Abstract
Design, synthesis and properties of polycyclic aromatic hydrocarbons (PAHs) has historically attracted a considerable interdisciplinary interest from both fundamental as well as applied viewpoint on account of their wonderful optoelectronic properties. The scientific interest in two-dimensional star-shaped PAHs particularly in truxene architectures arises because of their high thermal stability, exceptional solubility and ease with which they can be constructed and modified. Therefore, bearing in mind a wide range of applications of truxene and its congeners, herein we reveal three novel distinctly different routes for the generation of C3-symmetric pyrrole-based truxene architectures by means of cyclotrimerization, ring-closing metathesis (RCM), Clauson–Kaas and Ullmann-type coupling reactions as key steps. Moreover, we have also assembled some other interesting heterocyclic systems possessing oxazole, imidazole, benzimidazole, and benzoxazole in the framework of truxene. Additionally, the preliminary photophysical properties (absorption and emission) for these versatile systems has been revealed.
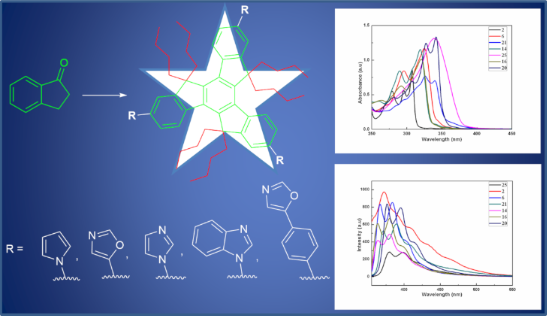
Graphical Abstract
Introduction
The two-dimensional phenylene-based π-conjugated star-shaped architectures has recently attracted a tremendous attention of the researchers across the globe because of their inimitable physiochemical properties, processability and availability [1-4]. Interestingly, compared to the π-conjugated one-dimensional rod-shaped architectures, two-dimensional star-shaped systems hold, superior solubility, improve morphological, optical, electrical and film-forming properties because of the involvement of extra dimensionality. Amongst them, the heptacyclic truxene scaffold possessing three overlapping fluorene units and its congeners are of archetypal interest due to their potential applications ranging from organic thin‐film transistors (OTFTs), organic light-emitting diodes (OLEDs), dye‐sensitized solar cells (DSSCs), fluorescent probes, organic photovoltaics (OPVs) to the high hole mobility semiconductors, blue light-emitting materials, nonlinear optical materials and so forth [5-7]. More interestingly, the high fluorescence quantum yield as well as the excellent photoluminescence quantum efficiency in addition to extraordinary thermal stability are the most significant characteristics of these systems.
Interestingly, this versatile C3-symmetric aromatic framework can also formally be considered as a 1,3,5‐triphenylbenzene derivative enjoying with three methylene units clipped in such a manner that all the four benzene rings are in conjugation with coplanar arrangement, resulting strong π–π stacking in addition to the strong electron‐donating ability which in turns provide a hint for its utility as novel building block for the development of a range of advanced functional materials for innumerable applications [8-16]. Moreover, truxene has also been vividly used for the construction of fullerenes and their bowl-shaped fragments besides their applicability in the synthesis of dendrimers as well as C3-tripods and also in the host–guest chemistry [17-21]. This architecturally simple yet effective rigid system was first prepared in 1894 by Kipping [22] involving the trimerization of 3‐phenylpropionic acid under acidic conditions but surprisingly its futuristic applications in a myriad of areas boomed in recent past only. It is really amazing that how time has passed and a plethora of novel synthetic breakthroughs, coupled with a range of revolutionary applications has drastically transformed this realm of modern science and technology. Not surprisingly, till date limitless functionalized truxenes and their isomeric isotruxene derivatives have been reported in the literature [23-27]. Since, the chemistry of truxenes is undoubtedly of great interest, therefore there is always a pressing need to develop simple yet powerful synthetic strategies and also novel truxene-based functional materials with unique properties. Bearing in mind the importance of the truxene framework, herein, we report diverse C3-symmetric heterocyclic systems possessing the truxene scaffold in their structures by means of cyclotrimerization, ring-closing metathesis, Ullmann-type coupling, Clauson–Kaas, and Van Leusen reactions as crucial transformations.
Synthetic Strategy
Basically, our endeavor toward the synthesis of truxene-based systems has been rooted in the development of new synthetic methodologies to assemble diverse polycyclic as well as spirocyclic structures. The present strategy to construct the pyrrole-based star-shaped truxene derivative 6 involve the three different retrosynthetic pathways A, B and C (Scheme 1). As can be seen from an inspection of Scheme 1, path A involves the transformation of the N-hexa-allylated system 3 to the required C3-symmetric tripyrroltruxene 6 by means of RCM in the presence of Grubbs′ first generation catalyst (G-I, 9) followed by self-aromatization without the involvement of any oxidizing agent. Compound 3 was produced from the hexabutylated truxene system 2 involving nitration, reduction and N-allylation sequences, which in turn was assembled from the commercially available 1-indanone (1) utilizing cyclotrimerization and subsequent alkylation reactions. On the other hand, the same compound 6 was generated form triaminotruxene 4 using three-fold Clauson–Kaas reaction which in turn was assembled from the same starting material 1 by employing the acid-catalyzed cyclotrimerization, alkylation and subsequent reduction reaction sequences (path B, Scheme 1). In sharp contrast, as can be inspected from path C (Scheme 1) title compound 6 was also synthesized from the tribromotruxene derivative 5 by virtue of the Ullmann-type coupling reaction. The tribromotruxene derivative 5 was prepared from the same starting point 1 in three steps (cyclotrimerization, bromination and alkylation) using the literature reported procedures (Scheme 1). Hopefully, these three distinctly different crucial strategies could open-up the challenging opportunities to magnify the chemical space not only for the pyrrole derivatives but also of the truxene-based architectures, and the present contribution would definitely add value to both heterocyclic systems as well as to the truxene chemistry.
