. Chem. 2014, 12, 2606–2614. doi:10.1039/c3ob42251h
Return to citation in text: [1] [2] -
Kotha, S.; Brahmachary, E. J. Org. Chem. 2000, 65, 1359–1365. doi:10.1021/jo991387v
Return to citation in text: [1] -
Kotha, S.; Sreenivasachary, N.; Mohanraja, K.; Durani, S. Bioorg. Med. Chem. Lett. 2001, 11, 1421–1423. doi:10.1016/s0960-894x(01)00227-x
Return to citation in text: [1] -
Kotha, S. Acc. Chem. Res. 2003, 36, 342–351. doi:10.1021/ar020147q
Return to citation in text: [1] -
Balaram, P. Curr. Opin. Struct. Biol. 1992, 2, 845–851. doi:10.1016/0959-440x(92)90110-s
Return to citation in text: [1] -
Casabona, D.; Cativiela, C. Synthesis 2006, 2440–2443. doi:10.1055/s-2006-942458
Return to citation in text: [1] -
Kotha, S.; Halder, S. Synlett 2010, 337–354. doi:10.1055/s-0029-1219149
Return to citation in text: [1] -
Kotha, S.; Ghosh, A. K. Tetrahedron 2004, 60, 10833–10841. doi:10.1016/j.tet.2004.09.051
Return to citation in text: [1] -
Kotha, S.; Todeti, S.; Das, T.; Datta, A. Tetrahedron Lett. 2018, 59, 1023–1027. doi:10.1016/j.tetlet.2018.01.084
Return to citation in text: [1] -
Dash, B. P.; Satapathy, R.; Maguire, J. A.; Hosmane, N. S. Org. Lett. 2008, 10, 2247–2250. doi:10.1021/ol8005248
Return to citation in text: [1] -
Kashiki, T.; Kohara, M.; Osaka, I.; Miyazaki, E.; Takimiya, K. J. Org. Chem. 2011, 76, 4061–4070. doi:10.1021/jo2005044
Return to citation in text: [1] -
Mbyas Saroukou, M. S.; Skalski, T.; Skene, W. G.; Lubell, W. D. Tetrahedron 2014, 70, 450–458. doi:10.1016/j.tet.2013.11.043
Return to citation in text: [1] -
Dash, J.; Trawny, D.; Rabe, J. P.; Reissig, H.-U. Synlett 2015, 26, 1486–1489. doi:10.1055/s-0034-1380716
Return to citation in text: [1] -
Preis, E.; Dong, W.; Brunklaus, G.; Scherf, U. J. Mater. Chem. C 2015, 3, 1582–1587. doi:10.1039/c4tc02664k
Return to citation in text: [1] -
Shah, S. R.; Thakore, R. R.; Vyas, T. A.; Sridhar, B. Synlett 2016, 27, 294–300. doi:10.1055/s-0035-1560576
Return to citation in text: [1] -
Kotha, S.; Todeti, S.; Gopal, M. B.; Datta, A. ACS Omega 2017, 2, 6291–6297. doi:10.1021/acsomega.7b00941
Return to citation in text: [1] -
Kotha, S.; Todeti, S.; Das, T.; Datta, A. ChemistrySelect 2018, 3, 136–141. doi:10.1002/slct.201702675
Return to citation in text: [1] -
Kotha, S.; Chakraborty, K.; Brahmachary, E. Synlett 1999, 1621–1623. doi:10.1055/s-1999-2895
Return to citation in text: [1] -
Thallapally, P. K.; Chakraborty, K.; Carrell, H. L.; Kotha, S.; Desiraju, G. R. Tetrahedron 2000, 56, 6721–6728. doi:10.1016/s0040-4020(00)00493-2
Return to citation in text: [1] -
El-Bendary, M.; Priest, F. G.; Charles, J.-F.; Mitchell, W. J. FEMS Microbiol. Lett. 2005, 252, 51–56. doi:10.1016/j.femsle.2005.08.027
Return to citation in text: [1] -
Mitchell, W. J.; Kopidakis, N.; Rumbles, G.; Ginley, D. S.; Shaheen, S. E. J. Mater. Chem. 2005, 15, 4518–4528. doi:10.1039/b508683c
Return to citation in text: [1] -
