

Abstract
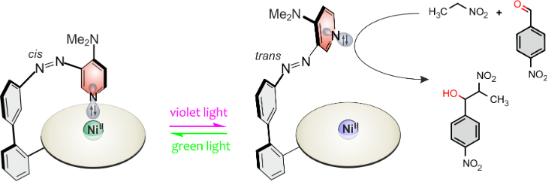
We present the synthesis and the photochemical and catalytic switching properties of an azopyridine as a photoswitchable ligand, covalently attached to a Ni(II)-porphyrin. Upon irradiation with 530 nm (green light), the azopyridine switches to the cis configuration and coordinates with the Ni2+ ion. Light of 435 nm (violet) isomerizes the ligand back to the trans configuration, which decoordinates for steric reasons. This so-called record player design has been used previously to switch the spin state of Ni2+ between singlet and triplet. We now use the coordination/decoordination process to switch the catalytic activity of the dimethylaminopyridine (DMAP) unit. DMAP is a known catalyst in the nitroaldol (Henry) reaction. Upon coordination to the Ni2+ ion, the basicity of the pyridine lone pair is attenuated and hence the catalytic activity is reduced. Decoordination restores the catalytic activity. The rate constants in the two switching states differ by a factor of 2.2, and the catalytic switching is reversible.
Graphical Abstract
Introduction
Photoswitchable catalysis has been realized following several approaches using a variety of photochromic systems. Feringa et al. recently published a review including systems based on double bond isomerizations [1]. An earlier review from the same group summarized light and redox responsive catalytic systems including azobenzenes, diarylethenes, spiropyranes, and stilbenes [2].
Diarylethenes were reported in the context of photoswitchable catalysis as inhibitors of the Karstedt´s catalyst [3] and for pKa modulation in acid–base-controlled processes [4]. Molecular motors for stereodivergent anion binding catalysis [5], azopeptides for the acetylation of sugars [6], enzyme mimics [7], and the utilization of intermolecular cooperative effects [8] are further applications of photoswitchable catalysis.
Particularly interesting and close to our approach is an early work by Inoue et al. who achieved control of the transformation of CO2 and 1,2-epoxypropane to propylene carbonate using an aluminum porphyrin and a photoresponsive ligand. The catalytic activity of the metal porphyrin depended on the axial coordination of an azostilbene and coordination of the latter ligand was controlled by photoisomerization of the stilbene unit [9].
Hecht et al. reported on the photoswitching of the basicity of a piperidine nitrogen by reversible steric shielding of the nitrogen lone pair. The photoswitchable base was applied as a nucleophilic catalyst in the nitroaldol (Henry) reaction. Attached to a sterically demanding azobenzene unit, the lone pair was shielded in the cis configuration and therefore catalytically inactive. The basicity was restored by isomerization to the trans state leading to a rapid conversion to the β-nitroalcohol, the product of the Henry reaction [10-12].
We now present a photoswitchable catalyst whose basicity is controlled by coordination/decoordination to the Ni2+ ion in a Ni-porphyrin. So-called record player molecules, including a Ni(II)-porphyrin as the square planar base complex and azopyridines as photoswitchable axial ligands, were previously investigated for spin switching applications [13-26]. In the present study, the basicity change linked to the coordination/decoordination process is explored. If the azo substituent is in cis configuration, the lone pair of the pyridine unit coordinates to the central Ni2+ ion, reducing the nucleophilic power and the basicity of the pyridine (Figure 1). In the trans configuration intramolecular coordination is prohibited and nucleophilicity and basicity are restored. Reversible switching between the two states is achieved by irradiation with green (trans→cis) and violet (cis→trans) light.
![[1860-5397-16-179-1]](/bjoc/content/figures/1860-5397-16-179-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Basicity and nucleophilicity switching of the 4-(N,N-dimethylamino)pyridine “record player” molecule 1.
Figure 1: Basicity and nucleophilicity switching of the 4-(N,N-dimethylamino)pyridine “record player” molecul...
The title compound of this study is 4-(N,N-dimethylamino)pyridine derivative 1 (Figure 1). The parent 4-(N,N-dimethylamino)pyridine (DMAP, 2) is known as a nucleophilic catalyst in a number of reactions, for example the Baylis–Hillman reaction [27] and the Steglich esterification [28,29]. To achieve control of the catalytic activity of DMAP, dimethylamino record player 1 was investigated as a photoswitchable DMAP catalyst in the nitroaldol reaction of 4-nitrobenzaldehyde and nitroethane (Henry reaction) as a model reaction.