![[1860-5397-17-96-i1]](/bjoc/content/inline/1860-5397-17-96-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic pathways to the pyrrole-based C3-symmetric truxene derivative 6.
Scheme 1: Retrosynthetic pathways to the pyrrole-based C3-symmetric truxene derivative 6.
Results and Discussion
Inspired from the available potential applications, and also bearing in mind the applicability of truxene-based materials from future perspective in addition to as a part of our major program on the synthesis of PAHs in general and functionalized truxenes in particular, our voyage for the synthesis of truxene-based heterocyclic systems commence with the construction of tripyrrolotruxene derivative 6 (Scheme 2). Basically, our interest to generate pyrrole-based C3-symmetric truxene derivatives was because of a range of applications of the pyrrole moiety in diverse fields, for instance, a wide variety of medicinally active as well as smart functional materials possessing the pyrrole as a fundamental subunit [28-30]. Additionally, 3,4-disubstituted pyrrole derivatives are versatile building blocks for the production of diverse bioactive molecules like co-enzyme, alkaloids, porphyrins and other related macrocycles [31,32]. Moreover, polypyrrolic systems are of extensive interest in numerous areas namely optics, medicine, and supramolecular chemistry in addition to the materials sciences and technology. Fascinatingly, in luminescence as well as fluorescence-based sensors, pyrrole derivatives play a vital role as donors in the intramolecular partial charge-transfer cascade to transfer the extra electronic density. Apart from pure affection besides many more reasons for the constant fascination and utility of this beautifully simple yet much effective heteroaromatic system, this unique nitrogen-containing five-membered aromatic pyrrole scaffold will continue to play a significant role in the development of novel anionic receptors [33,34]. Therefore, as outlined in Scheme 2, our synthetic strategy towards the construction of the target pyrrole-based truxene derivative 6 initiated with the acid-catalyzed cyclotrimerization of the easily accessible 1-indanone (1) to furnish truxene (7) in 80% yield. Having the pristine truxene (7) in our hands, it was then subjected to hexaalkylation using t-BuOK/n-BuBr in DMSO to afford the required hexabutylated truxene derivative 2 in 95% yield (Scheme 2). Next, compound 2 was subjected to nitration followed by reduction to deliver triaminotruxene 4 in excellent yield. Later, compound 4 was subjected to six-fold N-allylation in the presence of NaH/allyl bromide to provide the required compound 3 which was directly treated with G-I catalyst (9) to afford the desired tripyrrolotruxene 6 in 68% yield (two steps) in a single-pot without further addition of any oxidizing agent, may be due immediate conversion of intermediatory non-aromatized 2,5-dihydro-1H-pyrrole derivatives to more stable aromatic pyrrole units (Scheme 2). On the other hand, we have prepared the same compound 6 from the triamine 4 utilizing a three-fold Clauson–Kaas reaction in the presence of 2,5-dimethoxytetrahydrofuran (10) under acetic acid reflux conditions (Scheme 3). Alternatively, the pyrrole-based system 6 has also been prepared starting from truxene (7) in three steps, involving the bromination using Br2/CH2Cl2 and then subsequent alkylation followed by Ullmann-type copper-mediated cross-coupling reaction in overall good yield (Scheme 4).
![[1860-5397-17-96-i2]](/bjoc/content/inline/1860-5397-17-96-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tripyrrolotruxene 6 via cyclotrimerization and RCM as crucial steps.
Scheme 2: Synthesis of tripyrrolotruxene 6 via cyclotrimerization and RCM as crucial steps.
![[1860-5397-17-96-i3]](/bjoc/content/inline/1860-5397-17-96-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of star-shaped molecule 6 utilizing the Clauson–Kaas pyrrole strategy.
Scheme 3: Synthesis of star-shaped molecule 6 utilizing the Clauson–Kaas pyrrole strategy.
![[1860-5397-17-96-i4]](/bjoc/content/inline/1860-5397-17-96-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of truxene derivative 6 involving Ullmann-type cross-coupling reaction.
Scheme 4: Synthesis of truxene derivative 6 involving Ullmann-type cross-coupling reaction.
On the other hand, imidazole and benzimidazole containing C3-symmetric truxene-based molecules (14 and 16) have also been assembled from the hexaalkylated tribromotruxene 5 by means of Ullmann-type reaction in the presence of copper powder using pristine imidazole (13) and benzimidazole (15), respectively (Scheme 5). Moreover, we have successfully constructed the C3-symmetric oxazole containing truxene derivative 20 along with bisoxazole truxene derivative 21 by virtue of Van Leusen reaction as a key step (Scheme 6). To this context, we first treated the hexabutylated derivative 2 with 1,1-dichlorodimethyl ether acting as a formyl source in the presence of Lewis acid such as titanium tetrachloride (TiCl4) in Rieche manner to yield the required triformyltruxene 19 as a major product along with a minor amount of diformyltruxene 18 (Scheme 6). Later, both the di- and triformylated truxene systems 18/19 were subjected to oxazole formation involving p-toluenesulfonylmethyl isocyanide (TosMIC) in Van Leusen fashion to provide the corresponding desired truxene-based oxazole derivatives (Scheme 6). Additionally, to enhance the conjugation which in turn would undoubtedly tune the properties of the truxene-based heterocyclic systems, we also design and synthesized the benzene-bridged oxazole derivative 25 in three steps (Scheme 7). As can be inspected from Scheme 7, our journey in this regard stem from the iodination of 2 using H5IO6/I2/H2SO4 in acetic acid–water solvent system to afford the desired triiodotruxene derivative 22 in 50% yield. Furthermore, Suzuki–Miyaura cross-coupling reaction of 22 with 4-formylphenylboronic acid (23) furnished the triformyltruxene 24 (Scheme 7). Finally, the crude product 24 was directly treated with TosMIC under Van Leusen oxazole conditions to provide the required product 25 in 23% yield (two steps) as depicted in Scheme 7.
