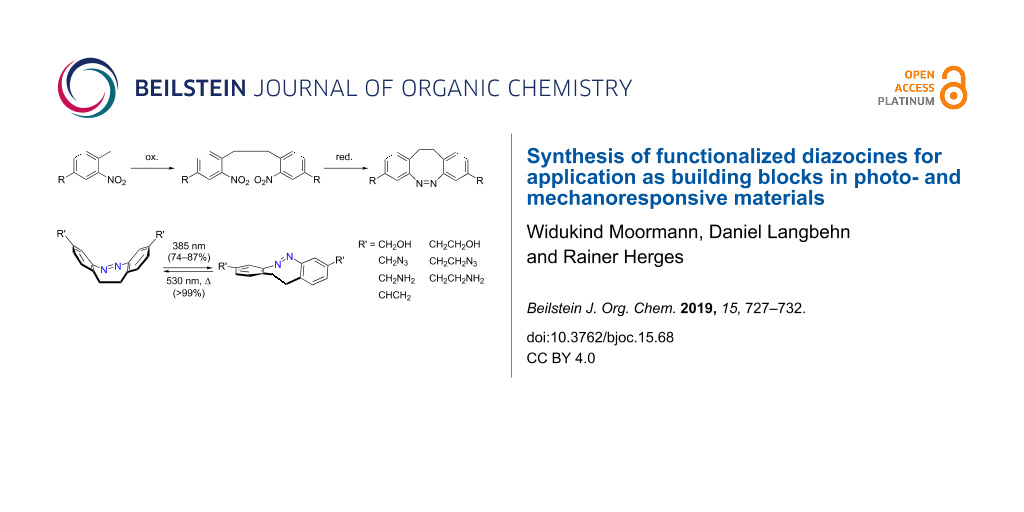
Abstract
Seven symmetrically 3,3’-substituted diazocines were synthesized. Functional groups include alcohol, azide, amine and vinyl groups, which are suitable for polymer synthesis. Upon irradiation at 385 and 530 nm the diazocines perform a reversible, pincer-type movement switching the 3,3’-distance between 6.1 Å (cis, stable isomer) and 8.2 Å (trans, metastable isomer). Key reactions in the synthesis are an oxidative C–C coupling of 2-nitrotoluenes (75–82% yield) and a reductive ring closure to form the diazocines (56–60% yield). The cyclization of the dinitro compound to the azo compound was improved in yield and reproducibility, by over-reduction to the hydrazine and reoxidation to the azo unit. In contrast to 3,3’- and 4,4’-diaminodiazocine, which have been implemented in macromolecules for conformation switching, our compounds exhibit improved photophysical properties (photostationary states, separation of absorption bands in the cis and trans configuration). Hence they are promising candidates as molecular switches in photo and mechanoresponsive macromolecules and other smart materials.

Graphical Abstract
Introduction
The field of photoresponsive materials is of growing interest [1-3]. Several mechanophores such as azobenzene [4-8], diarylethene [9-13] and spiropyrans [14-18] have been investigated as photoswitchable building blocks. Bridged azobenzenes also known as diazocines exhibit excellent photochemical properties but applications are limited and suitably functionalized compounds are rare [19-23]. In contrary to azobenzenes, diazocines 1 are stable in their cis configuration. The bent cis isomer is less prone to π–π stacking which is known to reduce the switching efficiency (Figure 1a) [19,24]. The reverse stability of the cis and trans isomers in azobenzenes and diazocines should allow reciprocal applications in mechanoresponsive materials and in photopharmacology [25]. Another advantage of diazocines over azobenzenes is their switchability in the visible range (400 nm cis → trans, 530 nm trans → cis) preventing deterioration of the material or tissue damage by UV light [19]. Well separated absorption bands, high switching efficiency and high quantum yields are further advantages regarding their application as switches in photoresponsive materials [19,24,26]. In contrast to spiropyrans which have been frequently used as photoswitches in materials, diazocines are stable over several thousand switching cycles under air [19,24,26,27]. Notwithstanding their excellent properties, to date only 3,3’- and 4,4’- functionalized diazocine 2 and 3 have been implemented in polymers [19] and proteins [28]. Unfortunately, similar to azobenzenes, aminosubstitution at the phenyl rings reduces switching efficiency [29]. In contrast to the parent system, separation of absorption bands of the cis and trans isomer in 2 and 3 is poor. Upon irradiation of the corresponding cis-configured compounds at 385 nm only 30% of the trans isomer of 3,3’-diazocine 2 and 25% of the 4,4’-diazocine 3 are formed (Figure 1b). Applications of diazocines 2 and 3 are further hampered by the low yields of their synthesis [19,20].