Results and Discussion
Synthesis
For the synthesis of 4-N,N-dimethylamino record player molecule 1, a modular approach described by Heitmann et al. was chosen [30] since the alternative mixed aldehyde route failed for related porphyrin derivatives [17]. Ni-porphyrin precursor 3 and 4-(N,N-dimethylaminoazo)pyridine 4 were synthesized according to literature procedures [17,30].
Following the conditions for the Suzuki cross-coupling reaction published for a similar system [30], decomposition products and only 25% yield were obtained. Optimization by lowering the reaction temperature increased the yield to 91% (Scheme 1).
![[1860-5397-16-179-i1]](/bjoc/content/inline/1860-5397-16-179-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 4-N,N-dimethylamino record player molecule 1 by Suzuki reaction between Ni-porphyrin precursor 3 and bromo-substituted 4-(N,N-dimethylaminoazo)pyridine 4.
Scheme 1: Synthesis of 4-N,N-dimethylamino record player molecule 1 by Suzuki reaction between Ni-porphyrin p...
Switching properties of porphyrin 1
Preliminary investigations on the photochemical switching properties of porphyrin 1 were performed to optimize the reaction conditions for the catalytic process. Among the different switching wavelengths investigated, 530 nm (trans→cis) and 435 nm (cis→trans) provided the largest conversion rates (Figure S1 in Supporting Information File 1). The influence of the solvent on the switching efficiency was evaluated by UV–vis (Figure S2, Supporting Information File 1) and NMR experiments (Figure 2). It is important to note that there are two ways to define the switching efficiency of record player molecules: 1. the cis–trans isomerization of the azopyridine unit, and 2. the coordination–decoordination process. Both switching processes are connected by the molecular design; however, they are not necessarily equally efficient because the cis isomer might not coordinate completely and there might be coordination in the trans state due to intermolecular coordination. On this account, isomerization and coordination were investigated separately by 1H NMR spectroscopy. The protons meta to the azo group in the azopyridine unit (H-11) are responsive to the configuration of the azo group (cis or trans) and the chemical shifts of the pyrrole protons of the porphyrin ring are strongly dependent on the axial coordination at the central Ni2+ ion (Figure 3, and for details see Tables S1 and S2 in Supporting Information File 1). The analysis of the NMR data revealed that 98.8–99.9% of the cis isomer is coordinated in all solvents, which is expectable because DMAP is a very strong ligand with respect to axial coordination to Ni-porphyrins [25]. According to 1H NMR studies, however, the trans isomer exhibits concentration-dependent coordination, which clearly indicates intermolecular coordination. Again, this is due to the very strong coordination power of the DMAP ligand. At a total concentration of 4 mM of the Ni-porphyrin, after irradiation with 435 nm (THF-d8, 25 °C) 46% of the molecules are in cis and 54% are in trans configuration. Whereas the cis isomers are almost completely coordinated, the trans porphyrins still exhibit 7% coordination, which is due to a fast intermolecular ligand exchange [14]. At a total porphyrin concentration of 40 mM, the intermolecular coordination of the trans species increases to 25%. Taking the incomplete photochemical conversion to the trans configuration into account (60%), only 45% of the Ni-porphyrin species should be catalytically active, if a solution of 40 mM total porphyrin concentration is applied.
![[1860-5397-16-179-2]](/bjoc/content/figures/1860-5397-16-179-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Composition of the different states of porphyrin 1 (1 mM) in the PSS at 530 nm and 435 nm, determined by 1H NMR spectroscopy (600 MHz; for details, see Supporting Information File 1).
Figure 2: Composition of the different states of porphyrin 1 (1 mM) in the PSS at 530 nm and 435 nm, determin...
![[1860-5397-16-179-3]](/bjoc/content/figures/1860-5397-16-179-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: a) UV–vis cuvette with a solution of porphyrin 1 (13.1 µM in THF) and the corresponding UV–vis spectra at 25 °C in the PSS at 530 nm (green) and 435 nm (blue). b) Signals of proton H-11 (1H NMR, 600 MHz, THF-d8, 318 K), which are an indicator for the cis–trans isomerization of the azo unit of porphyrin 1, in the PSS at 530 nm (green) and 435 nm (violet-blue). The signal at 6.54 ppm belongs to the cis species while the signal at 6.41 represents the trans isomer. c) 1H NMR signals (1H NMR, 600 MHz, THF-d8, 300 K) of the pyrrole protons in the PSS at 530 nm and 435 nm. Coordination/decoordination is fast on the NMR timescale and signal averages for the pyrrole protons are observed. 50.40 ppm corresponds to 100% coordinated (paramagnetic) Ni-porphyrin and 10.83 ppm is the chemical shift of the pure decoordinated (diamagnetic) system. The signals are broad because of paramagnetic line broadening of the coordinated species and fast ligand exchange.