![[1860-5397-17-96-i5]](/bjoc/content/inline/1860-5397-17-96-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of imidazole and benzimidazole containing truxene derivatives 14 and 16.
Scheme 5: Synthesis of imidazole and benzimidazole containing truxene derivatives 14 and 16.
![[1860-5397-17-96-i6]](/bjoc/content/inline/1860-5397-17-96-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Construction of truxene-based di- and trioxazole derivatives 21 and 20.
Scheme 6: Construction of truxene-based di- and trioxazole derivatives 21 and 20.
![[1860-5397-17-96-i7]](/bjoc/content/inline/1860-5397-17-96-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of benzene-bridged rings containing trioxazolotruxene system 25.
Scheme 7: Synthesis of benzene-bridged rings containing trioxazolotruxene system 25.
Absorption and emission spectra
The UV–vis absorption as well as fluorescence spectra of the newly synthesized truxene derivatives 2, 6, 14, 16, 20, 21 and 25 were recorded in chloroform using a 7.04 × 10−6 molar concentration of the solution at room temperature (Figure 1). As can be inspected from Figure 1, these truxene systems exhibit absorption bands between 278–342 nm corresponding to the π–π* transitions. The absorption spectra of the parent hexa-alkylated truxene derivative 2 exhibited a strong band at 307.27 nm along with two more bands at ca. 295.62 nm, and 278.44 nm. On the other hand, a strong band with absorption maxima at 325.48 and a less intense band at 295.62 nm were observed for tripyrrolotruxene 6. Similarly, two bands for example at 318.91 nm and 290.55 nm were noticed in the case of imidazole containing truxene derivative 14. Interestingly, to our expectation, in both the cases, i.e., in truxenes 6 and 14, we found almost similar types of spectrum possessing red-shift absorption maxima as compared to the parent compound 2, may be due to increased conjugation in these truxene systems with the incorporation of three heterocyclic units in each case (Figure 1). Moreover, the absorption spectrum of compound 16 exhibited slightly red-shifted maxima at ca. 320.41 nm and 291.07 nm in addition to an extra band at 263.22 nm due to the fusion of three benzene rings onto the imidazole moieties. In sharp contrast, more red-shifted absorption bands at 341.64 nm and 326.99 nm along with two shoulders at ca. 306.75 nm, and 279.94 nm were found with truxene having oxazole moiety 20 in its structure. As expected, in the case of dioxazole truxene system 21, the bands were noticed at slightly lower wavelengths (340.13 nm and 325.96 nm) as compared to truxene 20 having three oxazole units, due to lack of one conjugated oxazole subunit. Finally and more surprisingly, compound 25 exhibited only a single broad band at ca. 339.10 nm as can be inspected from Figure 1. To our expectation, as can be seen from an inspection of Figure 1, emission spectra of all the synthesized truxenes lies at longer wavelengths as compared to the absorption spectra for the corresponding truxene derivatives. The fluorescence spectrum of the parent truxene 2 exhibit a broad band at ca. 371.88 nm while as all the other truxenes displayed two bands in the range of 364–399 nm with a very small shoulder in each case, which shows well-defined vibronic features (Figure 1). The exact values of the fluorescence maxima for these compounds are as follow; 6 (366.47 and 383.73), 14 (363.50 and 379.75), 16 (363.50 and 380.25), 20 (370.32 and 395.54), 21 (371.37 and 389.64), and 25 (379.26 and 398.52).
![[1860-5397-17-96-1]](/bjoc/content/figures/1860-5397-17-96-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Normalized absorption (left); fluorescence spectra (right) of the synthesized truxene derivatives (7.04 × 10−6 M concentration) in chloroform.
Figure 1: Normalized absorption (left); fluorescence spectra (right) of the synthesized truxene derivatives (...
Conclusion
In summary, the scope of applicability of truxene and its congeners is continuously exploding day-by-day, thanks to its easy construction, and solubility in common organic solvents in addition to remarkable structural modifications of the truxene framework. Interestingly, the area of truxene is now fully matured, scientist’s imagination is the only limit for designing novel truxene-based functional materials and the opportunities are now open. Herein, we have assembled diverse truxene scaffolds containing heterocyclic systems involving trimerization, ring-closing metathesis, Clauson–Kaas and Van Leusen reactions as vital steps. Moreover, we have revealed the preliminary photophysical properties (absorption and emission spectra) of the newly synthesized molecules and the detailed studies for these systems would be published in due course. We are pretty sure that this particular work may offer guidance for further expansion of these materials with enhanced potential for optoelectronic applications and other, if any in material science and technology.
Experimental
General: All commercially available reagents were used without further purification and purchased from Sigma-Aldrich, Alfa Aesar, TCI, GLR, Avera or Spectrochem. Solvents were purified according to reported standard procedures. Pyrrole was used after distillation. Analytical thin-layer chromatography (TLC) was performed on aluminium plates coated with silica gel by using a suitable mixture of EtOAc and petroleum ether for development. Column chromatography was performed by using silica gel (100–200 mesh) with an appropriate mixture of EtOAc and petroleum ether. Characterization of the intermediates and final compounds were performed using 1H and 13C NMR, mass spectrometry and IR spectroscopy. FTIR spectra of the compounds were recorded on an Aligent FTIR spectrometer (ATR module of Cary 630 FTIR, Agilent Technologies) from 600 to 4000 cm−1. UV–vis spectra were recorded on a UV-1800 Shimadzu spectrophotometer and the fluorescence spectra were recorded on an Agilent Carry Eclipse spectrofluorimeter.
