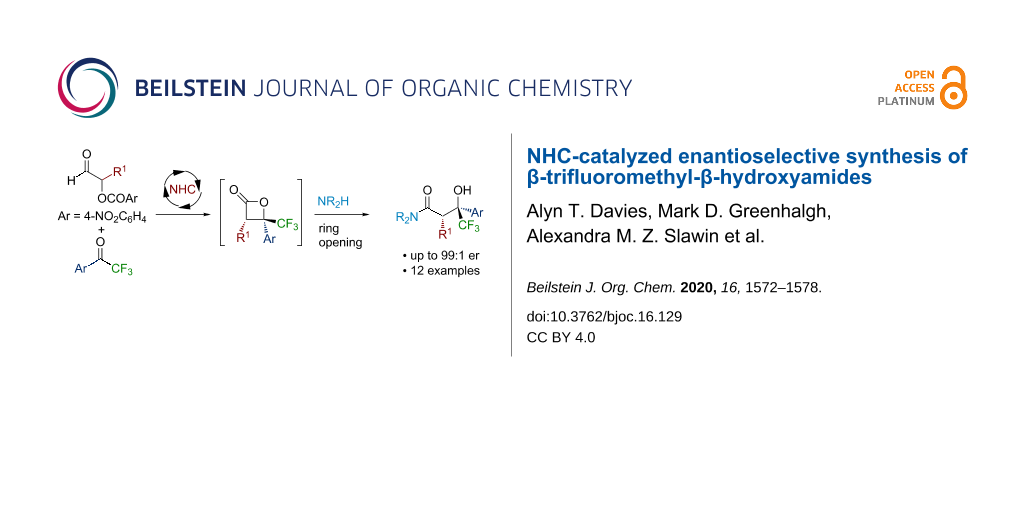
Abstract
The N-heterocyclic carbene (NHC)-catalyzed formal [2 + 2] cycloaddition between α-aroyloxyaldehydes and trifluoroacetophenones, followed by ring opening with an amine or a reducing agent is described. The resulting β-trifluoromethyl-β-hydroxyamide and alcohol products are produced with reasonable diastereocontrol (typically ≈70:30 dr) and excellent enantioselectivity, and they can be isolated in moderate to good yield as a single diastereoisomer.

Graphical Abstract
Introduction
The trifluoromethyl unit holds a prominent and privileged position within organic chemistry [1-6]. The incorporation of this motif is widely employed within the pharmaceutical, agrochemical, and materials industries as it can be used strategically to increase lipophilicity as well as to enhance metabolic stability and binding selectivity [7-9]. In this context, the generation of effective and practical methodologies capable of the enantioselective incorporation of the trifluoromethyl unit into organic molecules has been developed widely [10-12], with a series of elegant organocatalytic strategies utilized toward these aims [13]. One general strategy for the organocatalytic construction of stereogenic trifluoromethyl centers is through enantioselective addition of enolates or their equivalents to prochiral trifluoromethyl ketones (Figure 1A). Within this area, a common catalytic approach has utilized aliphatic ketones as enolate equivalents using prolinamide, cinchona, or hybrid catalysts that proceed via enamine intermediates (Figure 1B) [14-19]. The state of the art within this area has been recently demonstrated by Dixon and co-workers, who showed that bifunctional BIMP catalysts could promote the enantioselective addition of typically recalcitrant aryl ketones to trifluoromethyl ketones (Figure 1C) [20].
![[1860-5397-16-129-1]](/bjoc/content/figures/1860-5397-16-129-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Organocatalytic enantioselective aldol approaches using trifluoroacetophenone derivatives.
Figure 1: Organocatalytic enantioselective aldol approaches using trifluoroacetophenone derivatives.
Over the last twenty years, NHCs have been widely exploited as highly efficient organocatalysts that have found use in numerous applications and were the subject of many extensive reviews [21-26]. Among the most common reactive intermediates generated using NHCs, the azolium enolate has been widely used [27,28]. Methods to generate azolium enolates from a number of precursors have been reported, including the use of ketenes [29,30], α-functionalized aldehydes [31-35], enals [36-38], aryl esters [39-42], or aldehydes [43-45] in the presence of an oxidant. As representative examples of the use of azolium enolates in reactions with trifluoroacetophenone derivatives, Ye and co-workers have shown that using disubstituted ketenes as azolium enolate precursors and NHC precatalyst 1 allowed access to trifluoromethyl-substituted β-lactone products in a high yield, diastereo-, and enantioselectivity (Figure 2A) [46]. Chi and co-workers have reported limited examples of oxidative [2 + 2] cycloadditions using hydrocinnamaldehyde as an azolium enolate precursor and the NHC precatalyst 2, giving β-lactone products in high enantioselectivity but requiring superstoichiometric quantities of quinone as an oxidant (Figure 2B) [47]. In our previous work, we have developed α-aroyloxyaldehydes as reactive, bench-stable precursors of azolium enolates [48-53], which can be synthesized in a single step from the desired aldehyde [54]. We have previously applied these precursors in the NHC-catalyzed formal [2 + 2] cycloadditions between α-aroyloxyaldehydes and perfluoroalkyl-substituted ketones using NHC precatalyst 3 to produce polyfluorinated oxetanes and β-perfluoroalkyl-β-hydroxyamides after derivatization [55]. In this paper, this formal [2 + 2] protocol is extended to the use of trifluoroacetophenones to develop an enantioselective route to β-trifluoromethyl-β-hydroxy carboxylic acid derivatives (Figure 2C).