Figure 3: a) UV–vis cuvette with a solution of porphyrin 1 (13.1 µM in THF) and the corresponding UV–vis spec...
The photostationary states after irradiation with 435 and 530 nm are strongly solvent dependent (Figure 2). THF turned out to provide the largest conversion rate (96%) to the coordinated (cis) state [31]. The back-reaction with 56% decoordination (trans isomer) is less efficient. At the other end of the scale, in acetone only 69% coordination (530 nm) and 75% decoordination (435 nm) is achieved. It should be noted that a high conversion to the inactive state (in photocatalysis as well as photopharmacology) is more important than achieving a high conversion to the active state because the latter problem can be compensated by increasing the concentration, whereas incomplete deactivation is a problem, which cannot be circumvented. Hence, THF was chosen as the solvent of choice for the catalysis experiments.
A decreasing switching efficiency of 1 at 530 nm from trans to cis was observed at higher concentrations (83% cis-1 at 40 mM total porphyrin), whereas the switching to the trans state is almost unaffected (60%, Table S3 in Supporting Information File 1). The reduced switching efficiency from the trans to the cis isomer at higher concentrations can be explained by the intermolecular coordination, which stabilizes the trans isomer.
Long-term switching experiments were performed to investigate the fatigue resistance of 1. In contrast to other record player molecules, which are stable up to 100 000 switching cycles, the switching efficiency slowly decreased over 100 switching cycles to ≈80% of the original value (see Supporting Information File 1, Figure S4). A thermal decomposition at 40 °C in THF was observed by a color change from red to green. Hence, the catalytic experiments were limited to 25 °C.
At last, the thermal relaxation time for the cis→trans isomerization in THF was determined by 1H NMR spectroscopy. Similar to previous record player systems a long half-life of the metastable cis isomer of about 505 d (25 °C, THF-d8) was determined (Figure S5 in Supporting Information File 1). Thus, the thermal relaxation of the coordinated cis state is insignificant for our catalytic studies.
Experiments using porphyrin 1 as a catalyst
The Henry reaction (Scheme 2) was chosen as a model reaction to investigate the catalytic activity of porphyrin 1 in the two different switching states because the rate of the background reaction without catalyst is low, and the influence of the solvent on the reaction rate is small [10,32].
![[1860-5397-16-179-i2]](/bjoc/content/inline/1860-5397-16-179-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General scheme of the nitroaldol (Henry) reaction that was used to investigate photoswitchable catalysis. The following reaction conditions were applied: (0.4 M 4-nitrobenzaldehyde (6), 12 equivalents of nitroethane (5), and 4 mM or 40 mM of catalyst (see Scheme 3) at 25 °C in THF-d8) to yield 7. For details see Tables S4 and S5 in Supporting Information File 1.
Scheme 2: General scheme of the nitroaldol (Henry) reaction that was used to investigate photoswitchable cata...
No catalytic effect for the N=N functional group of azobenzenes was previously observed in similar systems [10]. Ni-porphyrin Ni-TPPF20 8 (without azopyridine substitution) does not catalyze the reaction either (Figure S6, Supporting Information File 1). Thus, it should be obvious that any catalytic effect, if detected, must be due to the DMAP unit.
In our kinetic experiments, the following compounds were used as catalysts or reference compounds: record player 1 in the PSS at 530 nm (cis/trans ratio: 93:7 at 4 mM and 83:17 at 40 mM total concentration of 1) and 435 nm (cis/trans ratio: 46:54 for 4 mM and 40:60 at 40 mM), DMAP 2, azopyridine 4, and Ni-porphyrin 8 (Scheme 3 and Figures S8 and S9, Supporting Information File 1).
![[1860-5397-16-179-i3]](/bjoc/content/inline/1860-5397-16-179-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: DMAP (2), azopyridine trans-4, record player trans- and cis-1 and Ni-porphyrin 8 were used in kinetic experiments.
Scheme 3: DMAP (2), azopyridine trans-4, record player trans- and cis-1 and Ni-porphyrin 8 were used in kinet...
All catalysts and reference compounds were used in two concentrations 4 mM (1 mol % with respect to 4-nitrobenzaldehyde (6, Figure 4a) and 40 mM (10 mol %, Figure 4b). As stated above, intermolecular coordination of trans-1 increases at higher concentrations (7% at 4 mM and 25% at 40 mM) and the trans→cis conversion drops from 93 to 83% at 40 mM concentration of 1. So one can expect that the catalytic switching efficiency of 1 should drop at higher concentrations.