Belton, C. R.; Kanibolotsky, A. L.; Kirkpatrick, J.; Orofino, C.; Elmasly, S. E. T.; Stavrinou, P. N.; Skabara, P. J.; Bradley, D. D. C. Adv. Funct. Mater. 2013, 23, 2792–2804. doi:10.1002/adfm.201202644
Return to citation in text: [1] -
Lai, W.-Y.; He, Q.-Y.; Zhu, R.; Chen, Q.-Q.; Huang, W. Adv. Funct. Mater. 2008, 18, 265–276. doi:10.1002/adfm.200700224
Return to citation in text: [1] -
Hoang, M. H.; Cho, M. J.; Kim, D. C.; Kim, K. H.; Shin, J. W.; Cho, M. Y.; Joo, J.-s.; Choi, D. H. Org. Electron. 2009, 10, 607–617. doi:10.1016/j.orgel.2009.02.021
Return to citation in text: [1] -
Ponomarenko, S. A.; Kirchmeyer, S.; Elschner, A.; Huisman, B.-H.; Karbach, A.; Drechsler, D. Adv. Funct. Mater. 2003, 13, 591–596. doi:10.1002/adfm.200304363
Return to citation in text: [1] -
Kinoshita, M.; Shirota, Y. Chem. Lett. 2001, 30, 614–615. doi:10.1246/cl.2001.614
Return to citation in text: [1] -
Kotha, S.; Shah, V. R. Amino Acids 2008, 35, 83–88. doi:10.1007/s00726-007-0626-9
Return to citation in text: [1] -
Pieters, R. J.; Cuntze, J.; Bonnet, M.; Diederich, F. J. Chem. Soc., Perkin Trans. 2 1997, 1891–1900. doi:10.1039/a702627g
Return to citation in text: [1] -
Gutiérrez-Abad, R.; Illa, O.; Ortuño, R. M. Org. Lett. 2010, 12, 3148–3151. doi:10.1021/ol1010664
Return to citation in text: [1] -
King, A. O.; Okukado, N.; Negishi, E.-i. J. Chem. Soc., Chem. Commun. 1977, 683–684. doi:10.1039/c39770000683
Return to citation in text: [1] -
Brittain, W. D. G.; Cobb, S. L. Org. Biomol. Chem. 2018, 16, 10–20. doi:10.1039/c7ob02682j
Return to citation in text: [1] -
Oswald, C. L.; Carrillo-Márquez, T.; Caggiano, L.; Jackson, R. F. W. Tetrahedron 2008, 64, 681–687. doi:10.1016/j.tet.2007.11.031
Return to citation in text: [1] -
Rilatt, I.; Caggiano, L.; Jackson, R. F. W. Synlett 2005, 2701–2719. doi:10.1055/s-2005-918950
Return to citation in text: [1] -
de Loos, M.; van Esch, J. H.; Kellogg, R. M.; Feringa, B. L. Tetrahedron 2007, 63, 7285–7301. doi:10.1016/j.tet.2007.02.066
Return to citation in text: [1] -
Danner, P.; Morkunas, M.; Maier, M. E. Org. Lett. 2013, 15, 2474–2477. doi:10.1021/ol4009409
Return to citation in text: [1] -
Bender, A. M.; Griggs, N. W.; Gao, C.; Trask, T. J.; Traynor, J. R.; Mosberg, H. I. ACS Med. Chem. Lett. 2015, 6, 1199–1203. doi:10.1021/acsmedchemlett.5b00344
Return to citation in text: [1] [2] -
Li, Z.; Ke, F.; Deng, H.; Xu, H.; Xiang, H.; Zhou, X. Org. Biomol. Chem. 2013, 11, 2943–2946. doi:10.1039/c3ob40464a
Return to citation in text: [1] -
Rajwar, D.; Sun, X.; Cho, S. J.; Grimsdale, A. C.; Fichou, D. CrystEngComm 2012, 14, 5182–5187. doi:10.1039/c2ce25530h
Return to citation in text: [1] -
Zhao, S.; Kang, L.; Ge, H.; Yang, F.; Wang, C.; Li, C.; Wang, Q.; Zhao, M. Synth. Commun. 2012, 42, 3569–3578. doi:10.1080/00397911.2011.585731
Return to citation in text: [1]
| 45. | Rajwar, D.; Sun, X.; Cho, S. J.; Grimsdale, A. C.; Fichou, D. CrystEngComm 2012, 14, 5182–5187. doi:10.1039/c2ce25530h |