![[1860-5397-15-68-1]](/bjoc/content/figures/1860-5397-15-68-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Isomerization of parent diazocine 1. Distances of carbon atoms para to the ethylene bridge were determined at the B3LYP/6-31g* level of DFT [23]. (b) UV–vis spectra of parent diazocine 1 (left), 3,3’-diaminodiazocine 2 (center) and 4,4’-diaminodiazocine 3 (right). Continuous lines: 100% cis and dashed lines: PSS (385 nm) at 298.15 K in acetonitrile.
Figure 1: (a) Isomerization of parent diazocine 1. Distances of carbon atoms para to the ethylene bridge were...
To decouple the electronic influence of the functional groups from the azo switching process and to improve yields of the azo cyclization step, we separated the functional groups from the aromatic system by one or two methylene groups and restricted substitution to the position meta with respect to the azo group (Figure 2) [19,20].
![[1860-5397-15-68-2]](/bjoc/content/figures/1860-5397-15-68-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesized target diazocines 4–7 for applications in responsive materials.
Figure 2: Synthesized target diazocines 4–7 for applications in responsive materials.
Results and Discussion
The synthesis of the targeted diazocines 4–7 is based on two key reactions, an oxidative C–C coupling of nitrotoluenes and the reductive ring closure of the dinitro compounds (Scheme 1). We recently improved the yield of the C–C coupling through addition of bromine as an oxidizing agent [27]. The reaction times thus are reduced to several minutes as compared to several hours in previous procedures using oxygen and the yields are increased from 65% to 95% in the parent system [30].
![[1860-5397-15-68-i1]](/bjoc/content/inline/1860-5397-15-68-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Key reactions in diazocine synthesis.
Scheme 1: Key reactions in diazocine synthesis.
In a recent work we observed that the reduction of 2,2′-dinitrodibenzyl is difficult to stop at the azo stage because further reduction to the hydrazine is faster than the preceding cyclization reaction [27]. The hydrazine is quite stable towards reduction to the diamine, and can easily be reoxidized to the azo compound using CuCl2/O2. The yields are higher and more reproducible using the above reduction/reoxidation scheme. Previously applied reducing agents include Ba(OH)2/Zn [27], glucose/NaOH [20], Pb/NEt3/HCOOH [22,23], or the Baeyer–Mills reaction via Zn/NH4Cl [25]. We chose the Ba(OH)2/Zn method because it provided superior yields even at larger scales. The syntheses of the functionalized diazocines 4–7 started with (4-methyl-3-nitrophenyl)methanol (8a) and (4-methyl-3-nitrophenyl)ethanol (8b). In a first step the hydroxy groups in 8a and 8b were protected as tert-butyl ethers (Scheme 2) to prevent oxidation in the following oxidative C–C coupling [31]. The tert-butyl ether was chosen as the protecting group because it is stable towards the oxidizing conditions of the C–C coupling reactions and the reducing conditions of the azo cyclization. Moreover, the tert-butyl group can be conveniently removed under acidic conditions. As described in [27] potassium butoxide is used as a non-nucleophilic base to remove the α-toluene protons of 9a and 9b. By addition of bromine as an oxidizing agent dimers 10a and 10b are formed, most probably through radical intermediates. Then the dinitro compounds 10a and 10b were reduced with Ba(OH)2/Zn to the hydrazine intermediates and subsequently oxidized with CuCl2 and air in a two-step reductive azo cyclization in a similar manner as described in [27]. After deprotection with TiCl4 the hydroxy-functionalized diazocines 4a and 4b were obtained [32]. The hydroxy groups in 4a and 4b were successfully converted into azides using 2-azido-1,3-dimethylimidazolinium hexafluorophosphate (ADMP) and DBU [33]. The synthesis was completed with a Staudinger reaction to obtain the amino-functionalized diazocines 6a and 6b [34]. Additionally, the diazocine 4b was converted into the divinyldiazocine 7. Towards this end, the hydroxy groups were tosylated, followed by elimination with potassium butoxide [35,36].
