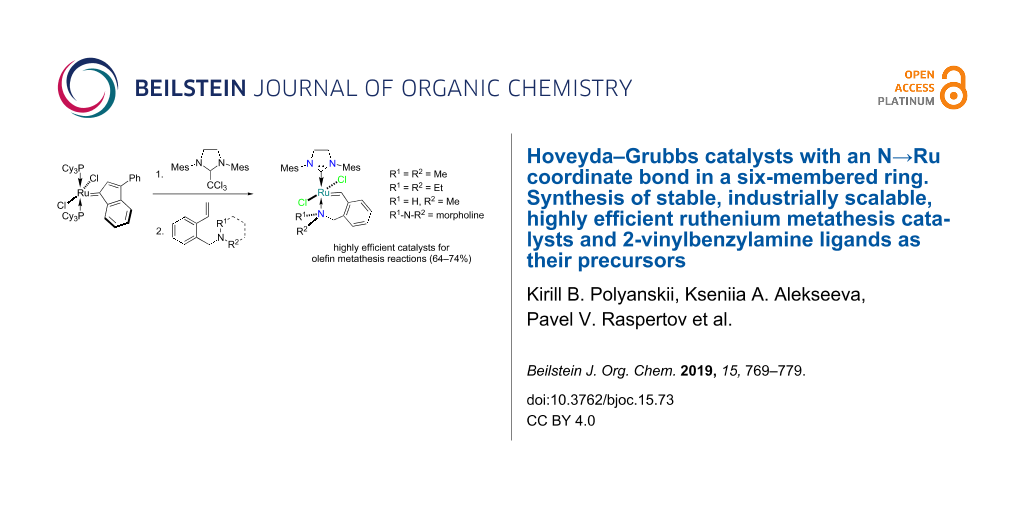
Abstract
A novel and efficient approach to the synthesis of 2-vinylbenzylamines is reported. This involves obtaining 2-vinylbenzylamine ligands from tetrahydroisoquinoline by alkylation and reduction followed by the Hofmann cleavage. The resultant 2-vinylbenzylamines allowed us to obtain new Hoveyda–Grubbs catalysts, which were thoroughly characterised by NMR, ESIMS, and X-ray crystallography. The utility of this chemistry is further demonstrated by the tests of the novel catalysts (up to 10−2 mol %) in different metathesis reactions such as cross metathesis (CM), ring-closing metathesis (RCM) and ring-opening cross metathesis (ROCM).

Graphical Abstract
Introduction
Ruthenium-catalysed olefin metathesis reactions have been playing an important role in various fields of organic synthesis in the past three decades. The significance of this transformation is confirmed by more than 20 reviews devoted to various aspects of metathesis reactions, which were published in last three years (2016–2018). In this paper, we mention only a few of them [1-12], including most popular recent books [13,14]. Obviously, the application of various catalysts is required to achieve the best results in each of the many directions of metathesis reactions such as cross metathesis – CM, ring-opening metathesis – ROM, ring-closing metathesis – RCM, ring-opening metathesis polymerization – ROMP and acyclic diene metathesis – ADMET. This motivates the investigations into the development of new, efficient, stable, and highly selective catalytic systems based on ruthenium complexes. However, in reality, a limited set of commercially available catalysts is used for the whole range of metathesis reactions most probably due to economical reasons. For example, in 2018, Merck offered more than 20 ruthenium metathesis catalysts. The most popular of them are shown in Figure 1.
![[1860-5397-15-73-1]](/bjoc/content/figures/1860-5397-15-73-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Commercially available ruthenium catalysts for metathesis reactions.
Figure 1: Commercially available ruthenium catalysts for metathesis reactions.
The framework of these catalysts consists of two main parts that surround the ruthenium centre – “the upper” one is the N-heterocyclic carbene (NHC) ligand and “the lower” one is the 2-alkoxybenzylidene ligand. These determine the principal catalytic properties of the ruthenium complexes. Many ligands were tested as the upper part in various publications, which concluded that NHCs groups, in particular, 1,3-bis(2,4,6-trimethylphenyl)imidazolidine are superior in terms of price/quality ratio. We suppose that further advances should be rather aimed at the lower part of the ruthenium complex [15-17]. This trend is confirmed partly by the Grubbs catalysts with a pyridine group (Figure 1) recently released to the market.
It has been experimentally established that the catalysts with an O-isopropyl group in their structure usually show the best combination of stability and activity. However, the oxygen atom having only one site for modification (i.e., the alkyl substituent) reduces the potential diversity of the chemical environment of the catalytic centre [18-21]. In our opinion, replacing the oxygen atom by the nitrogen atom in the lower part of the catalyst would enhance the variability for both steric and electronic effects of the substituents (Figure 1). This would enable a rational selection of the optimal catalysts for specific metathesis reactions.
It should be mentioned here that the idea of replacing the oxygen atom with nitrogen in the Grubbs catalyst is not new but only a limited number of examples are available in the literature [22-38]. Moreover, a small number of patents, which describe the applications of such type of catalysts in ROMP reactions, were published [25-28]. Among all these applications, the use of the catalysts for the polymerization of dicyclopentadiene has the greatest industrial importance [3,9,29-31]. Noteworthy, there are only rare and sporadic publications describing the synthesis and properties of the Grubbs catalysts with an N→Ru coordinate bond in a six-membered ring [32-38].
Thus, the present work opens a series of studies by our group, which will be devoted to the synthesis and reactivity of Hoveyda–Grubbs-type catalysts possessing a six-membered ruthenium-containing ring with the N→Ru, S→Ru, and Se (Te)→Ru coordinate bonds.
Results and Discussion
2-Vinylbenzylamine synthesis
The assembly of the nitrogen-containing ruthenium catalysts required preliminary synthesis of the imidazolium ligand and o-vinylbenzylamines (Figure 2). Whereas numerous methods for the preparation of the carbene precursor are known, no satisfactory suitable approach for the synthesis of ortho-substituted styrenes was found.
![[1860-5397-15-73-2]](/bjoc/content/figures/1860-5397-15-73-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthesis of the ruthenium catalysts.
Figure 2: Retrosynthesis of the ruthenium catalysts.
Several methods have been reported for the synthesis of 2-(N,N-dialkylaminomethyl)styrenes [39-46] relying on different approaches: i) from o-vinylbenzyl chloride [43], ii) by the Hofmann cleavage of quaternary tetrahydroisoquinoline salts under the action of silver oxide [39,42] and iii) by a reaction of 2-(2-bromoethyl)benzyl bromide with secondary amines under microwave irradiation followed by the decomposition of the products under the action of potassium tert-butoxide [46]. All of the above routes offer some advantages but they all are rather expensive.