Synthesis of 10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene (7): Truxene scaffold 7 was prepared according to a literature reported procedure [35]. l-Indanone (1, 6.8 g, 51.4 mmol) was added to a mixture of 60 mL acetic acid and 30 mL concentrated HCl, then the reaction mixture was stirred for 16 h at 100 °C. The solid precipitate was obtained by pouring the reaction mixture into crushed ice, washing with water, acetone followed by dichloromethane gave a white powder (4.7 g, 80%).
Synthesis of 5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene (2): Truxene (7, 4 g, 11.68 mmol), DMSO (35 mL), and t-BuOK (11.79 g, 105.12 mmol) were mixed in a two-necked round bottom flask (100 mL) under nitrogen atmosphere. The flask was cooled to 0 °C and stirred viciously after that n-BuBr (11.30 mL, 105.12 mmol) was slowly added to the flask, and the stirring was continued for 24 h at room temperature (rt), till the completion of the reaction (TLC monitoring). Then, the reaction was quenched with water and the crude product was extracted with ethyl acetate. The combined organic layer was washed with water (100 mL × 2) and dried over Na2SO4. After evaporation of the solvents, the residue was passed through silica gel to give the final product as a white powder (7.5 g, 95%). The 1H NMR spectrum was perfectly matching with the previously reported one [36].
Synthesis of 5,5,10,10,15,15-hexabutyl-2,7,12-trinitro-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene (8): To a solution of compound 2 (3.5 g, 5.15mmol) in dichloromethane (50 mL), conc. HNO3 (6.23 mL, 103 mmol) was slowly added and the resulting mixture was stirred for 2 h at 0 °C followed by 24 h at room temperature. Next, the reaction mixture was poured into ice cold water and neutralized with NaOH solution. Then the organic layer was extracted with ethyl acetate (50 mL × 3), dried over Na2SO4 and the crude product was purified with column chromatography to deliver the required compound 8 as yellow solid (4.15 g, 99%); Rf = 0.52 (10% EtOAc/petroleum ether); 1H NMR (300 MHz, CDCl3) δ 8.53 (d, J = 8.7 Hz, 3H), 8.37 (d, J = 9 Hz, 6H), 2.99–2.90 (m, 6H), 2.21–2.23 (m, 6H), 0.99–0.82 (m, 12H), 0.58-0.52 (m, 30H); 13C NMR (75 MHz, CDCl3) δ 154.86, 149.65, 147.08, 145.20, 137.48, 124.91, 122.66, 117.60, 56.57, 36.44, 26.54, 22.63, 13.71; IR (KBr): 2957, 2925, 2858, 1742, 1594, 1517, 1458 cm−1; MS (m/z): 814.00.
Synthesis of 5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene-2,7,12-triamine (4): To a 100 mL round bottom flask, SnCl2·2H2O (16.63 g, 73.5 mmol), compound 8 (2 g, 2.45 mmol) and dry ethyl acetate (60 mL) were added. Later, the resulting mixture was refluxed for 8 h and after completion of the reaction (TLC monitoring), the solution was quenched with aqueous saturated sodium bicarbonate solution within an ice bath. Then the reaction mixture was filtered and the organic layer was extracted with ethyl acetate (40 mL × 3), dried over Na2SO4 and after removing the solvent compound 4 was obtained (1.75 g, 99%); Rf = 0.36 (30% EtOAc/petroleum ether); 1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 8.4 Hz, 3H), 6.78 (s, 3H) 6.70 (d, J = 8.1 Hz, 3H), 3.69 (b–NH, 6H), 2.91–2.81 (m, 6H), 1.96–1.87 (m, 6H), 0.94–0.60 (m, 12H), 0.56–0.42 (m, 30H); 13C NMR (75 MHz, CDCl3) δ 155.72, 144.69, 141.19, 138.07, 132.30, 125.41, 113.23, 109.04, 55.05, 36.85, 26.50, 22.95, 13.91; IR (KBr): 3369, 3012, 2953, 2922, 2855, 1619, 1584, 1485 cm−1; MS (m/z): 724.66.
Synthesis of N2,N2,N7,N7,N12,N12-hexaallyl-5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene-2,7,12-triamine (3): To a suspension of sodium hydride (132 mg, 5.52 mmol) in dry DMF (5 mL), was added triamine derivative 4 (500 mg, 0.69 mmol) in DMF (5 mL) and allyl bromide (0.48 mL, 5.52 mmol) at 0 °C under nitrogen atmosphere. The mixture was stirred at rt for 12 h. After completion of the reaction (TLC monitoring), the reaction mixture was quenched with saturated NH4Cl and the aqueous layer was extracted with EtOAc (50 mL × 3), dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was directly used for the next step.
Synthesis of 1,1',1''-(5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene-2,7,12-triyl)tris(1H-pyrrole) (6): Method A: The solution of crude product 3 in dry dichloromethane (15 mL) was degassed with nitrogen for 15 min. After adding the Grubbs’ catalyst (15 mol %), the stirring was continued for 12 h at the same temperature. Next, the solvent was removed under reduced pressure followed by silica gel column chromatography (5% EtOAc/petroleum ether) provided the required product 6 (410 mg, 68%); Rf = 0.60 (5% ethyl acetate/petroleum ether); 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 8 Hz, 3H), 7.47–7.45 (m, 6H), 7.26–7.25 (m, 6H), 6.43–6.41 (m, 6H), 3.02–2.91 (m, 6H), 2.15–2.08 (m, 6H), 0.95–0.89 (m, 12H), 0.49–0.46 (m, 30H); 13C NMR (100 MHz, CDCl3) δ 155.42, 144.58, 139.38, 137.83, 137.70, 125.49, 119.39, 118.48, 114.03, 110.51, 110.33, 55.84, 36.76, 26.54, 22.83, 13.85; IR (KBr): 2953, 2922, 2855, 1962, 1608, 1497 cm−1; MS (m/z): 874.95.