![[1860-5397-16-129-2]](/bjoc/content/figures/1860-5397-16-129-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: NHC-catalyzed approaches to β-lactones using trifluoroacetophenone derivatives.
Figure 2: NHC-catalyzed approaches to β-lactones using trifluoroacetophenone derivatives.
Results and Discussion
Optimization
Optimization of the NHC-catalyzed formal [2 + 2] cycloaddition using the α-aroyloxyaldehyde 4 and trifluoromethylacetophenone (5) as reactants began using the NHC precatalyst 3, triethylamine as the base, and THF as the solvent (Table 1, entry 1). A moderate conversion (48%, as determined by NMR analysis) to the desired β-lactone product 6 as a 70:30 mixture of diastereoisomers was observed, which served as a basis for further optimization. Variation of the base showed caesium carbonate to be significantly more effective than the organic bases examined, giving 88% conversion to product 6 (Table 1, entries 2 and 3). Trialling a number of alternative solvents, including dichloromethane, diethyl ether, and toluene showed no improvement upon the yield obtained with THF (Table 1, entries 4–6). Unfortunately, attempts to isolate the desired β-lactone product 6 were unsuccessful, with 6 being unstable to chromatographic purification under a variety of different conditions. As such, following the NHC-catalyzed formal [2 + 2] cycloaddition in THF, a subsequent ring opening step with allylamine was investigated. Although the diastereomeric ratio of the resultant crude reaction mixture was 75:25 after chromatographic purification, as seen by NMR analysis, the corresponding β-trifluoromethyl-β-hydroxyamide 7 was isolated as a single diastereoisomer in 57% yield and >99:1 er (Table 1, entry 7).
Table 1: Optimization of the NHC-catalyzed formal [2 + 2] cycoaddition.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-129-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | base | drb | yield (%) | erc |
| 1 | THF | Et3N | 70:30 | 48d | – |
| 2 | THF | iPr2NEt | 70:30 | 50d | – |
| 3 | THF | Cs2CO3 | 75:25 | 88d | – |
| 4 | CH2Cl2 | Cs2CO3 | 80:20 | 61d | – |
| 5 | Et2O | Cs2CO3 | 70:30 | 58d | – |
| 6 | toluene | Cs2CO3 | 70:30 | 55d | – |
| 7 | THF | Cs2CO3 | 75:25 | 57e | >99:1 |
aThe data contained in this Table has previously been published as part of our previous communication in this area [55]. bDetermined by 1H NMR spectroscopic analysis of the crude reaction product mixture. cer of the major diastereoisomer (>95:5 dr), determined by GC analysis using a chiral support. dCombined yield of both diastereoisomers of 6, determined by the analysis of the crude 1H NMR spectra with reference to 2,5-dimethylfuran as an internal standard. eIsolated yield of product 7 (>95:5 dr).
Scope and limitations
Having optimized this process with a model system, further work probed the scope and limitations of this process through a sequential variation of the nucleophile used for the ring opening procedure, aryl and heteroaryl substitution of the trifluoromethylacetophenone, as well as variation of the α-aroyloxyaldehyde. Using α-aroyloxyaldehyde 4 and trifluoromethylacetophenone (5) as reactants, the variation of the nucleophile showed that a range of amine-based nucleophiles was readily tolerated in this process (Scheme 1). The use of benzylamine, pyrrolidine, and ammonia all gave the corresponding β-trifluoromethyl-β-hydroxyamide product in ≈75:25 dr, with purification giving 8–10, respectively, as a single diastereoisomer in 42–55% yield and excellent enantioselectivity. Using methanol as a nucleophile gave the corresponding ester, however, this product proved unstable to purification. Generation of the ester in situ, followed by subsequent reduction with LiAlH4, gave the diol 11 in 48% isolated yield as a single diastereoisomer in 96:4 er.