| 46. | Zhao, S.; Kang, L.; Ge, H.; Yang, F.; Wang, C.; Li, C.; Wang, Q.; Zhao, M. Synth. Commun. 2012, 42, 3569–3578. doi:10.1080/00397911.2011.585731 |
| 43. | Bender, A. M.; Griggs, N. W.; Gao, C.; Trask, T. J.; Traynor, J. R.; Mosberg, H. I. ACS Med. Chem. Lett. 2015, 6, 1199–1203. doi:10.1021/acsmedchemlett.5b00344 |
| 44. | Li, Z.; Ke, F.; Deng, H.; Xu, H.; Xiang, H.; Zhou, X. Org. Biomol. Chem. 2013, 11, 2943–2946. doi:10.1039/c3ob40464a |
| 43. | Bender, A. M.; Griggs, N. W.; Gao, C.; Trask, T. J.; Traynor, J. R.; Mosberg, H. I. ACS Med. Chem. Lett. 2015, 6, 1199–1203. doi:10.1021/acsmedchemlett.5b00344 |
| 1. | Dangel, B.; Clarke, M.; Haley, J.; Sames, D.; Polt, R. J. Am. Chem. Soc. 1997, 119, 10865–10866. doi:10.1021/ja972135j |
| 2. | Etayo, P.; Ayats, C.; Pericàs, M. A. Chem. Commun. 2016, 52, 1997–2010. doi:10.1039/c5cc08961a |
| 3. | Klajnert, B.; Bryszewska, M. Acta Biochim. Pol. 2001, 48, 199–208. |
| 4. | Romagnoli, B.; Hayes, W. J. Mater. Chem. 2002, 12, 767–799. doi:10.1039/b110218b |
| 8. | Berthelmann, A.; Lach, J.; Gräwert, M. A.; Groll, M.; Eichler, J. Org. Biomol. Chem. 2014, 12, 2606–2614. doi:10.1039/c3ob42251h |
| 9. | Kotha, S.; Brahmachary, E. J. Org. Chem. 2000, 65, 1359–1365. doi:10.1021/jo991387v |
| 10. | Kotha, S.; Sreenivasachary, N.; Mohanraja, K.; Durani, S. Bioorg. Med. Chem. Lett. 2001, 11, 1421–1423. doi:10.1016/s0960-894x(01)00227-x |
| 11. | Kotha, S. Acc. Chem. Res. 2003, 36, 342–351. doi:10.1021/ar020147q |
| 12. | Balaram, P. Curr. Opin. Struct. Biol. 1992, 2, 845–851. doi:10.1016/0959-440x(92)90110-s |
| 13. | Casabona, D.; Cativiela, C. Synthesis 2006, 2440–2443. doi:10.1055/s-2006-942458 |
| 14. | Kotha, S.; Halder, S. Synlett 2010, 337–354. doi:10.1055/s-0029-1219149 |
| 15. | Kotha, S.; Ghosh, A. K. Tetrahedron 2004, 60, 10833–10841. doi:10.1016/j.tet.2004.09.051 |
| 8. | Berthelmann, A.; Lach, J.; Gräwert, M. A.; Groll, M.; Eichler, J. Org. Biomol. Chem. 2014, 12, 2606–2614. doi:10.1039/c3ob42251h |
| 41. | de Loos, M.; van Esch, J. H.; Kellogg, R. M.; Feringa, B. L. Tetrahedron 2007, 63, 7285–7301. doi:10.1016/j.tet.2007.02.066 |
| 7. | Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J. D.; Grisé, H.; Ofek, G. A.; Taylor, K. A.; Roux, K. H. Nature 2006, 441, 847–852. doi:10.1038/nature04817 |
| 42. | Danner, P.; Morkunas, M.; Maier, M. E. Org. Lett. 2013, 15, 2474–2477. doi:10.1021/ol4009409 |
| 6. | Liu, J.; Bartesaghi, A.; Borgnia, M. J.; Sapiro, G.; Subramaniam, S. Nature 2008, 455, 109–113. doi:10.1038/nature07159 |
| 37. | King, A. O.; Okukado, N.; Negishi, E.-i. J. Chem. Soc., Chem. Commun. 1977, 683–684. doi:10.1039/c39770000683 |
| 38. | Brittain, W. D. G.; Cobb, S. L. Org. Biomol. Chem. 2018, 16, 10–20. doi:10.1039/c7ob02682j |
| 5. | Tansey, M. G.; Szymkowski, D. E. Drug Discovery Today 2009, 14, 1082–1088. doi:10.1016/j.drudis.2009.10.002 |
| 39. | Oswald, C. L.; Carrillo-Márquez, T.; Caggiano, L.; Jackson, R. F. W. Tetrahedron 2008, 64, 681–687. doi:10.1016/j.tet.2007.11.031 |