Method B: Triaminotruxene 4 (300 mg, 0.41 mmol) and 2,5-dimethoxytetrahydrofuran (10, 273.77 mg, 2.05 mmol) were dissolved in acetic acid (5 mL) and the reaction mixture was heated under reflux conditions for 4 h in nitrogen atmosphere. After completion of the reaction (TLC monitoring), the reaction mixture was cooled to room temperature and quenched with saturated NH4Cl solution. Then, the aqueous layer was extracted with EtOAc (30 mL × 3) and the organic portion was dried over Na2SO4. The solvent was removed at reduced pressure and the crude product was purified by silica gel column chromatography (5% EtOAc/petroleum ether) to afford tripyrrolotruxene 6 (155 mg, 43%) as a brown solid. The spectral data matched with the above data (method A).
Method C: Tribromotruxene derivative 5 (400 mg, 0.43 mmol), pyrrole (12, 102.5 mg, 1.50 mmol), K2CO3 (6.5 equiv), and copper power (3.5 equiv) were dissolved in dry DMSO (10 mL) and the reaction mixture was heated at 130 °C under nitrogen atmosphere for 2 days. After completion of the reaction (TLC monitoring), the reaction mixture was cooled to room temperature and the aqueous layer was extracted with EtOAc (10 mL × 3). Organic portion was dried over Na2SO4 and the solvent was removed at reduced pressure. Then, the crude product was purified by silica gel column chromatography (5% EtOAc/petroleum ether) to give the required product 6 (208 mg, 55%) as a brown solid. Spectral data was matched with the above compound obtained by method A.
Synthesis of 2,7,12‐tribromo‐10,15‐dihydro‐5H‐diindeno[1,2‐a;1′,2′‐c]fluorene (11): Compound 11 was prepared according to a literature reported procedure [37]. Bromine (2 mL, 29.2 mmol) was added dropwise to a suspension of truxene 7 (2 g, 5.84 mmol) in CH2Cl2 (15 mL) at rt. The reaction mixture was stirred for 16 h at the same temperature and the excess of bromine was removed by bubbling N2 through the solution. Then the solid was filtered, washed with CH2Cl2, acetone, and finally with ethyl acetate to give 11 (3.13 g, 92%) as a yellow solid.
Synthesis of 2,7,12-tribromo-5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene (5): Compound 11 (1.5 g, 2.59 mmol), dimethyl sulfoxide (20 mL), and potassium tert-butoxide (2.61 g, 23.31 mmol) were mixed in a three-necked round bottom flask (100 mL) under nitrogen atmosphere. The flask was cooled to 0 °C and stirred viciously. n-Butyl bromide (2.50 mL, 23.31 mmol) was then slowly added to the flask, and the reaction was returned to rt. The reaction was quenched with water and the organic layer was extracted with ethyl acetate. The combined organic layer was washed with water (50 mL × 3) and dried over Na2SO4. After evaporation of the solvent, the residue was passed through silica gel to provide the final product 5 as a white powder (1.10 g, 70%). The 1H NMR spectrum of compound 5 was matched with the literature reported (see Supporting Information File 1) [36]. 1H NMR (300 MHz) δ 8.21 (d, J = 8.6 Hz, 3H), 7.58 (s, 3H), 7.54 (d, J = 8.6 Hz, 3H), 2.93–2.83 (m, 6H), 2.11–2.01 (m, 6H), 0.99–0.84 (m, 12H), 0.48 (t, J = 8 Hz, 30H).
Synthesis of 1,1',1''-(5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene-2,7,12-triyl)tris(1H-imidazole) (14): A round-bottomed flask was loaded with compound 5 (800 mg, 0.87 mmol), K2CO3 (1.44 g, 10.4 mmol), imidazole (13, 0.35 g, 5.22 mmol), and Cu powder (0.33 g, 5.22 mmol) in DMSO (10 mL) and the reaction mixture was heated at 185 °C for 2 days. Then the organic layer was extracted with ethyl acetate and washed several times with water to remove the DMSO. The resulting organic solution was dried over Na2SO4, and then the solvent was removed under vacuum which on purification furnished the desired compound 14 (450 mg, 59%) as a yellow solid; Rf = 0.60 (8% MeOH/petroleum ether); 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 8 Hz, 3H), 7.96 (s, 3H), 7.40 (d, J = 12 Hz, 9H), 7.22 (s, 3H), 2.94–2.88 (m, 6H), 2.10–2.05 (m, 6H), 0.92–0.83 (m, 12H), 0.43–0.40 (t, J = 8 Hz, 30H); 13C NMR (101 MHz, CDCl3) δ 155.71, 145.45, 139.12, 137.68, 136.12, 135.67, 130.49, 125.74, 119.63, 118.33, 115.05, 56.05, 36.68, 26.54, 22.76, 13.81; IR (KBr): 2953, 2921, 2855, 1609, 1497 cm−1; MS (m/z): 877.79.
Synthesis of 1,1',1''-(5,5,10,10,15,15-hexabutyl-10,15-dihydro-5H-diindeno[1,2-a:1',2'-c]fluorene-2,7,12-triyl)tris(1H-benzo[d]imidazole) (16): A round-bottomed flask was loaded with compound 5 (500 mg, 0.54 mmol), K2CO3 (0.90 g, 6.48 mmol), benzimidazole (15, 387.0 mg, 3.24 mmol), and Cu powder (208 mg, 3.24 mmol) in DMSO (10 mL) and the reaction mixture was heated at 185 °C for 2 days. Then the organic layer was extracted with ethyl acetate and washed several times with water to remove the DMSO. The resulting organic solution was dried over Na2SO4, and then the solvent was removed under vacuum which on purification delivered the desired compound 16 (0.37 g, 66%) as an off white solid; 1H NMR (400 MHz, CDCl3) δ 8.58 (d, J = 8 Hz, 3H), 8.31 (s, 3H), 7.97–7.95 (m, 3H), 7.67 (d, J = 4 Hz, 6H), 7.64–7.61 (m, 3H), 7.43–7.41 (m, 6H) 3.08–3.02 (m, 6H), 2.26–2.19 (m, 6H), 1.06–0.99 (m, 12H), 0.58–0.54 (t, J = 8 Hz, 30H); 13C NMR (101 MHz, CDCl3) δ 155.84, 145.86, 144.20, 142.41, 139.55, 137.86, 135.11, 133.75, 125.95, 123.84, 123.00, 122.13, 120.83, 117.72, 110.58, 108.14, 56.17, 36.74, 26.67, 22.87, 13.91; IR (KBr): 2952, 2920, 2853, 1605, 1490, 1454 cm−1; MS (m/z): 1027.00.