Thus, the initial stage of this work included the development of a preparative scalable method for the synthesis of vinylbenzenes, which provided a wide range of o-aminomethylstyrenes from readily accessible reagents in good yields avoiding formation of byproducts. In this process, the synthesis of 2-vinylbenzylamines from isoquinolines (Scheme 1) involved the following steps: alkylation of isoquinolines to afford isoquinolinium salts 1, its reduction with formic acid giving rise to tetrahydroisoquinolines 2 in a nearly quantitative yield, which upon alkylation gave quaternary salts 3, finally these salts underwent the Hofmann elimination to form N,N-dialkylaminomethylstyrenes 4 in yields higher than 60% (Table 1). Our attempts to synthesise highly sterically hindered 2-vinyl-N,N-diisopropylbenzylamine by an analogous method failed at the stage of the quaternary salt 3.
![[1860-5397-15-73-i1]](/bjoc/content/inline/1860-5397-15-73-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Efficient multigram synthesis of N,N-dialkyl-2-vinylbenzylamines 4 (R1X = Me2SO4, Et2SO4 or BnCl, see Experimental part, Supporting Information File 1 and Table 1).
Scheme 1: Efficient multigram synthesis of N,N-dialkyl-2-vinylbenzylamines 4 (R1X = Me2SO4, Et2SO4 or BnCl, s...
Table 1: Yields of target 2-vinylbenzylamines 4 after four steps.
| entry | compound | R1 | R2 | R2X | yield, %а |
|---|---|---|---|---|---|
| 1 | 4a | Me | Me | Me2SO4 | 87 |
| 2 | 4b | Me | Et | Et2SO4 | 88 |
| 3 | 4c | Me | Bn | BnCl | 78 |
| 4 | 4d | Me | iPr | iPrI | 80 |
| 5 | 4e | Et | Et | Et2SO4 | 76 |
| 6 | 4f | Et | iPr | iPrI | 72 |
| 7 | 4g | Bn | Bn | BnCl | 60 |
aAll yields are given after flash column chromatography or vacuum distillation.
The application of terminal dihalogen derivatives afforded styrenes 5 with a cyclic tertiary amino group from 1,2,3,4-tetrahydroisoquinoline (Scheme 2). In this case, the initial isoquinoline was reduced in the presence of formic acid and then converted into the desired products 5 by a one-pot solvent-free reaction under the action of the corresponding dihalide in alkaline media in a total yield of 61–73% (Table 2).
![[1860-5397-15-73-i2]](/bjoc/content/inline/1860-5397-15-73-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of N-(2-ethenylbenzyl)heterocycles 5.
Scheme 2: Synthesis of N-(2-ethenylbenzyl)heterocycles 5.
Table 2: Structure and yields of N-(2-vinylbenzyl)heterocycles 5.
| entry | compound | structure | initial alkyl halide (HalCH2-R-CH2Hal) | yield, %a |
|---|---|---|---|---|
| 1 | 5a |
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-73-i7.svg?max-width=637&scale=1.0)
|
Br(CH2)4Br | 61 |
| 2 | 5b |
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-73-i8.svg?max-width=637&scale=1.0)
|
Br(CH2)5Br | 73 |
| 3 | 5c |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-73-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-73-i10.svg?max-width=637&scale=1.0)
|
72 |
| 4 | 5d |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-73-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-73-i12.svg?max-width=637&scale=1.0)
|
70 |
| 5 | 5e |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-73-i13.svg?max-width=637&scale=1.0)
|
(ClCH2CH2)2O | 72 |
aAll yields are given after column chromatography or vacuum distillation.
It should be noted that the above-described method was useful for the synthesis of styrenes in quantities of up to 100 g (or even more, if necessary). This scalability was purposefully demonstrated by the multigram synthesis of 5e (see Experimental part in Supporting Information File 1).
Despite a considerable scope for varying substituents at the nitrogen atom in styrenes 4 and 5, the developed procedures (Scheme 1 and Scheme 2) do not allow one to obtain benzylamines with a secondary nitrogen atom. The approach outlined in Scheme 3 makes it possible to overcome this problem [44].
![[1860-5397-15-73-i3]](/bjoc/content/inline/1860-5397-15-73-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of N-monoalkyl-2-vinylbenzylamine 7.
Scheme 3: Synthesis of N-monoalkyl-2-vinylbenzylamine 7.
Thus, the pathways described in Schemes 1–3 permit one to vary the steric volume of substituents at the nitrogen atom in a wide range enabling synthesis of selective Grubbs catalysts with different catalytic activity. These styrenes were used in the preparation of the target ruthenium complexes shown in Scheme 4. This transformation was carried out using known standard methods including the interaction of the indenylidene derivative 8 with 1,3-dimesityl-2-(trichloromethyl)imidazolidine (9) [47-50].
![[1860-5397-15-73-i4]](/bjoc/content/inline/1860-5397-15-73-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of Hoveyda–Grubbs-type catalysts 11.
Scheme 4: Synthesis of Hoveyda–Grubbs-type catalysts 11.
Several approaches have been described earlier for the preparation of the “chloroform adduct” (9) [51-55]. Even though these approaches provide good yields they have some drawbacks such as the use of expensive reagents, difficulties in the purification process, and data analysis. Here we propose an alternative reliable procedure for the synthesis of 2-(trichloromethyl)-1,3-bis(2,4,6-trimethylphenyl)imidazolidine (9), which was successfully scaled up to 15 g and 30 g (Scheme 5, see Experimental part in Supporting Information File 1). In this process, the key differences are the use of the Hung’s base at the cyclization stage and of granulated alkali in the last step, which provides the target high-purity imidazolidine in 85–87% yield. We stress that there is no need for the isolation and purification of intermediate substances.
![[1860-5397-15-73-i5]](/bjoc/content/inline/1860-5397-15-73-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the “chloroform adduct” 9.
Scheme 5: Synthesis of the “chloroform adduct” 9.
The introduction of Ru-indenylidene complex 8 in one-pot reaction with adduct 9 followed by reaction with styrenes 4a, 4e, 5e or 7 gave target Hoveyda–Grubbs-type catalysts 11 in moderate yields (Scheme 4). The products were light-green powders. The synthesised catalysts demonstrated prominent stability in air at room temperature for at least 4 years, which was proved by 1H NMR spectra. The simple spectrum recorded in 2014 and at the end of 2018 were identical, they did not show new signals. The catalysts have good solubility in CH2Cl2, CHCl3 and moderate solubility in benzene and toluene. Therefore, they can be used for almost any purpose.
Among all of the synthesised catalysts, only three catalysts 11a–c were obtained as good crystals suitable for X-ray diffraction analysis. Unfortunately, we were not able to obtain single crystals of satisfactory quality for the morpholino-containing catalyst 11d. Still, the accessible X-ray structural information is sufficient to correlate structure with catalytic activity as presented in the following section (Figure 3). According to the X-ray data, the molecules 11a–c comprise a heterocyclic system with a five-coordinated ruthenium atom having similar general geometrical features. Two chlorine atoms occupy an ordinary trans-position relative to the central ruthenium atom. The ruthenium-containing six-membered ring has a slightly distorted envelope conformation with a ca. 51° to 54° deviation of the nitrogen atom from the mean plane of other five atoms. The most important feature of the catalyst structure is the length of the ruthenium–nitrogen bond, which should have the strongest effect on the catalytic activity. With the increase of the steric volume of substituents at the nitrogen atom, the ruthenium–nitrogen coordinate bond is extended, which makes it weaker. That should, obviously, increase the activity of the catalyst towards metathesis reactions. An increase in the N→Ru bond length along the series 11c (2.193 Å) – 11a (2.243 Å) – 11b (2.297 Å) suggests that compound 11b (with the NEt2 substituent) is expected to be the most active as a catalyst.