![[1860-5397-15-68-i2]](/bjoc/content/inline/1860-5397-15-68-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syntheses of the functionalized diazocines 4–7. Reaction conditions: i) Isobutylene, sulfuric acid, DCM; ii) t-BuOK, Br2, THF; iii) Zn, Ba(OH)2 H2O/EtOH, and CuCl2/O2, NaOH/MeOH; iv) Ti(Cl)4, DCM; v) ADMP, DBU, THF; vi) PPh3, H2O, THF; viii) TsCl, DMAP, TEA, DCM, and t-BuOK, THF.
Scheme 2: Syntheses of the functionalized diazocines 4–7. Reaction conditions: i) Isobutylene, sulfuric acid,...
The photochemical and photophysical properties of compounds 4–7 were investigated by NMR and UV–vis spectroscopy and the results are listed in Table 1. Photostationary states (PSS) as well as half-lives (t1/2) were determined in acetonitrile at 300 K and 298.15 K, respectively. The (cis → trans) and (trans → cis) isomerization were achieved by irradiation into the appropriate n–π* bands at 385 and 530 nm. As a result of electronic decoupling the absorption bands are well separated and the photostationary states of diazocines 4–7 are considerably improved compared to 3,3’- (2) and 4,4’-diaminodiazocine 3 (Figure 3). The (cis → trans) isomerization of diazocines 4–7 was achieved after 2 min of irradiation at 385 nm in yields of 74–85%. All trans-diazocines were converted quantitatively to the cis-configuration either by thermal relaxation or by irradiation at 530 nm. In general, the (trans → cis) isomerization can be accomplished with wavelengths between 520 and 620 nm. The half-lives (t1/2) for the thermal relaxation (trans → cis) at 298.15 K of diazocines 4–7 in acetonitrile are between 10.2–16.7 h and thus are in the same order of magnitude as the parent system 1 (15.3 h). In comparison, the 3,3’-diaminodiazocine 2 has a much longer half-life (t1/2) of 24.5 h. The electronic decoupling of substituents in diazocines 4–7 has proven to retain the excellent photochemical properties in regard to PSS and half-life (t1/2) of the parent system 1.
Table 1: Photostationary states (300 K), absorption maxima and half-lives (298.15 K), determined by 1H NMR and UV–vis spectroscopy in acetonitrile.
| molecule |
PSS (385 nm)
[%] trans |
PSS (530 nm)
[%] cis |
λmax (cis)
[nm] |
λmax (trans)
[nm] |
t1/2 (UV) [h]
at 298.15 K |
| 1 | 87 | >99 | 402 | 486 | 15.3 |
| 2 | 30 | >99 | 401 | 487 | 24.8 |
| 3 | 25 | >99 | 400 | 475 | 20 |
| 4a | 83 | >99 | 404 | 485 | 11.4 |
| 4b | 81 | >99 | 405 | 487 | 16.7 |
| 5a | 85 | >99 | 402 | 484 | 11.2 |
| 5b | 82 | >99 | 405 | 489 | 14.0 |
| 6a | 81 | >99 | 405 | 488 | 14.7 |
| 6b | 78 | >99 | 400 | 485 | 10.1 |
| 7 | 74 | >99 | 403 | 484 | 13.1 |
| 11a | 82 | >99 | 403 | 487 | 10.2 |
| 11b | 81 | >99 | 405 | 488 | 15.9 |
![[1860-5397-15-68-3]](/bjoc/content/figures/1860-5397-15-68-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of 3,3’-diaminodiazocine 2 (left), 3,3’-di(aminomethyl)diazocine 6a (center), and 3,3’-di(aminoethyl)diazocine 6b (right). Continuous lines: 100% cis and dashed lines: PSS (385 nm) at 298.15 K in acetonitrile.