![[1860-5397-16-179-4]](/bjoc/content/figures/1860-5397-16-179-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Conversion of 4-nitrobenzaldehyde (6) in the Henry reaction with nitroethane (5) as a function of time, using (a, left) 4 mM concentration of catalyst or reference compound (1 mol % compared to 4-nitrobenzaldehyde) or 40 mM catalyst (10 mol % compared to 4-nitrobenzaldehyde) (b, right). Rate constants (ktot) are given for DMAP (2) (black dots), azopyridine trans-4 (orange), photoswitchable porphyrin 1 at PSS435nm (predominantly trans-1, blue), and 1 at PSS530nm (predominantly cis-1, green). The background reaction was determined with porphyrin 8 (cyan dots) and for a sample without catalyst or reference compounds (red stars).
Figure 4: Conversion of 4-nitrobenzaldehyde (6) in the Henry reaction with nitroethane (5) as a function of t...
As expected, DMAP 2 exhibits the highest catalytic activity. The azo-substituted DMAP trans-4 is slightly less effective because of the electron-withdrawing effect of the azo group. Surprisingly, however, the catalytic effect drops by a factor of 54 (4 mM pure trans-1 vs trans-4) or by a factor of 18 (40 mM pure trans-1 vs trans-4), if the azopyridine unit of trans-4 is attached to the porphyrin core as realized in trans-1. This is unexpected because any electron-withdrawing effect of the electron poor porphyrin should be weak. There are eight bonds between the porphyrin β-position and the pyridine ring including one meta-phenyl connection attenuating the through-bond conjugation. A closer look at the kinetic data in Table 1 (sample I, II, and VII) might give an explanation for the surprisingly low catalytic activity of trans-1. While the background reaction of the Henry reaction is very slow but still measurable, the addition of Ni-porphyrin 8 inhibits this reaction completely (see also Supporting Information File 1, Figure S8). We propose that the actual nucleophile in the Henry reaction, the nitroethane anion intermediate, coordinates to the Ni-porphyrin, which slows down nucleophilic addition to the nitrobenzaldehyde. Hence, the switchable catalyst 1 contains both, a catalytic center (nitrogen lone pair) and an inhibitor (Ni2+ ion). If the nitrogen lone pair coordinates to the Ni2+ ion in the cis configuration of 1, both reactive centers keep each other at bay. The isomerization to the trans configuration unleashes the catalytic as well as the inhibitory properties of 1.
Table 1: Pseudo-first order rate constants (k) of the Henry reaction (Scheme 2) in the presence of different catalysts and reference compounds. Calculated rate constants are based on the assumption of complete isomerization and no intermolecular coordination. The concentration of the catalyst is given in mol % (in parentheses) with respect to the initial nitrobenzaldehyde (6) concentration.
| sample | catalyst (mol %) | cis/trans ratio | k [s−1] |
| I | blank (0%) | – | 3.32 × 10−9 |
| II | Ni-TPPF20 8 (1%) | – | –a |
| III | DMAP 2 (1%) | – | 6.33 × 10−6 |
| IV | trans-NMe2-azopyridine 4 (1%) | 0:100 | 2.32 × 10−6 |
| V | 530 nm PSS: NMe2-record player 1 (1%) | 93:7 | 2.57 × 10−8 |
| VI | 435 nm PSS: NMe2-record player 1 (1%) | 46:54 | 3.38 × 10−8 |
| calc.b | cis-NMe2-record player 1 (1%) | 100:0 | (2.45 × 10−8)b |
| calc.b | trans-NMe2-record player 1 (1%) | 0:100 | (4.30 × 10−8)b |
| VII | Ni-TPPF20 8 (10%) | – | –a |
| VIII | DMAP 2 (10%) | – | 1.09 × 10−4 |
| IX | trans-NMe2-azopyridine 4 (10%) | 0:100 | 3.08 × 10−5 |
| X | 530 nm PSS: NMe2-record player 1 (10%) | 83:17 | 3.84 × 10−7 |
| XI | 435 nm PSS: NMe2-record player 1 (10%) | 40:60 | 8.62 × 10−7 |
| calc.b | cis-NMe2-record player 1 (10%) | 100:0 | (1.95 × 10−7)b |
| calc.b | trans-NMe2-record player 1 (10%) | 0:100 | (1.68 × 10−6)b |
aVery slow, not measurable; bassuming complete isomerization and no intermolecular coordination. For details see Supporting Information File 1, section IV.