![[1860-5397-15-73-3]](/bjoc/content/figures/1860-5397-15-73-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected X-ray data for ruthenium complexes 11a–c. All hydrogen atoms were deleted for clarity (except for the hydrogen atom belonging to the NH group in compound 11c).
Figure 3: Selected X-ray data for ruthenium complexes 11a–c. All hydrogen atoms were deleted for clarity (exc...
It should be mentioned that some of catalysts 11 have been synthesized recently in a more complex way in lower yields [32].
The third part of this study was devoted to the demonstration of catalytic properties of metallo-complexes 11 in “standard” metathesis reactions (Scheme 6, Table 3). As a model substrate we chose easily available alkenes and dienes, such as i) styrene (12) and allylbenzene (14) for CM reactions, ii) diethyl diallylmalonate (17) and diallyltosylamide (19) for RCM reactions, iii) norbornene (21) and styrene/hex-1-ene for ROCM metathesis reactions. This selection of model subtests for metathesis is also explained by the possibility to control the course of metathesis and the composition of the reaction mixtures by the GС–MS technique only (GC–MS was carried out using external calibration for CM and RCM reactions). The validity of quantitative GC–MS analysis was confirmed by additional LC–MS and 1H NMR analysis of selected reaction mixtures.
![[1860-5397-15-73-i6]](/bjoc/content/inline/1860-5397-15-73-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Catalytic activity of compounds 11 in the metathesis reactions.
Scheme 6: Catalytic activity of compounds 11 in the metathesis reactions.
Table 3: Reaction conditions and yields of the metathesis products.
| entry | starting compound | catalyst | catalyst concentration (mol %) | solventa (conditions) | yield (%), ratiob of the products |
|---|---|---|---|---|---|
| 1 | 12 | 11a | 1 | PhMe (Ar) | 13 (traces)c |
| 2 | 12 | 11b | 1 | PhMe (Ar) | 12/13 (43%), 51:49 |
| 3 | 12 | 11a | 1 | PhH (Ar) | 13 (traces)c |
| 4 | 12 | 11b | 1 | PhH (Ar) | 12/13 (52%), 40:60 |
| 5 | 12 | 11a | 1 | MeCN (Ar) | no productc |
| 6 | 12 | 11a | 1 | THF (Ar) | no productc |
| 7 | 12 | 11a | 1 | CH2Cl2 (Ar) | 13 (17%)d |
| 8 | 12 | 11a | 0.1 | CH2Cl2 (Ar) | 13 (traces)c |
| 9 | 12 | 11b | 1 | CH2Cl2 (Ar) | 13 (49%)d |
| 10 | 12 | 11b | 0.1 | CH2Cl2 (Ar) | 13 (79%)d |
| 11 | 12 | 11c | 1 | CH2Cl2 (Ar) | 12/13 (51%), 44:56 |
| 12 | 12 | 11d | 1 | CH2Cl2 (Ar) | 13 (91%)d |
| 13 | 12 | 11d | 0.1 | CH2Cl2 (Ar) | 13 (97%)d |
| 14 | 12 | 11d | 0.01 | CH2Cl2 (Ar) | 12/13 (93%), 69:31 |
| 15 | 12 | 11a | 1 | CHCl3 (Ar) | 13 (86%)d |
| 16 | 12 | 11a | 0.1 | CHCl3 (Ar) | 13 (95%)d |
| 17 | 12 | 11b | 1 | CHCl3 (Ar) | 13 (96%)d |
| 18 | 12 | 11b | 0.1 | CHCl3 (Ar) | 13 (97%)d |
| 19 | 12 | 11b | 0.01 | CHCl3 (Ar) | 13 (81%)d |
| 20 | 12 | 11b | 0.1 | CHCl3 (air) | 13 (89%)d |
| 21 | 12 | 11d | 1 | CHCl3 (Ar) | 13 (95%)d |
| 22 | 12 | 11d | 0.1 | CHCl3 (Ar) | 13 (99%)d |
| 23 | 12 | 11d | 0.01 | CHCl3 (Ar) | 12/13 (98%), 64:36 |
| 24 | 14 | 11b | 0.1 | CHCl3 (Ar) | 15/16 (93%), 64:36 |
| 25 | 17 | 11a | 0.1 | CHCl3 (air) | 17/18 (46%), 44:56 |
| 26 | 17 | 11b | 0.1 | CHCl3 (air) | 17/18 (58%), 42:58 |
| 27 | 17 | 11b | 0.01 | CHCl3 (Ar) | 18 (traces)e |
| 28 | 17 | 11b | 0.1 | CHCl3 (Ar) | 17/18 (98%), 2:98 |
| 29 | 17 | 11d | 0.1 | CHCl3 (air) | 17/18 (63%), 42:58 |
| 30 | 17 | 11d | 0.1 | CHCl3 (Ar) | 17/18 (96%), 4:96 |
| 31 | 17 | 11d | 0.01 | CHCl3 (air) | 18 (traces) |
| 32 | 19 | 11d | 0.1 | CHCl3 (Ar) | 19/20 (99%), 5:95 |
| 33 | 19 | 11d | 0.01 | CHCl3 (Ar) | 19/20 (75%), 23/77 |
| 34 | 21 + 2 equiv 12 | 11a | 0.1 | CHCl3 (Ar) | 13/22/23 (71%), ≈77/20/3f |
| 35 | 21 + 2 equiv 12 | 11d | 0.1 | CHCl3 (Ar) | 13/22/23 (78%), ≈81/18/1f |
| 36 | 21 + 2 equiv 24 | 11a | 0.1 | CHCl3 (Ar) | 25/26/27 (40%), ≈3/76/21f |
| 37 | 21 + 2 equiv 24 | 11b | 0.1 | CHCl3 (Ar) | 25/26/27 (52%), ≈2/72/26f |
aAll experiments were performed in boiling solvents (10 mL) for 4 h at stirring. bAccording to GС–MS analysis with external calibration. cOnly the starting styrene (12) was detected by GС–MS. Term “traces” was used if the product content in the resulting mixture was less than 1%. dIsolated yields are given. eThe starting diallyl diethyl malonate 17 was detected by GС–MS. fThe compositions of the reaction mixtures were determined by GC–MS without external calibration.
At the beginning of this part, we tested the stability of catalysts 11 in different solvents at different temperatures and conditions (10 mg of 11 in 10 mL of solvent). From this study, we concluded that all catalysts were stable in boiling dry toluene or benzene under argon atmosphere. The catalysts retained their green color for at least a week, which confirms the absence of decomposition. In more polar solvents like dichloromethane and chloroform (bp 40 and 61 °C) boiling under argon also does not cause visible decoloration. On the other hand, the catalysts readily decompose within 0.5–1 h upon boiling in CH2Cl2 or CHCl3 in the presence of air. Boiling in tetrahydrofuran or acetonitrile even under an argon atmosphere leads to a rapid decomposition of the catalysts within 5–10 minutes (the solutions turn black). Therefore, THF and MeCN were excluded from further studies.