Supporting Information
| Supporting Information File 1: Additional general procedures, experimental and analytical data as well as copies of NMR spectra. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Gámez-Valenzuela, S.; Echeverri, M.; Gómez-Lor, B.; Martínez, J. I.; Ruiz Delgado, M. C. J. Mater. Chem. C 2020, 8, 15416–15425. doi:10.1039/d0tc03139a
Return to citation in text: [1] -
Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718–747. doi:10.1021/cr068010r
Return to citation in text: [1] -
Zhang, H.; Wu, D.; Liu, S. H.; Yin, J. Curr. Org. Chem. 2012, 16, 2124–2158. doi:10.2174/138527212803532477
Return to citation in text: [1] -
Alvi, S.; Ali, R. Beilstein J. Org. Chem. 2020, 16, 2212–2259. doi:10.3762/bjoc.16.186
Return to citation in text: [1] -
Goubard, F.; Dumur, F. RSC Adv. 2015, 5, 3521–3551. doi:10.1039/c4ra11559g
Return to citation in text: [1] -
Wagay, S. A.; Rather, I. A.; Ali, R. ChemistrySelect 2019, 4, 12272–12288. doi:10.1002/slct.201903076
Return to citation in text: [1] -
Shi, K.; Wang, J.-Y.; Pei, J. Chem. Rec. 2015, 15, 52–72. doi:10.1002/tcr.201402071
Return to citation in text: [1] -
de Frutos, Ó.; Gómez-Lor, B.; Granier, T.; Monge, M. Á.; Gutiérrez-Puebla, E.; Echavarren, A. M. Angew. Chem., Int. Ed. 1999, 38, 204–207. doi:10.1002/(sici)1521-3773(19990115)38:1/2<204::aid-anie204>3.0.co;2-5
Return to citation in text: [1] -
Sharma, R.; Thomas, M. B.; Misra, R.; D'Souza, F. Angew. Chem. 2019, 131, 4394–4399. doi:10.1002/ange.201814388
Return to citation in text: [1] -
Sharma, R.; Volyniuk, D.; Popli, C.; Bezvikonnyi, O.; Grazulevicius, J. V.; Misra, R. J. Phys. Chem. C 2018, 122, 15614–15624. doi:10.1021/acs.jpcc.8b00777
Return to citation in text: [1] -
Misra, R.; Sharma, R.; Mobin, S. M. Dalton Trans. 2014, 43, 6891. doi:10.1039/c4dt00210e
Return to citation in text: [1] -
Sharma, R.; Maragani, R.; Misra, R. New J. Chem. 2018, 42, 882–890. doi:10.1039/c7nj03934d
Return to citation in text: [1] -
Such, B.; Trevethan, T.; Glatzel, T.; Kawai, S.; Zimmerli, L.; Meyer, E.; Shluger, A. L.; Amijs, C. H. M.; de Mendoza, P.; Echavarren, A. M. ACS Nano 2010, 4, 3429–3439. doi:10.1021/nn100424g
Return to citation in text: [1] -
de Frutos, Ó.; Granier, T.; Gómez-Lor, B.; Jiménez-Barbero, J.; Monge, Á.; Gutiérrez-Puebla, E.; Echavarren, A. M. Chem. – Eur. J. 2002, 8, 2879. doi:10.1002/1521-3765(20020703)8:13<2879::aid-chem2879>3.0.co;2-4
Return to citation in text: [1] -
González-Cantalapiedra, E.; Ruiz, M.; Gómez-Lor, B.; Alonso, B.; García-Cuadrado, D.; Cárdenas, D. J.; Echavarren, A. M. Eur. J. Org. Chem. 2005, 4127–4140. doi:10.1002/ejoc.200500260
Return to citation in text: [1] -
Ruiz, M.; Gómez-Lor, B.; Santos, A.; Echavarren, A. M. Eur. J. Org. Chem. 2004, 858–866. doi:10.1002/ejoc.200300632
Return to citation in text: [1] -
Amsharov, K. Y.; Jansen, M. J. Org. Chem. 2008, 73, 2931–2934. doi:10.1021/jo7027008
Return to citation in text: [1] -
Amsharov, K.; Jansen, M. Chem. Commun. 2009, 2691. doi:10.1039/b901496a
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Acc. Chem. Res. 2019, 52, 1812–1823. doi:10.1021/acs.accounts.9b00227
Return to citation in text: [1] -
Chen, Q.-Q.; Liu, F.; Ma, Z.; Peng, B.; Wei, W.; Huang, W. Chem. Lett. 2008, 37, 178–179. doi:10.1246/cl.2008.178
Return to citation in text: [1] -
Pérez, E. M.; Sierra, M.; Sánchez, L.; Torres, M. R.; Viruela, R.; Viruela, P. M.; Ortí, E.; Martín, N. Angew. Chem., Int. Ed. 2007, 46, 1847–1851. doi:10.1002/anie.200604327
Return to citation in text: [1] -
Kipping, F. S. J. Chem. Soc., Trans. 1894, 65, 269–290. doi:10.1039/ct8946500269
Return to citation in text: [1] -
Ali, R.; Alvi, S. Tetrahedron 2020, 76, 131345. doi:10.1016/j.tet.2020.131345
Return to citation in text: [1] -
Kotha, S.; Ali, R.; Panguluri, N. R.; Datta, A.; Kannaujiya, K. K. Tetrahedron Lett. 2018, 59, 4080–4085. doi:10.1016/j.tetlet.2018.10.005
Return to citation in text: [1] -
Kotha, S.; Ali, R.; Panguluri, N. R.; Deb, A. C. Indian J. Chem. 2018, 57B, 1489–1492.