Nevertheless, the reaction was faster after irradiation of the catalyst 1 (4 mM in THF-d8) with 435 nm (54% trans, 46% cis isomer) as compared to the catalyst 1 irradiated with 530 nm (7% trans, 93% cis isomer) by a factor of 1.3. At a concentration of 1 of 40 mM the photoswitchable catalyst at PSS435nm (60% trans, 40% cis) is 2.25 times more efficient as the catalyst at PSS530nm (17% trans, 83% cis) (Figure 4a and b). The theoretical reaction rate k0 of the nitroaldol reaction with 100% uncoordinated trans-1 (40 mM) is 8.6 times higher than the reaction rate with pure cis-1. Thus, incomplete conversion and intermolecular coordination are further reasons for the relatively low switching efficiency.
Conclusion
A photoswitchable base catalyst for the nitroaldol (Henry) reaction was synthesized and investigated. Principle of function is the control of the basicity of a DMAP derivative by light-induced coordination/decoordination of an azo-DMAP unit covalently bound to a Ni-porphyrin. Upon irradiation with 530 nm (green) light and 435 nm (violet) light, the nitroaldol reaction was accelerated/decelerated by a factor of 2.25. One of the factors reducing the catalytic activity and switching efficiency, obviously, was the deactivation of the nitroalkyl anion intermediate by coordination to the Ni-porphyrin. The DMAP-catalyzed acetylation of alcohols should not suffer from this drawback because the reactive intermediate (acylpyridinium cation) is positively charged and does not coordinate to the Ni2+ ion [33]. Another promising approach towards coordination-based, photoswitchable catalysts are photodissociable ligands. These ligands coordinate in their trans form and decoordinate in the cis configuration driven by steric hindrance [14,34,35].
Supporting Information
| Supporting Information File 1: General information, synthetic and photophysical procedures, and copies of NMR spectra. | ||
| Format: PDF | Size: 931.4 KB | Download |
References
-
Dorel, R.; Feringa, B. L. Chem. Commun. 2019, 55, 6477–6486. doi:10.1039/c9cc01891c
Return to citation in text: [1] -
Vlatković, M.; Collins, B. S. L.; Feringa, B. L. Chem. – Eur. J. 2016, 22, 17080–17111. doi:10.1002/chem.201602453
Return to citation in text: [1] -
Kaur, B.; Raza, R.; Stashick, M. J.; Branda, N. R. Org. Chem. Front. 2019, 6, 1253–1256. doi:10.1039/c9qo00221a
Return to citation in text: [1] -
Gurke, J.; Budzák, Š.; Schmidt, B. M.; Jacquemin, D.; Hecht, S. Angew. Chem., Int. Ed. 2018, 57, 4797–4801. doi:10.1002/anie.201801270
Return to citation in text: [1] -
Dorel, R.; Feringa, B. L. Angew. Chem., Int. Ed. 2020, 59, 785–789. doi:10.1002/anie.201913054
Return to citation in text: [1] -
Niedek, D.; Erb, F. R.; Topp, C.; Seitz, A.; Wende, R. C.; Eckhardt, A. K.; Kind, J.; Herold, D.; Thiele, C. M.; Schreiner, P. R. J. Org. Chem. 2020, 85, 1835–1846. doi:10.1021/acs.joc.9b01913
Return to citation in text: [1] -
Saha, M.; Bandyopadhyay, S. Chem. Commun. 2019, 55, 3294–3297. doi:10.1039/c9cc00018f
Return to citation in text: [1] -
Ren, C. Z.-J.; Solís Muñana, P.; Dupont, J.; Zhou, S. S.; Chen, J. L.-Y. Angew. Chem., Int. Ed. 2019, 58, 15254–15258. doi:10.1002/anie.201907078
Return to citation in text: [1] -
Sugimoto, H.; Kimura, T.; Inoue, S. J. Am. Chem. Soc. 1999, 121, 2325–2326. doi:10.1021/ja983960i
Return to citation in text: [1] -
Peters, M. V.; Stoll, R. S.; Kühn, A.; Hecht, S. Angew. Chem. 2008, 120, 6056–6060. doi:10.1002/ange.200802050
Return to citation in text: [1] [2] [3] -
Hecht, S.; Peters, M. V.; Stoll, R. S. Photoschaltbare Katalysatoren. German Patent DE102006057612.8, Dec 5, 2006.