In the test reactions, three concentrations of the catalysts (1.0, 0.1 and 0.01 mol %) were applied for the transformation of styrene (12) to trans-stilbene (13) by cross-metathesis reaction (Scheme 6). First experiments (entries 1–4, Table 3) revealed that the nonpolar solvents (PhH and PhMe) are not suitable for the CM reactions. Using of 1 mol % of catalyst 11a even under an argon atmosphere produced only traces of the target stilbene (13). In these conditions, catalyst 11b was more active than 11a, but also gave insufficient results. Thereupon, these two solvents were also abandoned in the course of the following investigations.
In this process, temperature also exerts a strong influence on the catalytic activity of metallo complexes 11. None of catalysts 11 were active at room temperature (19–23 °С) towards the styrene cross metathesis, meaning that all reactions required temperatures higher than 30–35 °С. After all observations, we concluded that dichloromethane is the best solvent for this reaction. At 40 °C, concentrations from 1.0 to 0.1 mol % of the morpholine-based complex 11d showed the best catalytic activity by providing styrene in 91–97% yields (entries 12 and 13 of Table 3). The somewhat less sterically loaded N,N-diethyl catalyst 11b also gave acceptable yields for concentrations from 1.0 to 0.1 mol % (Table 3, entries 9 and 10). Under the same conditions the N,N-dimethyl catalyst 11a provided lower yields (Table 3, entries 7 and 8). Similarly, the least sterically loaded N-methyl complex 11c gave a mixture of products 12/13 (Table 3, entry 11) in low yields. The metathesis reaction did not proceed completely, even in the presence of 2 mol % of the catalyst. As a result, we did not explore the catalytic activity of 11c hereinafter. These experimental observations are consistent with the Х-ray data obtained for catalysts 11a–c (Figure 3). In spite of the fact that we do not have the X-ray analysis for catalyst 11d, it is possible to assume that this complex should exhibit the longest coordination N→Ru bond (at least more than 2.30 Å). Interestingly, lower yields of stilbene from styrene in CH2Cl2 were obtained in the presence of 1.0 mol % of catalyst 11a,c,d as compared with 0.1 mol % of catalyst (cf. Table 3, entries 9/10, 12/13). Obviously, a high concentration of the catalysts accelerates the formation of undesirable products (oligomers and polymers of styrene). At 40 °C in dichloromethane, excellent yields of stilbene were obtained only under the action of metallo complex 11d. For this reason, we explored elevated temperatures for the metathesis reactions.
The following series of experiments was performed in CHCl3 (entries 15–23, Table 3) at 60–61 °C with different concentrations of catalysts. Ruthenium complex 11b was efficient in the 0.01 mol % concentration under argon atmosphere and in the 0.1 mol % concentration in air. In case of allylbenzene (14), the action of catalyst 11b (0.1 mol %) gave the mixture of isomeric 1,4-diphenylbutenes 15 and 16 in the ratio of 64:36 in 93% yield. Only a small amount of the starting compound 14 underwent polymerisation.
The utility of the catalyst for the RCM reaction was demonstrated by the cyclization of dienes 17 and 19 (Table 3, entries 25–33) in both air and argon atmosphere. The complexes 11a, 11b and 11d in the air atmosphere provided cyclic alkenes 18, 20 with a strong admixture of initial dienes (Table 3, entries 25, 26, 29). Under argon atmosphere, the same transformations provided good results in the presence of 0.1–0.01 mol % of catalysts 11b,d (Table 3, entries 28, 30–32).
Similarly, in the case of the ROCM reactions (Scheme 6), catalysts 11a,b did not provide high selectivity (Table 3, entries 34–37). Interactions of norbornene (21) with a two-fold excess of styrene (12) or hex-1-ene (24) was accompanied by the CM reaction, which provided products of the ring opening (22, 23, 25, 26) and significant amounts of byproducts due to the side cross metathesis (13 and 27). Moreover, sparingly soluble high molecular weight products were isolated from all reactions; according to gel permeation chromatography data, these solids are, presumably, norbornene oligomers (see, for example data for entry 35, Supporting Information File 1 and Supporting Information File 2). These four examples demonstrate the principal possibility of application of catalysts 11 in ROCM reactions.
It is known that metathesis reactions carried out in chloroform medium under similar conditions (see Table 3) can give products of the Kharasch radical addition of CHCl3 across olefins [56,57]. It is worth to note in the end of this part, that we did not detect formation of chlorine-containing products (molecular peaks with the isotopic distribution characteristic for chlorine were absent in GC–MS spectra).
Conclusion
The present work reports an efficient method for the synthesis of 2-(N,N-dialkylaminomethyl)styrenes. The resultant vinyl benzenes are excellent precursors for the synthesis of a new type of Hoveyda–Grubbs catalysts bearing an N→Ru coordinate bond in a six-membered ring. This process does not require the use of complex equipment, extremely expensive or toxic reagents. The structure of the catalysts were elucidated in detail by 2D NMR and X-ray crystallography. The high catalytic activity of the metallo complexes was demonstrated by several examples of cross metathesis (CM), ring-closing (RCM) and ring-opening cross metathesis (ROCM) reactions.
Furthermore, almost all steps of ligands’ and catalysts’ synthesis were accomplished in preparative and multigram scales.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 391.6 KB | Download |
| Supporting Information File 2: Copies of NMR spectra of synthesised compounds and selected GC–MS data of the metathesis products. Check-cif reports for compounds 11a–c. | ||
| Format: PDF | Size: 10.5 MB | Download |
References
-
Yu, M.; Lou, S.; Gonzalez-Bobes, F. Org. Process Res. Dev. 2018, 22, 918–946. doi:10.1021/acs.oprd.8b00093
Return to citation in text: [1] -
Ogba, O. M.; Warner, N. C.; O’Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510–4544. doi:10.1039/c8cs00027a
Return to citation in text: [1] -
Chen, Y.; Abdellatif, M. M.; Nomura, K. Tetrahedron 2018, 74, 619–643. doi:10.1016/j.tet.2017.12.041
Return to citation in text: [1] [2] -
Mukherjee, N.; Planer, S.; Grela, K. Org. Chem. Front. 2018, 5, 494–516. doi:10.1039/c7qo00800g
Return to citation in text: [1] -
Lecourt, C.; Dhambri, S.; Allievi, L.; Sanogo, Y.; Zeghbib, N.; Ben Othman, R.; Lannou, M.-I.; Sorin, G.; Ardisson, J. Nat. Prod. Rep. 2018, 35, 105–124. doi:10.1039/c7np00048k
Return to citation in text: [1] -
Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249–2295. doi:10.1021/acs.chemrev.7b00443
Return to citation in text: [1] -
Montgomery, T. P.; Johns, A. M.; Grubbs, R. H. Catalysts 2017, 7, 87. doi:10.3390/catal7030087
Return to citation in text: [1] -
Hughes, D.; Wheeler, P.; Ene, D. Org. Process Res. Dev. 2017, 21, 1938–1962. doi:10.1021/acs.oprd.7b00319
Return to citation in text: [1] -
Sinclair, F.; Alkattan, M.; Prunet, J.; Shaver, M. P. Polym. Chem. 2017, 8, 3385–3398. doi:10.1039/c7py00340d
Return to citation in text: [1] [2] -
Montgomery, T. P.; Ahmed, T. S.; Grubbs, R. H. Angew. Chem., Int. Ed. 2017, 56, 11024–11036. doi:10.1002/anie.201704686
Return to citation in text: [1] -
Paradiso, V.; Costabile, C.; Grisi, F. Molecules 2016, 21, No. 117. doi:10.3390/molecules21010117
Return to citation in text: [1] -
Higman, C. S.; Lummiss, J. A. M.; Fogg, D. E. Angew. Chem., Int. Ed. 2016, 55, 3552–3565. doi:10.1002/anie.201506846
Return to citation in text: [1] -
Grubbs, R. H.; Wenzel, A. G.; O’Leary, D. J.; Khosravi, E. Handbook of Metathesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2015.