![[1860-5397-16-129-i1]](/bjoc/content/inline/1860-5397-16-129-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction scope with respect to the nucleophile. aIsolated yield of the product in >95:5 dr. bDetermined by 1H NMR spectroscopic analysis of the crude reaction product mixture. cDetermined by HPLC analysis or GC analysis using a chiral support.
Scheme 1: Reaction scope with respect to the nucleophile. aIsolated yield of the product in >95:5 dr. bDeterm...
Subsequent variation of the electronic nature of the aromatic substituent within the trifluoroacetophenone component, followed by a ring opening with allylamine, showed that the introduction of electron-withdrawing groups (positive Hammett sigma constants) [56], such as p-bromo, p-fluoro, and p-trifluoromethyl groups, respectively, were well tolerated, giving a consistent diastereoselectivity of ≈75:25 dr (Scheme 2). Following chromatographic purification, the desired products 12–14, respectively, were isolated in 59–71% yield as single diastereoisomers with excellent enantioselectivity (95:5 to >99:1 er). The relative and absolute configuration of (2S,3S)-β-trifluoromethyl-β-hydroxyamide 12 was confirmed by single crystal X-ray crystallographic analysis, with all other products in this series assigned by analogy to 12 [57]. The introduction of an electron-donating p-tolyl substituent was also tolerated in the system, providing the corresponding β-trifluoromethyl-β-hydroxyamide 15 in a moderate yield of 46%, although the er of 15 could not be determined by either GC or HPLC analysis. The trifluoroacetophenone derivatives with stronger electron-donating p-methoxy and p-dimethylamino substituents proved unreactive, consistent with the expected lower electrophilicity. A heterocyclic substituent could also be incorporated using this methodology, with thiophene-substituted β-trifluoromethyl-β-hydroxyamide 16 being produced in 80:20 dr, with purification giving 16 in 51% yield as a single diastereoisomer and 96:4 er. The substrate scope was further investigated by variation of the α-aroyloxyaldehyde component of the reaction. Functionalized α-aroyloxyaldehydes containing a C2-benzyl and a C2-benzyloxy derivative were tested, producing the corresponding β-trifluoromethyl-β-hydroxyamides 17 and 18, respectively, in ≈70:30 dr after a ring opening with allylamine. Purification gave 17 and 18, respectively, in a moderate yield as single diastereoisomers and with a good enantioselectivity.
![[1860-5397-16-129-i2]](/bjoc/content/inline/1860-5397-16-129-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction scope with respect to the trifluoroacetophenone derivative and α-aroyloxyaldehyde. aIsolated yield of the product in >95:5 dr. bDetermined by 1H NMR spectroscopic analysis of the crude reaction product mixture. cer of the major diastereoisomer (>95:5 dr), determined by HPLC analysis or GC analysis using a chiral support. dMolecular representation of the X-ray crystal structure of 12. The unit cell of 12 contained two molecules of 12, with only one shown for clarity.
Scheme 2: Reaction scope with respect to the trifluoroacetophenone derivative and α-aroyloxyaldehyde. aIsolat...
The mechanism of this NHC redox process is believed to proceed through the following mechanism (Scheme 3): After deprotonation of the triazolium salt precatalyst 3 [58], reversible addition of the free NHC I to the aldehyde leads to adduct II [59]. A subsequent deprotonation allows access to Breslow intermediate III, which can eliminate para-nitrobenzoate to leave azolium enol IV. Deprotonation gives azolium enolate intermediate V, which undergoes a formal [2 + 2] cycloaddition with the trifluoroacetophenone derivative to give VI [60]. Elimination of the NHC catalyst completes the catalytic cycle and provides the corresponding β-lactone product. Subsequent ring opening with a suitable nucleophilic amine leads to the isolable β-trifluoromethyl-β-hydroxyamide (Scheme 3).
![[1860-5397-16-129-i3]](/bjoc/content/inline/1860-5397-16-129-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In this paper, we showed that azolium enolates generated from α-aroyloxyaldehydes can undergo NHC-catalyzed formal [2 + 2] cycloadditions with trifluoroacetophenone derivatives. Although the β-lactone products proved unstable to chromatographic purification, ring opening with amine nucleophiles allowed access to β-trifluoromethyl-β-hydroxyamides in moderate to good yield as single diastereoisomers in excellent er following purification [61].