Return to citation in text: [1] -
Stoll, R. S.; Peters, M. V.; Kuhn, A.; Heiles, S.; Goddard, R.; Bühl, M.; Thiele, C. M.; Hecht, S. J. Am. Chem. Soc. 2009, 131, 357–367. doi:10.1021/ja807694s
Return to citation in text: [1] -
Venkataramani, S.; Jana, U.; Dommaschk, M.; Sönnichsen, F. D.; Tuczek, F.; Herges, R. Science 2011, 331, 445–448. doi:10.1126/science.1201180
Return to citation in text: [1] -
Dommaschk, M.; Schütt, C.; Venkataramani, S.; Jana, U.; Näther, C.; Sönnichsen, F. D.; Herges, R. Dalton Trans. 2014, 43, 17395–17405. doi:10.1039/c4dt03048f
Return to citation in text: [1] [2] [3] -
Dommaschk, M.; Näther, C.; Herges, R. J. Org. Chem. 2015, 80, 8496–8500. doi:10.1021/acs.joc.5b01524
Return to citation in text: [1] -
Dommaschk, M.; Thoms, V.; Schütt, C.; Näther, C.; Puttreddy, R.; Rissanen, K.; Herges, R. Inorg. Chem. 2015, 54, 9390–9392. doi:10.1021/acs.inorgchem.5b01756
Return to citation in text: [1] -
Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929
Return to citation in text: [1] [2] [3] -
Ludwig, J.; Gröbner, J.; Dommaschk, M.; Huber, L. M.; Peters, M. K.; Hövener, J.-B.; Herges, R. J. Porphyrins Phthalocyanines 2020, 24, 480–488. doi:10.1142/s1088424619501803
Return to citation in text: [1] -
Herges, R.; Dommaschk, M.; Heitmann, G.; Gröbner, J. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. German Pat. Appl. DE102017119482A1, Feb 28, 2019.
Return to citation in text: [1] -
Herges, R.; Dommaschk, M.; Heitmann, G.; Gröbner, J.; Thoms, V. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. WO Pat. Appl. WO2019/038428A2, Feb 28, 2019.
Return to citation in text: [1] -
Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g
Return to citation in text: [1] -
Gutzeit, F.; Dommaschk, M.; Levin, N.; Buchholz, A.; Schaub, E.; Plass, W.; Näther, C.; Herges, R. Inorg. Chem. 2019, 58, 12542–12546. doi:10.1021/acs.inorgchem.9b00348
Return to citation in text: [1] -
Matino, F.; Schull, G.; Jana, U.; Köhler, F.; Berndt, R.; Herges, R. Chem. Commun. 2010, 46, 6780–6782. doi:10.1039/c0cc00959h
Return to citation in text: [1] -
Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F. D.; Näther, C.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2010, 16, 10074–10083. doi:10.1002/chem.201000603
Return to citation in text: [1] -
Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d
Return to citation in text: [1] [2] -
Köbke, A.; Gutzeit, F.; Röhricht, F.; Schlimm, A.; Grunwald, J.; Tuczek, F.; Studniarek, M.; Longo, D.; Choueikani, F.; Otero, E.; Ohresser, P.; Rohlf, S.; Johannsen, S.; Diekmann, F.; Rossnagel, K.; Weismann, A.; Jasper-Toennies, T.; Näther, C.; Herges, R.; Berndt, R.; Gruber, M. Nat. Nanotechnol. 2020, 15, 18–21. doi:10.1038/s41565-019-0594-8
Return to citation in text: [1] -
Octavio, R.; de Souza, M. A.; Vasconcellos, M. L. A. A. Synth. Commun. 2003, 33, 1383–1389. doi:10.1081/scc-120018699
Return to citation in text: [1] -
Steglich, W.; Höfle, G. Angew. Chem., Int. Ed. Engl. 1969, 8, 981. doi:10.1002/anie.196909811
Return to citation in text: [1] -
Neises, B.; Steglich, W. Org. Synth. 1985, 63, 183. doi:10.15227/orgsyn.063.0183
Return to citation in text: [1] -
Heitmann, G.; Dommaschk, M.; Löw, R.; Herges, R. Org. Lett. 2016, 18, 5228–5231. doi:10.1021/acs.orglett.6b02507
Return to citation in text: [1] [2] [3] -
Dommaschk, M. Intramolekularer Light-Driven-Coordination-Induced-Spin-State-Switch (LD-CISSS) in Nickel(II)porphyrinen zur Entwicklung photoschaltbarer MRT-Kontrastmittel”; Diplomarbeit, Christian-Abrechts-Universität zu Kiel, 2011; pp 45f.
Return to citation in text: [1] -
Kürti, L.; Czako, B. Strategic Applications of Named Reactions in Organic Synthesis; Academic Press: London, U.K., 2009; pp 202–203.