Return to citation in text: [1] -
Kotha, S.; Meshram, M.; Chakkapalli, C. Beilstein J. Org. Chem. 2018, 14, 2468–2481. doi:10.3762/bjoc.14.223
Return to citation in text: [1] -
Kotha, S.; Kashinath, D.; Lahiri, K.; Sunoj, R. B. Eur. J. Org. Chem. 2004, 4003–4013. doi:10.1002/ejoc.200400257
Return to citation in text: [1] -
Kotha, S.; Todeti, S.; Das, T.; Datta, A. Tetrahedron Lett. 2018, 59, 1023–1027. doi:10.1016/j.tetlet.2018.01.084
Return to citation in text: [1] -
Pak, J.; Ali, R.; Park, J. S. Bull. Korean Chem. Soc. 2016, 37, 732–735. doi:10.1002/bkcs.10732
Return to citation in text: [1] -
Fan, H.; Peng, J.; Hamann, M. T.; Hu, J.-F. Chem. Rev. 2008, 108, 264–287. doi:10.1021/cr078199m
Return to citation in text: [1] -
Lee, J. Y.; Root, H. D.; Ali, R.; An, W.; Lynch, V. M.; Bähring, S.; Kim, I. S.; Sessler, J. L.; Park, J. S. J. Am. Chem. Soc. 2020, 142, 19579–19587. doi:10.1021/jacs.0c08106
Return to citation in text: [1] -
Rather, I. A.; Wagay, S. A.; Hasnain, M. S.; Ali, R. RSC Adv. 2019, 9, 38309–38344. doi:10.1039/c9ra07399j
Return to citation in text: [1] -
Rather, I. A.; Wagay, S. A.; Ali, R. Coord. Chem. Rev. 2020, 415, 213327. doi:10.1016/j.ccr.2020.213327
Return to citation in text: [1] -
Kim, A.; Ali, R.; Park, S. H.; Kim, Y.-H.; Park, J. S. Chem. Commun. 2016, 52, 11139–11142. doi:10.1039/c6cc04562f
Return to citation in text: [1] -
Dehmlow, E. V.; Kelle, T. Synth. Commun. 1997, 27, 2021–2031. doi:10.1080/00397919708006804
Return to citation in text: [1] -
Wang, X.; Wang, Y.; Yang, H.; Fang, H.; Chen, R.; Sun, Y.; Zheng, N.; Tan, K.; Lu, X.; Tian, Z.; Cao, X. Nat. Commun. 2016, 7, 12469. doi:10.1038/ncomms12469
Return to citation in text: [1] [2] -
Gómez-Lor, B.; de Frutos, Ó.; Ceballos, P. A.; Granier, T.; Echavarren, A. M. Eur. J. Org. Chem. 2001, 2107–2114. doi:10.1002/1099-0690(200106)2001:11<2107::aid-ejoc2107>3.0.co;2-f
Return to citation in text: [1]
| 1. | Gámez-Valenzuela, S.; Echeverri, M.; Gómez-Lor, B.; Martínez, J. I.; Ruiz Delgado, M. C. J. Mater. Chem. C 2020, 8, 15416–15425. doi:10.1039/d0tc03139a |
| 2. | Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718–747. doi:10.1021/cr068010r |
| 3. | Zhang, H.; Wu, D.; Liu, S. H.; Yin, J. Curr. Org. Chem. 2012, 16, 2124–2158. doi:10.2174/138527212803532477 |
| 4. | Alvi, S.; Ali, R. Beilstein J. Org. Chem. 2020, 16, 2212–2259. doi:10.3762/bjoc.16.186 |
| 22. | Kipping, F. S. J. Chem. Soc., Trans. 1894, 65, 269–290. doi:10.1039/ct8946500269 |
| 17. | Amsharov, K. Y.; Jansen, M. J. Org. Chem. 2008, 73, 2931–2934. doi:10.1021/jo7027008 |
| 18. | Amsharov, K.; Jansen, M. Chem. Commun. 2009, 2691. doi:10.1039/b901496a |
| 19. | Dorel, R.; Echavarren, A. M. Acc. Chem. Res. 2019, 52, 1812–1823. doi:10.1021/acs.accounts.9b00227 |
| 20. | Chen, Q.-Q.; Liu, F.; Ma, Z.; Peng, B.; Wei, W.; Huang, W. Chem. Lett. 2008, 37, 178–179. doi:10.1246/cl.2008.178 |
| 21. | Pérez, E. M.; Sierra, M.; Sánchez, L.; Torres, M. R.; Viruela, R.; Viruela, P. M.; Ortí, E.; Martín, N. Angew. Chem., Int. Ed. 2007, 46, 1847–1851. doi:10.1002/anie.200604327 |
| 8. | de Frutos, Ó.; Gómez-Lor, B.; Granier, T.; Monge, M. Á.; Gutiérrez-Puebla, E.; Echavarren, A. M. Angew. Chem., Int. Ed. 1999, 38, 204–207. doi:10.1002/(sici)1521-3773(19990115)38:1/2<204::aid-anie204>3.0.co;2-5 |
| 9. | Sharma, R.; Thomas, M. B.; Misra, R.; D'Souza, F. Angew. Chem. 2019, 131, 4394–4399. doi:10.1002/ange.201814388 |