Return to citation in text: [1] -
van Lierop, B. J.; Lummiss, J. A. M.; Fogg, D. E. Ring-Closing Metathesis. Olefin Metathesis; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2014; pp 85–152. doi:10.1002/9781118711613.ch3
Return to citation in text: [1] -
Scholl, M.; Trnka, T. M.; Morgan, J. P.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 2247–2250. doi:10.1016/s0040-4039(99)00217-8
Return to citation in text: [1] -
Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q
Return to citation in text: [1] -
Fürstner, A.; Ackermann, L.; Gabor, B.; Goddard, R.; Lehmann, C. W.; Mynott, R.; Stelzer, F.; Thiel, O. R. Chem. – Eur. J. 2001, 7, 3236–3253. doi:10.1002/1521-3765(20010803)7:15<3236::aid-chem3236>3.0.co;2-s
Return to citation in text: [1] -
Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J. Am. Chem. Soc. 2004, 126, 9318–9325. doi:10.1021/ja048794v
Return to citation in text: [1] -
Barbasiewicz, M.; Bieniek, M.; Michrowska, A.; Szadkowska, A.; Makal, A.; Woźniak, K.; Grela, K. Adv. Synth. Catal. 2007, 349, 193–203. doi:10.1002/adsc.200600478
Return to citation in text: [1] -
Kos, P.; Savka, R.; Plenio, H. Adv. Synth. Catal. 2013, 355, 439–447. doi:10.1002/adsc.201200956
Return to citation in text: [1] -
Engle, K. M.; Lu, G.; Luo, S.-X.; Henling, L. M.; Takase, M. K.; Liu, P.; Houk, K. N.; Grubbs, R. H. J. Am. Chem. Soc. 2015, 137, 5782–5792. doi:10.1021/jacs.5b01144
Return to citation in text: [1] -
Duan, Y.; Wang, T.; Xie, Q.; Yu, X.; Guo, W.; Wang, J.; Liu, G. Dalton Trans. 2016, 45, 19441–19448. doi:10.1039/c6dt03899a
Return to citation in text: [1] -
Occhipinti, G.; Bjørsvik, H.-R.; Törnroos, W. K.; Jensen, V. R. Organometallics 2007, 26, 5803–5814. doi:10.1021/om070219n
Return to citation in text: [1] -
Tzur, E.; Szadkowska, A.; Ben-Asuly, A.; Makal, A.; Goldberg, I.; Woźniak, K.; Grela, K.; Lemcoff, N. G. Chem. – Eur. J. 2010, 16, 8726–8737. doi:10.1002/chem.200903457
Return to citation in text: [1] -
Zannan Science and Technology (Shanghai) Co., Ltd.; Zheng-Yun, Z. Metal complex ligand, metal complex, preparation method and use of metal complex, polymers and preparation method and use of polymers. CN Patent 104262403A, Sept 27, 2008.
Return to citation in text: [1] [2] -
Francis Walter Cornelius Verpoort; Xia, L. Group 8 transition metal catalysts and method for making same and process for use of same in olefin disproportionation reactions. WO Patent WO2017185324 A1, April 29, 2016.
Return to citation in text: [1] [2] -
Zannan Scitech Co., Ltd.; Zhan, J. Z.-Y. Highly active metathesis catalysts selective for ROMP and RCM reactions. WO Patent WO201179439 A1, Dec 30, 2009.
Return to citation in text: [1] [2] -
PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanas'ev, V. V.; Bespalova, N. B. Dicyclopentadiene metathesis polymerization catalyst in the form of a ruthenium complex and method for producing same. WO Patent WO2015115937 A1, Jan 29, 2014.
Return to citation in text: [1] [2] -
PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanasev, V. V.; Zemtsov, D. B.; Panov, D. M.; Bespalova, N. B. Method of producing catalyst for metathesis polymerisation of dicyclopentadiene. RU Patent RU2577252 C1, Feb 26, 2015.
Return to citation in text: [1] [2] -
PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanas'ev, V. V.; Zemtsov, D. B.; Panov, D. M.; Bespalova, N. B. Catalyst of metathesis polymerisation of dicyclopentadiene in form of ruthenium complex and method of obtaining thereof. RU Patent RU2545179 C1, Jan 29, 2014.
Return to citation in text: [1] [2] -
Ivin, K. J.; Mol, J. C. Olefin metathesis and metathesis polymerization; Academic Press: London, 1997; p 204.