Return to citation in text: [1] -
Xu, S.; Held, I.; Kempf, B.; Mayr, H.; Steglich, W.; Zipse, H. Chem. – Eur. J. 2005, 11, 4751–4757. doi:10.1002/chem.200500398
Return to citation in text: [1] -
Thies, S.; Sell, H.; Bornholdt, C.; Schütt, C.; Köhler, F.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2012, 18, 16358–16368. doi:10.1002/chem.201201698
Return to citation in text: [1] -
Peters, M. K.; Hamer, S.; Jäkel, T.; Röhricht, F.; Sönnichsen, F. D.; von Essen, C.; Lahtinen, M.; Naether, C.; Rissanen, K.; Herges, R. Inorg. Chem. 2019, 58, 5265–5272. doi:10.1021/acs.inorgchem.9b00349
Return to citation in text: [1]
| 14. | Dommaschk, M.; Schütt, C.; Venkataramani, S.; Jana, U.; Näther, C.; Sönnichsen, F. D.; Herges, R. Dalton Trans. 2014, 43, 17395–17405. doi:10.1039/c4dt03048f |
| 30. | Heitmann, G.; Dommaschk, M.; Löw, R.; Herges, R. Org. Lett. 2016, 18, 5228–5231. doi:10.1021/acs.orglett.6b02507 |
| 25. | Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d |
| 1. | Dorel, R.; Feringa, B. L. Chem. Commun. 2019, 55, 6477–6486. doi:10.1039/c9cc01891c |
| 5. | Dorel, R.; Feringa, B. L. Angew. Chem., Int. Ed. 2020, 59, 785–789. doi:10.1002/anie.201913054 |
| 17. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 4. | Gurke, J.; Budzák, Š.; Schmidt, B. M.; Jacquemin, D.; Hecht, S. Angew. Chem., Int. Ed. 2018, 57, 4797–4801. doi:10.1002/anie.201801270 |
| 17. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 30. | Heitmann, G.; Dommaschk, M.; Löw, R.; Herges, R. Org. Lett. 2016, 18, 5228–5231. doi:10.1021/acs.orglett.6b02507 |
| 3. | Kaur, B.; Raza, R.; Stashick, M. J.; Branda, N. R. Org. Chem. Front. 2019, 6, 1253–1256. doi:10.1039/c9qo00221a |
| 28. | Steglich, W.; Höfle, G. Angew. Chem., Int. Ed. Engl. 1969, 8, 981. doi:10.1002/anie.196909811 |
| 29. | Neises, B.; Steglich, W. Org. Synth. 1985, 63, 183. doi:10.15227/orgsyn.063.0183 |
| 14. | Dommaschk, M.; Schütt, C.; Venkataramani, S.; Jana, U.; Näther, C.; Sönnichsen, F. D.; Herges, R. Dalton Trans. 2014, 43, 17395–17405. doi:10.1039/c4dt03048f |
| 34. | Thies, S.; Sell, H.; Bornholdt, C.; Schütt, C.; Köhler, F.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2012, 18, 16358–16368. doi:10.1002/chem.201201698 |
| 35. | Peters, M. K.; Hamer, S.; Jäkel, T.; Röhricht, F.; Sönnichsen, F. D.; von Essen, C.; Lahtinen, M.; Naether, C.; Rissanen, K.; Herges, R. Inorg. Chem. 2019, 58, 5265–5272. doi:10.1021/acs.inorgchem.9b00349 |
| 2. | Vlatković, M.; Collins, B. S. L.; Feringa, B. L. Chem. – Eur. J. 2016, 22, 17080–17111. doi:10.1002/chem.201602453 |
| 30. | Heitmann, G.; Dommaschk, M.; Löw, R.; Herges, R. Org. Lett. 2016, 18, 5228–5231. doi:10.1021/acs.orglett.6b02507 |
| 9. | Sugimoto, H.; Kimura, T.; Inoue, S. J. Am. Chem. Soc. 1999, 121, 2325–2326. doi:10.1021/ja983960i |
| 13. | Venkataramani, S.; Jana, U.; Dommaschk, M.; Sönnichsen, F. D.; Tuczek, F.; Herges, R. Science 2011, 331, 445–448. doi:10.1126/science.1201180 |
| 14. | Dommaschk, M.; Schütt, C.; Venkataramani, S.; Jana, U.; Näther, C.; Sönnichsen, F. D.; Herges, R. Dalton Trans. 2014, 43, 17395–17405. doi:10.1039/c4dt03048f |
| 15. | Dommaschk, M.; Näther, C.; Herges, R. J. Org. Chem. 2015, 80, 8496–8500. doi:10.1021/acs.joc.5b01524 |
| 16. | Dommaschk, M.; Thoms, V.; Schütt, C.; Näther, C.; Puttreddy, R.; Rissanen, K.; Herges, R. Inorg. Chem. 2015, 54, 9390–9392. doi:10.1021/acs.inorgchem.5b01756 |