| 10. | Sharma, R.; Volyniuk, D.; Popli, C.; Bezvikonnyi, O.; Grazulevicius, J. V.; Misra, R. J. Phys. Chem. C 2018, 122, 15614–15624. doi:10.1021/acs.jpcc.8b00777 |
| 11. | Misra, R.; Sharma, R.; Mobin, S. M. Dalton Trans. 2014, 43, 6891. doi:10.1039/c4dt00210e |
| 12. | Sharma, R.; Maragani, R.; Misra, R. New J. Chem. 2018, 42, 882–890. doi:10.1039/c7nj03934d |
| 13. | Such, B.; Trevethan, T.; Glatzel, T.; Kawai, S.; Zimmerli, L.; Meyer, E.; Shluger, A. L.; Amijs, C. H. M.; de Mendoza, P.; Echavarren, A. M. ACS Nano 2010, 4, 3429–3439. doi:10.1021/nn100424g |
| 14. | de Frutos, Ó.; Granier, T.; Gómez-Lor, B.; Jiménez-Barbero, J.; Monge, Á.; Gutiérrez-Puebla, E.; Echavarren, A. M. Chem. – Eur. J. 2002, 8, 2879. doi:10.1002/1521-3765(20020703)8:13<2879::aid-chem2879>3.0.co;2-4 |
| 15. | González-Cantalapiedra, E.; Ruiz, M.; Gómez-Lor, B.; Alonso, B.; García-Cuadrado, D.; Cárdenas, D. J.; Echavarren, A. M. Eur. J. Org. Chem. 2005, 4127–4140. doi:10.1002/ejoc.200500260 |
| 16. | Ruiz, M.; Gómez-Lor, B.; Santos, A.; Echavarren, A. M. Eur. J. Org. Chem. 2004, 858–866. doi:10.1002/ejoc.200300632 |
| 36. | Wang, X.; Wang, Y.; Yang, H.; Fang, H.; Chen, R.; Sun, Y.; Zheng, N.; Tan, K.; Lu, X.; Tian, Z.; Cao, X. Nat. Commun. 2016, 7, 12469. doi:10.1038/ncomms12469 |
| 5. | Goubard, F.; Dumur, F. RSC Adv. 2015, 5, 3521–3551. doi:10.1039/c4ra11559g |
| 6. | Wagay, S. A.; Rather, I. A.; Ali, R. ChemistrySelect 2019, 4, 12272–12288. doi:10.1002/slct.201903076 |
| 7. | Shi, K.; Wang, J.-Y.; Pei, J. Chem. Rec. 2015, 15, 52–72. doi:10.1002/tcr.201402071 |
| 33. | Rather, I. A.; Wagay, S. A.; Ali, R. Coord. Chem. Rev. 2020, 415, 213327. doi:10.1016/j.ccr.2020.213327 |
| 34. | Kim, A.; Ali, R.; Park, S. H.; Kim, Y.-H.; Park, J. S. Chem. Commun. 2016, 52, 11139–11142. doi:10.1039/c6cc04562f |
| 36. | Wang, X.; Wang, Y.; Yang, H.; Fang, H.; Chen, R.; Sun, Y.; Zheng, N.; Tan, K.; Lu, X.; Tian, Z.; Cao, X. Nat. Commun. 2016, 7, 12469. doi:10.1038/ncomms12469 |
| 31. | Lee, J. Y.; Root, H. D.; Ali, R.; An, W.; Lynch, V. M.; Bähring, S.; Kim, I. S.; Sessler, J. L.; Park, J. S. J. Am. Chem. Soc. 2020, 142, 19579–19587. doi:10.1021/jacs.0c08106 |
| 32. | Rather, I. A.; Wagay, S. A.; Hasnain, M. S.; Ali, R. RSC Adv. 2019, 9, 38309–38344. doi:10.1039/c9ra07399j |
| 37. | Gómez-Lor, B.; de Frutos, Ó.; Ceballos, P. A.; Granier, T.; Echavarren, A. M. Eur. J. Org. Chem. 2001, 2107–2114. doi:10.1002/1099-0690(200106)2001:11<2107::aid-ejoc2107>3.0.co;2-f |
| 28. | Kotha, S.; Todeti, S.; Das, T.; Datta, A. Tetrahedron Lett. 2018, 59, 1023–1027. doi:10.1016/j.tetlet.2018.01.084 |
| 29. | Pak, J.; Ali, R.; Park, J. S. Bull. Korean Chem. Soc. 2016, 37, 732–735. doi:10.1002/bkcs.10732 |
| 30. | Fan, H.; Peng, J.; Hamann, M. T.; Hu, J.-F. Chem. Rev. 2008, 108, 264–287. doi:10.1021/cr078199m |
| 23. | Ali, R.; Alvi, S. Tetrahedron 2020, 76, 131345. doi:10.1016/j.tet.2020.131345 |
| 24. | Kotha, S.; Ali, R.; Panguluri, N. R.; Datta, A.; Kannaujiya, K. K. Tetrahedron Lett. 2018, 59, 4080–4085. doi:10.1016/j.tetlet.2018.10.005 |
| 25. | Kotha, S.; Ali, R.; Panguluri, N. R.; Deb, A. C. Indian J. Chem. 2018, 57B, 1489–1492. |
| 26. | Kotha, S.; Meshram, M.; Chakkapalli, C. Beilstein J. Org. Chem. 2018, 14, 2468–2481. doi:10.3762/bjoc.14.223 |
| 27. | Kotha, S.; Kashinath, D.; Lahiri, K.; Sunoj, R. B. Eur. J. Org. Chem. 2004, 4003–4013. doi:10.1002/ejoc.200400257 |
| 35. | Dehmlow, E. V.; Kelle, T. Synth. Commun. 1997, 27, 2021–2031. doi:10.1080/00397919708006804 |
© 2021 Alvi and Ali; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)