Return to citation in text: [1] [2] -
Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. Chem. Bull. 2016, 65, 490–497. doi:10.1007/s11172-016-1327-x
Return to citation in text: [1] [2] [3] -
Kiselev, S. A.; Lenev, D. A.; Lyapkov, A. A.; Semakin, S. V.; Bozhenkova, G.; Verpoort, F.; Ashirov, R. V. RSC Adv. 2016, 6, 5177–5183. doi:10.1039/c5ra25197d
Return to citation in text: [1] [2] -
Pump, E.; Leitgeb, A.; Kozłowska, A.; Torvisco, A.; Falivene, L.; Cavallo, L.; Grela, K.; Slugovc, C. Organometallics 2015, 34, 5383–5392. doi:10.1021/acs.organomet.5b00715
Return to citation in text: [1] [2] -
Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. J. Org. Chem. 2015, 51, 907–909. doi:10.1134/s1070428015070015
Return to citation in text: [1] [2] -
Grudzień, K.; Żukowska, K.; Malińska, M.; Woźniak, K.; Barbasiewicz, M. Chem. – Eur. J. 2014, 20, 2819–2828. doi:10.1002/chem.201303826
Return to citation in text: [1] [2] -
Ashirov, R. V.; Zemlyakov, D. I.; Lyapkov, A. A.; Kiselev, S. A. Kinet. Catal. 2013, 54, 469–474. doi:10.1134/s0023158413040010
Return to citation in text: [1] [2] -
Szadkowska, A.; Gstrein, X.; Burtscher, D.; Jarzembska, K.; Woźniak, K.; Slugovc, C.; Grela, K. Organometallics 2010, 29, 117–124. doi:10.1021/om900857w
Return to citation in text: [1] [2] -
Rheiner, A., Jr.; Brossi, A. Helv. Chim. Acta 1962, 45, 2590–2600. doi:10.1002/hlca.19620450728
Return to citation in text: [1] [2] -
Ito, Y.; Nakatsuka, M.; Saegusa, T. J. Am. Chem. Soc. 1982, 104, 7609–7622. doi:10.1021/ja00390a036
Return to citation in text: [1] -
Suzuki, T.; Takamoto, M.; Okamoto, T.; Takayama, H. Chem. Pharm. Bull. 1986, 34, 1888–1900. doi:10.1248/cpb.34.1888
Return to citation in text: [1] -
Kafka, S.; Trška, P.; Kytner, J.; Taufmann, P.; Ferles, M. Collect. Czech. Chem. Commun. 1987, 52, 2047–2056. doi:10.1135/cccc19872047
Return to citation in text: [1] [2] -
Padwa, A.; Dent, W. J. Org. Chem. 1987, 52, 235–244. doi:10.1021/jo00378a013
Return to citation in text: [1] [2] -
Molander, G. A.; Pack, S. K. Tetrahedron 2003, 59, 10581–10591. doi:10.1016/j.tet.2003.08.071
Return to citation in text: [1] [2] -
Segal, I.; Zablotskaya, A.; Lukevics, E. Chem. Heterocycl. Compd. 2005, 41, 613–624. doi:10.1007/s10593-005-0192-6
Return to citation in text: [1] -
Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V. Russ. J. Org. Chem. 2013, 49, 1329–1334. doi:10.1134/s1070428013090145
Return to citation in text: [1] [2] -
Dorta, R.; Kelly, R. A., III; Nolan, S. P. Adv. Synth. Catal. 2004, 346, 917–920. doi:10.1002/adsc.200404047
Return to citation in text: [1] -
Fürstner, A.; Guth, O.; Düffels, A.; Seidel, G.; Liebl, M.; Gabor, B.; Mynott, R. Chem. – Eur. J. 2001, 7, 4811–4820. doi:10.1002/1521-3765(20011119)7:22<4811::aid-chem4811>3.0.co;2-p
Return to citation in text: [1] -
Jimenez, L. R.; Tolentino, D. R.; Gallon, B. J.; Schrodi, Y. Molecules 2012, 17, 5675–5689. doi:10.3390/molecules17055675
Return to citation in text: [1] -
Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Organometallics 2015, 34, 3107–3111. doi:10.1021/om501246q
Return to citation in text: [1] -
Musengimana, E.; Fatakanwa, C. Orient. J. Chem. 2013, 29, 1489–1496. doi:10.13005/ojc/290426
Return to citation in text: [1] -
Trnka, T. M.; Morgan, J. P.; Sanford, M. S.; Wilhelm, T. E.; Scholl, M.; Choi, T.-L.; Ding, S.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 2546–2558. doi:10.1021/ja021146w
Return to citation in text: [1] -
Nyce, G. W.; Csihony, S.; Waymouth, R. M.; Hedrick, J. L. Chem. – Eur. J. 2004, 10, 4073–4079. doi:10.1002/chem.200400196
Return to citation in text: [1] -
Bell, A.; Grubbs, R. H.; Morgan, J. P.; Moore, J. L. High activity metal carbene metathesis catalysts generated using a thermally activated N-heterocyclic carbene precursor. US Patent US6838489 B2, March 23, 2001.
Return to citation in text: [1] -
Arduengo, A. J., III; Calabrese, J. C.; Davidson, F.; Rasika Dias, H. V.; Goerlich, J. R.; Krafezyk, R.; Marshall, W. J.; Tamm, M.; Schmutzler, R. Helv. Chim. Acta 1999, 82, 2348–2364. doi:10.1002/(sici)1522-2675(19991215)82:12<2348::aid-hlca2348>3.0.co;2-m
Return to citation in text: [1] -
Tallarico, J. A.; Malnick, L. M.; Snapper, M. L. J. Org. Chem. 1999, 64, 344–345. doi:10.1021/jo982349z
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Luna, A. Chem. Rev. 2009, 109, 3817–3858. doi:10.1021/cr9001512
Return to citation in text: [1]
| 56. | Tallarico, J. A.; Malnick, L. M.; Snapper, M. L. J. Org. Chem. 1999, 64, 344–345. doi:10.1021/jo982349z |
| 57. | Alcaide, B.; Almendros, P.; Luna, A. Chem. Rev. 2009, 109, 3817–3858. doi:10.1021/cr9001512 |
| 1. | Yu, M.; Lou, S.; Gonzalez-Bobes, F. Org. Process Res. Dev. 2018, 22, 918–946. doi:10.1021/acs.oprd.8b00093 |
| 2. | Ogba, O. M.; Warner, N. C.; O’Leary, D. J.; Grubbs, R. H. Chem. Soc. Rev. 2018, 47, 4510–4544. doi:10.1039/c8cs00027a |
| 3. | Chen, Y.; Abdellatif, M. M.; Nomura, K. Tetrahedron 2018, 74, 619–643. doi:10.1016/j.tet.2017.12.041 |
| 4. | Mukherjee, N.; Planer, S.; Grela, K. Org. Chem. Front. 2018, 5, 494–516. doi:10.1039/c7qo00800g |
| 5. | Lecourt, C.; Dhambri, S.; Allievi, L.; Sanogo, Y.; Zeghbib, N.; Ben Othman, R.; Lannou, M.-I.; Sorin, G.; Ardisson, J. Nat. Prod. Rep. 2018, 35, 105–124. doi:10.1039/c7np00048k |
| 6. | Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249–2295. doi:10.1021/acs.chemrev.7b00443 |
| 7. | Montgomery, T. P.; Johns, A. M.; Grubbs, R. H. Catalysts 2017, 7, 87. doi:10.3390/catal7030087 |