| 17. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 18. | Ludwig, J.; Gröbner, J.; Dommaschk, M.; Huber, L. M.; Peters, M. K.; Hövener, J.-B.; Herges, R. J. Porphyrins Phthalocyanines 2020, 24, 480–488. doi:10.1142/s1088424619501803 |
| 19. | Herges, R.; Dommaschk, M.; Heitmann, G.; Gröbner, J. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. German Pat. Appl. DE102017119482A1, Feb 28, 2019. |
| 20. | Herges, R.; Dommaschk, M.; Heitmann, G.; Gröbner, J.; Thoms, V. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. WO Pat. Appl. WO2019/038428A2, Feb 28, 2019. |
| 21. | Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g |
| 22. | Gutzeit, F.; Dommaschk, M.; Levin, N.; Buchholz, A.; Schaub, E.; Plass, W.; Näther, C.; Herges, R. Inorg. Chem. 2019, 58, 12542–12546. doi:10.1021/acs.inorgchem.9b00348 |
| 23. | Matino, F.; Schull, G.; Jana, U.; Köhler, F.; Berndt, R.; Herges, R. Chem. Commun. 2010, 46, 6780–6782. doi:10.1039/c0cc00959h |
| 24. | Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F. D.; Näther, C.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2010, 16, 10074–10083. doi:10.1002/chem.201000603 |
| 25. | Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d |
| 26. | Köbke, A.; Gutzeit, F.; Röhricht, F.; Schlimm, A.; Grunwald, J.; Tuczek, F.; Studniarek, M.; Longo, D.; Choueikani, F.; Otero, E.; Ohresser, P.; Rohlf, S.; Johannsen, S.; Diekmann, F.; Rossnagel, K.; Weismann, A.; Jasper-Toennies, T.; Näther, C.; Herges, R.; Berndt, R.; Gruber, M. Nat. Nanotechnol. 2020, 15, 18–21. doi:10.1038/s41565-019-0594-8 |
| 10. | Peters, M. V.; Stoll, R. S.; Kühn, A.; Hecht, S. Angew. Chem. 2008, 120, 6056–6060. doi:10.1002/ange.200802050 |
| 8. | Ren, C. Z.-J.; Solís Muñana, P.; Dupont, J.; Zhou, S. S.; Chen, J. L.-Y. Angew. Chem., Int. Ed. 2019, 58, 15254–15258. doi:10.1002/anie.201907078 |
| 27. | Octavio, R.; de Souza, M. A.; Vasconcellos, M. L. A. A. Synth. Commun. 2003, 33, 1383–1389. doi:10.1081/scc-120018699 |
| 33. | Xu, S.; Held, I.; Kempf, B.; Mayr, H.; Steglich, W.; Zipse, H. Chem. – Eur. J. 2005, 11, 4751–4757. doi:10.1002/chem.200500398 |
| 7. | Saha, M.; Bandyopadhyay, S. Chem. Commun. 2019, 55, 3294–3297. doi:10.1039/c9cc00018f |
| 31. | Dommaschk, M. Intramolekularer Light-Driven-Coordination-Induced-Spin-State-Switch (LD-CISSS) in Nickel(II)porphyrinen zur Entwicklung photoschaltbarer MRT-Kontrastmittel”; Diplomarbeit, Christian-Abrechts-Universität zu Kiel, 2011; pp 45f. |
| 6. | Niedek, D.; Erb, F. R.; Topp, C.; Seitz, A.; Wende, R. C.; Eckhardt, A. K.; Kind, J.; Herold, D.; Thiele, C. M.; Schreiner, P. R. J. Org. Chem. 2020, 85, 1835–1846. doi:10.1021/acs.joc.9b01913 |
| 10. | Peters, M. V.; Stoll, R. S.; Kühn, A.; Hecht, S. Angew. Chem. 2008, 120, 6056–6060. doi:10.1002/ange.200802050 |
| 11. | Hecht, S.; Peters, M. V.; Stoll, R. S. Photoschaltbare Katalysatoren. German Patent DE102006057612.8, Dec 5, 2006. |
| 12. | Stoll, R. S.; Peters, M. V.; Kuhn, A.; Heiles, S.; Goddard, R.; Bühl, M.; Thiele, C. M.; Hecht, S. J. Am. Chem. Soc. 2009, 131, 357–367. doi:10.1021/ja807694s |
| 10. | Peters, M. V.; Stoll, R. S.; Kühn, A.; Hecht, S. Angew. Chem. 2008, 120, 6056–6060. doi:10.1002/ange.200802050 |
| 32. | Kürti, L.; Czako, B. Strategic Applications of Named Reactions in Organic Synthesis; Academic Press: London, U.K., 2009; pp 202–203. |
© 2020 Ludwig et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