| 40. | Rilatt, I.; Caggiano, L.; Jackson, R. F. W. Synlett 2005, 2701–2719. doi:10.1055/s-2005-918950 |
| 31. | Hoang, M. H.; Cho, M. J.; Kim, D. C.; Kim, K. H.; Shin, J. W.; Cho, M. Y.; Joo, J.-s.; Choi, D. H. Org. Electron. 2009, 10, 607–617. doi:10.1016/j.orgel.2009.02.021 |
| 32. | Ponomarenko, S. A.; Kirchmeyer, S.; Elschner, A.; Huisman, B.-H.; Karbach, A.; Drechsler, D. Adv. Funct. Mater. 2003, 13, 591–596. doi:10.1002/adfm.200304363 |
| 34. | Kotha, S.; Shah, V. R. Amino Acids 2008, 35, 83–88. doi:10.1007/s00726-007-0626-9 |
| 29. | Belton, C. R.; Kanibolotsky, A. L.; Kirkpatrick, J.; Orofino, C.; Elmasly, S. E. T.; Stavrinou, P. N.; Skabara, P. J.; Bradley, D. D. C. Adv. Funct. Mater. 2013, 23, 2792–2804. doi:10.1002/adfm.201202644 |
| 30. | Lai, W.-Y.; He, Q.-Y.; Zhu, R.; Chen, Q.-Q.; Huang, W. Adv. Funct. Mater. 2008, 18, 265–276. doi:10.1002/adfm.200700224 |
| 35. | Pieters, R. J.; Cuntze, J.; Bonnet, M.; Diederich, F. J. Chem. Soc., Perkin Trans. 2 1997, 1891–1900. doi:10.1039/a702627g |
| 36. | Gutiérrez-Abad, R.; Illa, O.; Ortuño, R. M. Org. Lett. 2010, 12, 3148–3151. doi:10.1021/ol1010664 |
| 27. | El-Bendary, M.; Priest, F. G.; Charles, J.-F.; Mitchell, W. J. FEMS Microbiol. Lett. 2005, 252, 51–56. doi:10.1016/j.femsle.2005.08.027 |
| 28. | Mitchell, W. J.; Kopidakis, N.; Rumbles, G.; Ginley, D. S.; Shaheen, S. E. J. Mater. Chem. 2005, 15, 4518–4528. doi:10.1039/b508683c |
| 16. | Kotha, S.; Todeti, S.; Das, T.; Datta, A. Tetrahedron Lett. 2018, 59, 1023–1027. doi:10.1016/j.tetlet.2018.01.084 |
| 17. | Dash, B. P.; Satapathy, R.; Maguire, J. A.; Hosmane, N. S. Org. Lett. 2008, 10, 2247–2250. doi:10.1021/ol8005248 |
| 18. | Kashiki, T.; Kohara, M.; Osaka, I.; Miyazaki, E.; Takimiya, K. J. Org. Chem. 2011, 76, 4061–4070. doi:10.1021/jo2005044 |
| 19. | Mbyas Saroukou, M. S.; Skalski, T.; Skene, W. G.; Lubell, W. D. Tetrahedron 2014, 70, 450–458. doi:10.1016/j.tet.2013.11.043 |
| 20. | Dash, J.; Trawny, D.; Rabe, J. P.; Reissig, H.-U. Synlett 2015, 26, 1486–1489. doi:10.1055/s-0034-1380716 |
| 21. | Preis, E.; Dong, W.; Brunklaus, G.; Scherf, U. J. Mater. Chem. C 2015, 3, 1582–1587. doi:10.1039/c4tc02664k |
| 22. | Shah, S. R.; Thakore, R. R.; Vyas, T. A.; Sridhar, B. Synlett 2016, 27, 294–300. doi:10.1055/s-0035-1560576 |
| 23. | Kotha, S.; Todeti, S.; Gopal, M. B.; Datta, A. ACS Omega 2017, 2, 6291–6297. doi:10.1021/acsomega.7b00941 |
| 24. | Kotha, S.; Todeti, S.; Das, T.; Datta, A. ChemistrySelect 2018, 3, 136–141. doi:10.1002/slct.201702675 |
| 25. | Kotha, S.; Chakraborty, K.; Brahmachary, E. Synlett 1999, 1621–1623. doi:10.1055/s-1999-2895 |
| 26. | Thallapally, P. K.; Chakraborty, K.; Carrell, H. L.; Kotha, S.; Desiraju, G. R. Tetrahedron 2000, 56, 6721–6728. doi:10.1016/s0040-4020(00)00493-2 |
| 33. | Kinoshita, M.; Shirota, Y. Chem. Lett. 2001, 30, 614–615. doi:10.1246/cl.2001.614 |
© 2019 Kotha and Todeti; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)