| 8. | Hughes, D.; Wheeler, P.; Ene, D. Org. Process Res. Dev. 2017, 21, 1938–1962. doi:10.1021/acs.oprd.7b00319 |
| 9. | Sinclair, F.; Alkattan, M.; Prunet, J.; Shaver, M. P. Polym. Chem. 2017, 8, 3385–3398. doi:10.1039/c7py00340d |
| 10. | Montgomery, T. P.; Ahmed, T. S.; Grubbs, R. H. Angew. Chem., Int. Ed. 2017, 56, 11024–11036. doi:10.1002/anie.201704686 |
| 11. | Paradiso, V.; Costabile, C.; Grisi, F. Molecules 2016, 21, No. 117. doi:10.3390/molecules21010117 |
| 12. | Higman, C. S.; Lummiss, J. A. M.; Fogg, D. E. Angew. Chem., Int. Ed. 2016, 55, 3552–3565. doi:10.1002/anie.201506846 |
| 22. | Duan, Y.; Wang, T.; Xie, Q.; Yu, X.; Guo, W.; Wang, J.; Liu, G. Dalton Trans. 2016, 45, 19441–19448. doi:10.1039/c6dt03899a |
| 23. | Occhipinti, G.; Bjørsvik, H.-R.; Törnroos, W. K.; Jensen, V. R. Organometallics 2007, 26, 5803–5814. doi:10.1021/om070219n |
| 24. | Tzur, E.; Szadkowska, A.; Ben-Asuly, A.; Makal, A.; Goldberg, I.; Woźniak, K.; Grela, K.; Lemcoff, N. G. Chem. – Eur. J. 2010, 16, 8726–8737. doi:10.1002/chem.200903457 |
| 25. | Zannan Science and Technology (Shanghai) Co., Ltd.; Zheng-Yun, Z. Metal complex ligand, metal complex, preparation method and use of metal complex, polymers and preparation method and use of polymers. CN Patent 104262403A, Sept 27, 2008. |
| 26. | Francis Walter Cornelius Verpoort; Xia, L. Group 8 transition metal catalysts and method for making same and process for use of same in olefin disproportionation reactions. WO Patent WO2017185324 A1, April 29, 2016. |
| 27. | Zannan Scitech Co., Ltd.; Zhan, J. Z.-Y. Highly active metathesis catalysts selective for ROMP and RCM reactions. WO Patent WO201179439 A1, Dec 30, 2009. |
| 28. | PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanas'ev, V. V.; Bespalova, N. B. Dicyclopentadiene metathesis polymerization catalyst in the form of a ruthenium complex and method for producing same. WO Patent WO2015115937 A1, Jan 29, 2014. |
| 29. | PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanasev, V. V.; Zemtsov, D. B.; Panov, D. M.; Bespalova, N. B. Method of producing catalyst for metathesis polymerisation of dicyclopentadiene. RU Patent RU2577252 C1, Feb 26, 2015. |
| 30. | PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanas'ev, V. V.; Zemtsov, D. B.; Panov, D. M.; Bespalova, N. B. Catalyst of metathesis polymerisation of dicyclopentadiene in form of ruthenium complex and method of obtaining thereof. RU Patent RU2545179 C1, Jan 29, 2014. |
| 31. | Ivin, K. J.; Mol, J. C. Olefin metathesis and metathesis polymerization; Academic Press: London, 1997; p 204. |
| 32. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. Chem. Bull. 2016, 65, 490–497. doi:10.1007/s11172-016-1327-x |
| 33. | Kiselev, S. A.; Lenev, D. A.; Lyapkov, A. A.; Semakin, S. V.; Bozhenkova, G.; Verpoort, F.; Ashirov, R. V. RSC Adv. 2016, 6, 5177–5183. doi:10.1039/c5ra25197d |
| 34. | Pump, E.; Leitgeb, A.; Kozłowska, A.; Torvisco, A.; Falivene, L.; Cavallo, L.; Grela, K.; Slugovc, C. Organometallics 2015, 34, 5383–5392. doi:10.1021/acs.organomet.5b00715 |
| 35. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. J. Org. Chem. 2015, 51, 907–909. doi:10.1134/s1070428015070015 |
| 36. | Grudzień, K.; Żukowska, K.; Malińska, M.; Woźniak, K.; Barbasiewicz, M. Chem. – Eur. J. 2014, 20, 2819–2828. doi:10.1002/chem.201303826 |
| 37. | Ashirov, R. V.; Zemlyakov, D. I.; Lyapkov, A. A.; Kiselev, S. A. Kinet. Catal. 2013, 54, 469–474. doi:10.1134/s0023158413040010 |
| 38. | Szadkowska, A.; Gstrein, X.; Burtscher, D.; Jarzembska, K.; Woźniak, K.; Slugovc, C.; Grela, K. Organometallics 2010, 29, 117–124. doi:10.1021/om900857w |
| 51. | Musengimana, E.; Fatakanwa, C. Orient. J. Chem. 2013, 29, 1489–1496. doi:10.13005/ojc/290426 |
| 52. | Trnka, T. M.; Morgan, J. P.; Sanford, M. S.; Wilhelm, T. E.; Scholl, M.; Choi, T.-L.; Ding, S.; Day, M. W.; Grubbs, R. H. J. Am. Chem. Soc. 2003, 125, 2546–2558. doi:10.1021/ja021146w |
| 53. | Nyce, G. W.; Csihony, S.; Waymouth, R. M.; Hedrick, J. L. Chem. – Eur. J. 2004, 10, 4073–4079. doi:10.1002/chem.200400196 |
| 54. | Bell, A.; Grubbs, R. H.; Morgan, J. P.; Moore, J. L. High activity metal carbene metathesis catalysts generated using a thermally activated N-heterocyclic carbene precursor. US Patent US6838489 B2, March 23, 2001. |
| 55. | Arduengo, A. J., III; Calabrese, J. C.; Davidson, F.; Rasika Dias, H. V.; Goerlich, J. R.; Krafezyk, R.; Marshall, W. J.; Tamm, M.; Schmutzler, R. Helv. Chim. Acta 1999, 82, 2348–2364. doi:10.1002/(sici)1522-2675(19991215)82:12<2348::aid-hlca2348>3.0.co;2-m |
| 18. | Michrowska, A.; Bujok, R.; Harutyunyan, S.; Sashuk, V.; Dolgonos, G.; Grela, K. J. Am. Chem. Soc. 2004, 126, 9318–9325. doi:10.1021/ja048794v |
| 19. | Barbasiewicz, M.; Bieniek, M.; Michrowska, A.; Szadkowska, A.; Makal, A.; Woźniak, K.; Grela, K. Adv. Synth. Catal. 2007, 349, 193–203. doi:10.1002/adsc.200600478 |
| 20. | Kos, P.; Savka, R.; Plenio, H. Adv. Synth. Catal. 2013, 355, 439–447. doi:10.1002/adsc.201200956 |
| 21. | Engle, K. M.; Lu, G.; Luo, S.-X.; Henling, L. M.; Takase, M. K.; Liu, P.; Houk, K. N.; Grubbs, R. H. J. Am. Chem. Soc. 2015, 137, 5782–5792. doi:10.1021/jacs.5b01144 |
| 32. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. Chem. Bull. 2016, 65, 490–497. doi:10.1007/s11172-016-1327-x |
| 15. | Scholl, M.; Trnka, T. M.; Morgan, J. P.; Grubbs, R. H. Tetrahedron Lett. 1999, 40, 2247–2250. doi:10.1016/s0040-4039(99)00217-8 |
| 16. | Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org. Lett. 1999, 1, 953–956. doi:10.1021/ol990909q |