Figure 3: UV–vis spectra of 3,3’-diaminodiazocine 2 (left), 3,3’-di(aminomethyl)diazocine 6a (center), and 3,...
Conclusion
Seven symmetrically substituted diazocines 4–7 were synthesized and characterized. Oxidative C–C coupling and reductive azo condensation proved to be reliable key steps in the synthesis of these substituted diazocines. The photophysical properties of compounds 4–7 were investigated by NMR and UV–vis experiments. The previously investigated 3,3’-diaminodiazocine 2 and 4,4’-diaminodiazocine 3 exhibited poor photostationary states (PSS (385 nm): 25–30% trans). The electronic decoupling of the azobenzene unit and the oxygen and nitrogen containing functional groups (OH, OR, N3, NH2) was achieved by insertion of one or two CH2 groups. Thereby, the switching efficiencies were increased by about a factor of two (PSS (385 nm): 74–85% trans), and thus are close to the parent system 1 (87%). Moreover, the yields of the two synthetic key steps, the oxidative C–C coupling and the azo cyclization have been improved. Diazocines 4–7 are easily accessible and valuable building blocks for the synthesis of photo- and mechanoresponsive polymers such as polyurethanes, polyesters, polyamides, polyureas and polyolefines.
Supporting Information
| Supporting Information File 1: Analytical equipment, experimental procedures, NMR and UV–vis spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Ikeda, T.; Mamiya, J.-i.; Yu, Y. Angew. Chem., Int. Ed. 2007, 46, 506–528. doi:10.1002/anie.200602372
Return to citation in text: [1] -
Davis, D. A.; Hamilton, A.; Yang, J.; Cremar, L. D.; Van Gough, D.; Potisek, S. L.; Ong, M. T.; Braun, P. V.; Martínez, T. J.; White, S. R.; Moore, J. S.; Sottos, N. R. Nature 2009, 459, 68–72. doi:10.1038/nature07970
Return to citation in text: [1] -
Potisek, S. L.; Davis, D. A.; Sottos, N. R.; White, S. R.; Moore, J. S. J. Am. Chem. Soc. 2007, 129, 13808–13809. doi:10.1021/ja076189x
Return to citation in text: [1] -
Kim, S.-J.; Reneker, D. H. Polym. Bull. 1993, 31, 367–374. doi:10.1007/bf00692965
Return to citation in text: [1] -
Hosono, N.; Kajitani, T.; Fukushima, T.; Ito, K.; Sasaki, S.; Takata, M.; Aida, T. Science 2010, 330, 808–811. doi:10.1126/science.1195302
Return to citation in text: [1] -
Kim, C. B.; Wistrom, J. C.; Ha, H.; Zhou, S. X.; Katsumata, R.; Jones, A. R.; Janes, D. W.; Miller, K. M.; Ellison, C. J. Macromolecules 2016, 49, 7069–7076. doi:10.1021/acs.macromol.6b01848
Return to citation in text: [1] -
Yu, Y.; Nakano, M.; Ikeda, T. Nature 2003, 425, 145. doi:10.1038/425145a
Return to citation in text: [1] -
Bandara, H. M. D.; Burdette, S. C. Chem. Soc. Rev. 2012, 41, 1809–1825. doi:10.1039/c1cs15179g
Return to citation in text: [1] -
Luo, Q.; Cheng, H.; Tian, H. Polym. Chem. 2011, 2, 2435–2443. doi:10.1039/c1py00167a
Return to citation in text: [1] -
Ryo, S.; Ishibashi, Y.; Murakami, M.; Miyasaka, H.; Kobatake, S.; Irie, M. J. Phys. Org. Chem. 2007, 20, 953–959. doi:10.1002/poc.1190
Return to citation in text: [1] -
Corredor, C. C.; Huang, Z.-L.; Belfield, K. D.; Morales, A. R.; Bondar, M. V. Chem. Mater. 2007, 19, 5165–5173. doi:10.1021/cm071336b
Return to citation in text: [1] -
Stellacci, F.; Bertarelli, C.; Toscano, F.; Gallazzi, M. C.; Zotti, G.; Zerbi, G. Adv. Mater. (Weinheim, Ger.) 1999, 11, 292–295. doi:10.1002/(sici)1521-4095(199903)11:4<292::aid-adma292>3.0.co;2-v
Return to citation in text: [1] -
Pu, S.-Z.; Sun, Q.; Fan, C.-B.; Wang, R.-J.; Liu, G. J. Mater. Chem. C 2016, 4, 3075–3093. doi:10.1039/c6tc00110f
Return to citation in text: [1] -
Gossweiler, G. R.; Hewage, G. B.; Soriano, G.; Wang, Q.; Welshofer, G. W.; Zhao, X.; Craig, S. L. ACS Macro Lett. 2014, 3, 216–219. doi:10.1021/mz500031q
Return to citation in text: [1] -
Celestine, A.-D. N.; Beiermann, B. A.; May, P. A.; Moore, J. S.; Sottos, N. R.; White, S. R. Polymer 2014, 55, 4164–4171. doi:10.1016/j.polymer.2014.06.019
Return to citation in text: [1] -
Lee, C. K.; Beiermann, B. A.; Silberstein, M. N.; Wang, J.; Moore, J. S.; Sottos, N. R.; Braun, P. V. Macromolecules 2013, 46, 3746–3752. doi:10.1021/ma4005428
Return to citation in text: [1] -
Beiermann, B. A.; Davis, D. A.; Kramer, S. L. B.; Moore, J. S.; Sottos, N. R.; White, S. R. J. Mater. Chem. 2011, 21, 8443. doi:10.1039/c0jm03967e
Return to citation in text: [1] -
Klajn, R. Chem. Soc. Rev. 2014, 43, 148–184. doi:10.1039/c3cs60181a
Return to citation in text: [1] -
Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1
Return to citation in text: [1] [2] [3] [4] -
Tellkamp, T.; Shen, J.; Okamoto, Y.; Herges, R. Eur. J. Org. Chem. 2014, 5456–5461. doi:10.1002/ejoc.201402541
Return to citation in text: [1] -
Joshi, D. K.; Mitchell, M. J.; Bruce, D.; Lough, A. J.; Yan, H. Tetrahedron 2012, 68, 8670–8676. doi:10.1016/j.tet.2012.06.007
Return to citation in text: [1] [2] -
Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846
Return to citation in text: [1] [2] [3] -
Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d
Return to citation in text: [1] [2] [3] -
Schehr, M.; Hugenbusch, D.; Moje, T.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2018, 14, 2799–2804. doi:10.3762/bjoc.14.257
Return to citation in text: [1] [2] -
Siewertsen, R.; Schönborn, J. B.; Hartke, B.; Renth, F.; Temps, F. Phys. Chem. Chem. Phys. 2011, 13, 1054–1063. doi:10.1039/c0cp01148g
Return to citation in text: [1] [2] -
Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383
Return to citation in text: [1] -
Rau, H. Azo Compounds. Photochromism: Molecules and Systems, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp 165–192. doi:10.1016/b978-044451322-9/50008-7
Return to citation in text: [1] -
Chaudhuri, N. K.; Ball, T. J. J. Labelled Compd. Radiopharm. 1981, 18, 1189–1196. doi:10.1002/jlcr.2580180814
Return to citation in text: [1] -
Beyerman, H. C.; Bontekoe, J. S. Proc. Chem. Soc., London 1961, 249.