| 17. | Fürstner, A.; Ackermann, L.; Gabor, B.; Goddard, R.; Lehmann, C. W.; Mynott, R.; Stelzer, F.; Thiel, O. R. Chem. – Eur. J. 2001, 7, 3236–3253. doi:10.1002/1521-3765(20010803)7:15<3236::aid-chem3236>3.0.co;2-s |
| 44. | Molander, G. A.; Pack, S. K. Tetrahedron 2003, 59, 10581–10591. doi:10.1016/j.tet.2003.08.071 |
| 13. | Grubbs, R. H.; Wenzel, A. G.; O’Leary, D. J.; Khosravi, E. Handbook of Metathesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2015. |
| 14. | van Lierop, B. J.; Lummiss, J. A. M.; Fogg, D. E. Ring-Closing Metathesis. Olefin Metathesis; John Wiley & Sons, Inc.: Hoboken, NJ, U.S.A., 2014; pp 85–152. doi:10.1002/9781118711613.ch3 |
| 47. | Dorta, R.; Kelly, R. A., III; Nolan, S. P. Adv. Synth. Catal. 2004, 346, 917–920. doi:10.1002/adsc.200404047 |
| 48. | Fürstner, A.; Guth, O.; Düffels, A.; Seidel, G.; Liebl, M.; Gabor, B.; Mynott, R. Chem. – Eur. J. 2001, 7, 4811–4820. doi:10.1002/1521-3765(20011119)7:22<4811::aid-chem4811>3.0.co;2-p |
| 49. | Jimenez, L. R.; Tolentino, D. R.; Gallon, B. J.; Schrodi, Y. Molecules 2012, 17, 5675–5689. doi:10.3390/molecules17055675 |
| 50. | Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Organometallics 2015, 34, 3107–3111. doi:10.1021/om501246q |
| 39. | Rheiner, A., Jr.; Brossi, A. Helv. Chim. Acta 1962, 45, 2590–2600. doi:10.1002/hlca.19620450728 |
| 40. | Ito, Y.; Nakatsuka, M.; Saegusa, T. J. Am. Chem. Soc. 1982, 104, 7609–7622. doi:10.1021/ja00390a036 |
| 41. | Suzuki, T.; Takamoto, M.; Okamoto, T.; Takayama, H. Chem. Pharm. Bull. 1986, 34, 1888–1900. doi:10.1248/cpb.34.1888 |
| 42. | Kafka, S.; Trška, P.; Kytner, J.; Taufmann, P.; Ferles, M. Collect. Czech. Chem. Commun. 1987, 52, 2047–2056. doi:10.1135/cccc19872047 |
| 43. | Padwa, A.; Dent, W. J. Org. Chem. 1987, 52, 235–244. doi:10.1021/jo00378a013 |
| 44. | Molander, G. A.; Pack, S. K. Tetrahedron 2003, 59, 10581–10591. doi:10.1016/j.tet.2003.08.071 |
| 45. | Segal, I.; Zablotskaya, A.; Lukevics, E. Chem. Heterocycl. Compd. 2005, 41, 613–624. doi:10.1007/s10593-005-0192-6 |
| 46. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V. Russ. J. Org. Chem. 2013, 49, 1329–1334. doi:10.1134/s1070428013090145 |
| 39. | Rheiner, A., Jr.; Brossi, A. Helv. Chim. Acta 1962, 45, 2590–2600. doi:10.1002/hlca.19620450728 |
| 42. | Kafka, S.; Trška, P.; Kytner, J.; Taufmann, P.; Ferles, M. Collect. Czech. Chem. Commun. 1987, 52, 2047–2056. doi:10.1135/cccc19872047 |
| 32. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. Chem. Bull. 2016, 65, 490–497. doi:10.1007/s11172-016-1327-x |
| 33. | Kiselev, S. A.; Lenev, D. A.; Lyapkov, A. A.; Semakin, S. V.; Bozhenkova, G.; Verpoort, F.; Ashirov, R. V. RSC Adv. 2016, 6, 5177–5183. doi:10.1039/c5ra25197d |
| 34. | Pump, E.; Leitgeb, A.; Kozłowska, A.; Torvisco, A.; Falivene, L.; Cavallo, L.; Grela, K.; Slugovc, C. Organometallics 2015, 34, 5383–5392. doi:10.1021/acs.organomet.5b00715 |
| 35. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V.; Krasnokutskaya, E. A. Russ. J. Org. Chem. 2015, 51, 907–909. doi:10.1134/s1070428015070015 |
| 36. | Grudzień, K.; Żukowska, K.; Malińska, M.; Woźniak, K.; Barbasiewicz, M. Chem. – Eur. J. 2014, 20, 2819–2828. doi:10.1002/chem.201303826 |
| 37. | Ashirov, R. V.; Zemlyakov, D. I.; Lyapkov, A. A.; Kiselev, S. A. Kinet. Catal. 2013, 54, 469–474. doi:10.1134/s0023158413040010 |
| 38. | Szadkowska, A.; Gstrein, X.; Burtscher, D.; Jarzembska, K.; Woźniak, K.; Slugovc, C.; Grela, K. Organometallics 2010, 29, 117–124. doi:10.1021/om900857w |
| 46. | Shcheglova, N. M.; Kolesnik, V. D.; Ashirov, R. V. Russ. J. Org. Chem. 2013, 49, 1329–1334. doi:10.1134/s1070428013090145 |
| 3. | Chen, Y.; Abdellatif, M. M.; Nomura, K. Tetrahedron 2018, 74, 619–643. doi:10.1016/j.tet.2017.12.041 |
| 9. | Sinclair, F.; Alkattan, M.; Prunet, J.; Shaver, M. P. Polym. Chem. 2017, 8, 3385–3398. doi:10.1039/c7py00340d |
| 29. | PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanasev, V. V.; Zemtsov, D. B.; Panov, D. M.; Bespalova, N. B. Method of producing catalyst for metathesis polymerisation of dicyclopentadiene. RU Patent RU2577252 C1, Feb 26, 2015. |
| 30. | PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanas'ev, V. V.; Zemtsov, D. B.; Panov, D. M.; Bespalova, N. B. Catalyst of metathesis polymerisation of dicyclopentadiene in form of ruthenium complex and method of obtaining thereof. RU Patent RU2545179 C1, Jan 29, 2014. |
| 31. | Ivin, K. J.; Mol, J. C. Olefin metathesis and metathesis polymerization; Academic Press: London, 1997; p 204. |
| 25. | Zannan Science and Technology (Shanghai) Co., Ltd.; Zheng-Yun, Z. Metal complex ligand, metal complex, preparation method and use of metal complex, polymers and preparation method and use of polymers. CN Patent 104262403A, Sept 27, 2008. |
| 26. | Francis Walter Cornelius Verpoort; Xia, L. Group 8 transition metal catalysts and method for making same and process for use of same in olefin disproportionation reactions. WO Patent WO2017185324 A1, April 29, 2016. |
| 27. | Zannan Scitech Co., Ltd.; Zhan, J. Z.-Y. Highly active metathesis catalysts selective for ROMP and RCM reactions. WO Patent WO201179439 A1, Dec 30, 2009. |
| 28. | PJSC Rosneft Oil Co.; Polyanskii, K. B.; Afanas'ev, V. V.; Bespalova, N. B. Dicyclopentadiene metathesis polymerization catalyst in the form of a ruthenium complex and method for producing same. WO Patent WO2015115937 A1, Jan 29, 2014. |
© 2019 Polyanskii et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)