Return to citation in text: [1] -
Schlessinger, R. H.; Nugent, R. A. J. Am. Chem. Soc. 1982, 104, 1116–1118. doi:10.1021/ja00368a044
Return to citation in text: [1] -
Kitamura, M.; Koga, T.; Yano, M.; Okauchi, T. Synlett 2012, 23, 1335–1338. doi:10.1055/s-0031-1290958
Return to citation in text: [1] -
Meguro, T.; Yoshida, S.; Hosoya, T. Chem. Lett. 2017, 46, 473–476. doi:10.1246/cl.161159
Return to citation in text: [1] -
Stokes, B. J.; Opra, S. M.; Sigman, M. S. J. Am. Chem. Soc. 2012, 134, 11408–11411. doi:10.1021/ja305403s
Return to citation in text: [1] -
Thiraporn, A.; Rukachaisirikul, V.; Iawsipo, P.; Somwang, T.; Tadpetch, K. Eur. J. Org. Chem. 2017, 7133–7147. doi:10.1002/ejoc.201701272
Return to citation in text: [1]
| 33. | Kitamura, M.; Koga, T.; Yano, M.; Okauchi, T. Synlett 2012, 23, 1335–1338. doi:10.1055/s-0031-1290958 |
| 34. | Meguro, T.; Yoshida, S.; Hosoya, T. Chem. Lett. 2017, 46, 473–476. doi:10.1246/cl.161159 |
| 35. | Stokes, B. J.; Opra, S. M.; Sigman, M. S. J. Am. Chem. Soc. 2012, 134, 11408–11411. doi:10.1021/ja305403s |
| 36. | Thiraporn, A.; Rukachaisirikul, V.; Iawsipo, P.; Somwang, T.; Tadpetch, K. Eur. J. Org. Chem. 2017, 7133–7147. doi:10.1002/ejoc.201701272 |
| 1. | Ikeda, T.; Mamiya, J.-i.; Yu, Y. Angew. Chem., Int. Ed. 2007, 46, 506–528. doi:10.1002/anie.200602372 |
| 2. | Davis, D. A.; Hamilton, A.; Yang, J.; Cremar, L. D.; Van Gough, D.; Potisek, S. L.; Ong, M. T.; Braun, P. V.; Martínez, T. J.; White, S. R.; Moore, J. S.; Sottos, N. R. Nature 2009, 459, 68–72. doi:10.1038/nature07970 |
| 3. | Potisek, S. L.; Davis, D. A.; Sottos, N. R.; White, S. R.; Moore, J. S. J. Am. Chem. Soc. 2007, 129, 13808–13809. doi:10.1021/ja076189x |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 20. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
| 21. | Tellkamp, T.; Shen, J.; Okamoto, Y.; Herges, R. Eur. J. Org. Chem. 2014, 5456–5461. doi:10.1002/ejoc.201402541 |
| 22. | Joshi, D. K.; Mitchell, M. J.; Bruce, D.; Lough, A. J.; Yan, H. Tetrahedron 2012, 68, 8670–8676. doi:10.1016/j.tet.2012.06.007 |
| 23. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 23. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 14. | Gossweiler, G. R.; Hewage, G. B.; Soriano, G.; Wang, Q.; Welshofer, G. W.; Zhao, X.; Craig, S. L. ACS Macro Lett. 2014, 3, 216–219. doi:10.1021/mz500031q |
| 15. | Celestine, A.-D. N.; Beiermann, B. A.; May, P. A.; Moore, J. S.; Sottos, N. R.; White, S. R. Polymer 2014, 55, 4164–4171. doi:10.1016/j.polymer.2014.06.019 |
| 16. | Lee, C. K.; Beiermann, B. A.; Silberstein, M. N.; Wang, J.; Moore, J. S.; Sottos, N. R.; Braun, P. V. Macromolecules 2013, 46, 3746–3752. doi:10.1021/ma4005428 |
| 17. | Beiermann, B. A.; Davis, D. A.; Kramer, S. L. B.; Moore, J. S.; Sottos, N. R.; White, S. R. J. Mater. Chem. 2011, 21, 8443. doi:10.1039/c0jm03967e |
| 18. | Klajn, R. Chem. Soc. Rev. 2014, 43, 148–184. doi:10.1039/c3cs60181a |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 20. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
| 9. | Luo, Q.; Cheng, H.; Tian, H. Polym. Chem. 2011, 2, 2435–2443. doi:10.1039/c1py00167a |
| 10. | Ryo, S.; Ishibashi, Y.; Murakami, M.; Miyasaka, H.; Kobatake, S.; Irie, M. J. Phys. Org. Chem. 2007, 20, 953–959. doi:10.1002/poc.1190 |
| 11. | Corredor, C. C.; Huang, Z.-L.; Belfield, K. D.; Morales, A. R.; Bondar, M. V. Chem. Mater. 2007, 19, 5165–5173. doi:10.1021/cm071336b |
| 12. | Stellacci, F.; Bertarelli, C.; Toscano, F.; Gallazzi, M. C.; Zotti, G.; Zerbi, G. Adv. Mater. (Weinheim, Ger.) 1999, 11, 292–295. doi:10.1002/(sici)1521-4095(199903)11:4<292::aid-adma292>3.0.co;2-v |
| 13. | Pu, S.-Z.; Sun, Q.; Fan, C.-B.; Wang, R.-J.; Liu, G. J. Mater. Chem. C 2016, 4, 3075–3093. doi:10.1039/c6tc00110f |
| 29. | Rau, H. Azo Compounds. Photochromism: Molecules and Systems, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp 165–192. doi:10.1016/b978-044451322-9/50008-7 |
| 4. | Kim, S.-J.; Reneker, D. H. Polym. Bull. 1993, 31, 367–374. doi:10.1007/bf00692965 |
| 5. | Hosono, N.; Kajitani, T.; Fukushima, T.; Ito, K.; Sasaki, S.; Takata, M.; Aida, T. Science 2010, 330, 808–811. doi:10.1126/science.1195302 |
| 6. | Kim, C. B.; Wistrom, J. C.; Ha, H.; Zhou, S. X.; Katsumata, R.; Jones, A. R.; Janes, D. W.; Miller, K. M.; Ellison, C. J. Macromolecules 2016, 49, 7069–7076. doi:10.1021/acs.macromol.6b01848 |
| 7. | Yu, Y.; Nakano, M.; Ikeda, T. Nature 2003, 425, 145. doi:10.1038/425145a |
| 8. | Bandara, H. M. D.; Burdette, S. C. Chem. Soc. Rev. 2012, 41, 1809–1825. doi:10.1039/c1cs15179g |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 20. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 24. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 26. | Siewertsen, R.; Schönborn, J. B.; Hartke, B.; Renth, F.; Temps, F. Phys. Chem. Chem. Phys. 2011, 13, 1054–1063. doi:10.1039/c0cp01148g |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 28. | Samanta, S.; Qin, C.; Lough, A. J.; Woolley, G. A. Angew. Chem. 2012, 124, 6558–6561. doi:10.1002/ange.201202383 |
| 25. | Schehr, M.; Hugenbusch, D.; Moje, T.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2018, 14, 2799–2804. doi:10.3762/bjoc.14.257 |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 24. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 19. | Li, S.; Han, G.; Zhang, W. Macromolecules 2018, 51, 4290–4297. doi:10.1021/acs.macromol.8b00687 |
| 24. | Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. J. Am. Chem. Soc. 2009, 131, 15594–15595. doi:10.1021/ja906547d |
| 26. | Siewertsen, R.; Schönborn, J. B.; Hartke, B.; Renth, F.; Temps, F. Phys. Chem. Chem. Phys. 2011, 13, 1054–1063. doi:10.1039/c0cp01148g |
| 27. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 27. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 27. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 30. | Chaudhuri, N. K.; Ball, T. J. J. Labelled Compd. Radiopharm. 1981, 18, 1189–1196. doi:10.1002/jlcr.2580180814 |
| 27. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 32. | Schlessinger, R. H.; Nugent, R. A. J. Am. Chem. Soc. 1982, 104, 1116–1118. doi:10.1021/ja00368a044 |
| 27. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 22. | Joshi, D. K.; Mitchell, M. J.; Bruce, D.; Lough, A. J.; Yan, H. Tetrahedron 2012, 68, 8670–8676. doi:10.1016/j.tet.2012.06.007 |
| 23. | Hammerich, M.; Schütt, C.; Stähler, C.; Lentes, P.; Röhricht, F.; Höppner, R.; Herges, R. J. Am. Chem. Soc. 2016, 138, 13111–13114. doi:10.1021/jacs.6b05846 |
| 25. | Schehr, M.; Hugenbusch, D.; Moje, T.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2018, 14, 2799–2804. doi:10.3762/bjoc.14.257 |
| 27. | Moormann, W.; Langbehn, D.; Herges, R. Synthesis 2017, 49, 3471–3475. doi:10.1055/s-0036-1590685 |
| 20. | Sell, H.; Näther, C.; Herges, R. Beilstein J. Org. Chem. 2013, 9, 1–7. doi:10.3762/bjoc.9.1 |
© 2019 Moormann et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)