Supporting Information
| Supporting Information File 1: Experimental procedures, product characterization data (mp, NMR, IR, HRMS, [α]D, HPLC), and spectra (1H, 13C, and 19F NMR, HPLC). | ||
| Format: PDF | Size: 4.2 MB | Download |
| Supporting Information File 2: Crystallographic details for 12. | ||
| Format: CIF | Size: 390.3 KB | Download |
References
-
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950–8958. doi:10.1002/anie.201202624
Return to citation in text: [1] -
Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293
Return to citation in text: [1] -
Dilman, A. D.; Levin, V. V. Eur. J. Org. Chem. 2011, 831–841. doi:10.1002/ejoc.201001558
Return to citation in text: [1] -
Liu, H.; Gu, Z.; Jiang, X. Adv. Synth. Catal. 2013, 355, 617–626. doi:10.1002/adsc.201200764
Return to citation in text: [1] -
Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f
Return to citation in text: [1] -
Sani, M.; Volonterio, A.; Zanda, M. ChemMedChem 2007, 2, 1693–1700. doi:10.1002/cmdc.200700156
Return to citation in text: [1] -
Léger, S.; Bayly, C. I.; Black, W. C.; Desmarais, S.; Falgueyret, J.-P.; Massé, F.; Percival, M. D.; Truchon, J.-F. Bioorg. Med. Chem. Lett. 2007, 17, 4328–4332. doi:10.1016/j.bmcl.2007.05.024
Return to citation in text: [1] -
Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a
Return to citation in text: [1] -
Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65
Return to citation in text: [1] -
Ma, J.-A.; Cahard, D. Chem. Rev. 2004, 104, 6119–6146. doi:10.1021/cr030143e
Return to citation in text: [1] -
Valero, G.; Companyó, X.; Rios, R. Chem. – Eur. J. 2011, 17, 2018–2037. doi:10.1002/chem.201001546
See for a review.
Return to citation in text: [1] -
Yang, W.; Cui, Y.-M.; Zhou, W.; Li, L.; Yang, K.-F.; Zheng, Z.-J.; Lu, Y.; Xu, L.-W. Synlett 2014, 25, 1461–1465. doi:10.1055/s-0033-1341281
Return to citation in text: [1] -
Vlasserou, I.; Sfetsa, M.; Gerokonstantis, D.-T.; Kokotos, C. G.; Moutevelis-Minakakis, P. Tetrahedron 2018, 74, 2338–2349. doi:10.1016/j.tet.2018.03.054
Return to citation in text: [1] -
Duangdee, N.; Harnying, W.; Rulli, G.; Neudörfl, J.-M.; Gröger, H.; Berkessel, A. J. Am. Chem. Soc. 2012, 134, 11196–11205. doi:10.1021/ja302511t
Return to citation in text: [1] -
Hara, N.; Tamura, R.; Funahashi, Y.; Nakamura, S. Org. Lett. 2011, 13, 1662–1665. doi:10.1021/ol2001039
Return to citation in text: [1] -
Kokotos, C. G. J. Org. Chem. 2012, 77, 1131–1135. doi:10.1021/jo2020104
Return to citation in text: [1] -
Qui, L.-H.; Shen, Z.-X.; Shi, C.-Q.; Liu, Y.-H.; Zhang, Y.-W. Chin. J. Chem. 2005, 23, 584–588. doi:10.1002/cjoc.200590584
Return to citation in text: [1] -
Thomson, C. J.; Barber, D. M.; Dixon, D. J. Angew. Chem., Int. Ed. 2020, 59, 5359–5364. doi:10.1002/anie.201916129
Return to citation in text: [1] -
Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77–144.
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380
Return to citation in text: [1] -
Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z
Return to citation in text: [1] -
Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384
Return to citation in text: [1] -
Ryan, S. J.; Candish, L.; Lupton, D. W. Chem. Soc. Rev. 2013, 42, 4906–4917. doi:10.1039/c3cs35522e
Return to citation in text: [1] -
Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307–9387. doi:10.1021/acs.chemrev.5b00060
Return to citation in text: [1] -
Vora, H. U.; Wheeler, P.; Rovis, T. Adv. Synth. Catal. 2012, 354, 1617–1639. doi:10.1002/adsc.201200031
Return to citation in text: [1] -
Douglas, J.; Churchill, G.; Smith, A. D. Synthesis 2012, 44, 2295–2309. doi:10.1055/s-0031-1289788
Return to citation in text: [1] -
Zhang, Y.-R.; He, L.; Wu, X.; Shao, P.-L.; Ye, S. Org. Lett. 2008, 10, 277–280. doi:10.1021/ol702759b
Return to citation in text: [1] -
Duguet, N.; Campbell, C. D.; Slawin, A. M. Z.; Smith, A. D. Org. Biomol. Chem. 2008, 6, 1108–1113. doi:10.1039/b800857b
Return to citation in text: [1] -
He, M.; Uc, G. J.; Bode, J. W. J. Am. Chem. Soc. 2006, 128, 15088–15089. doi:10.1021/ja066380r
Return to citation in text: [1] -
He, M.; Beahm, B. J.; Bode, J. W. Org. Lett. 2008, 10, 3817–3820. doi:10.1021/ol801502h
Return to citation in text: [1] -
Kobayashi, S.; Kinoshita, T.; Uehara, H.; Sudo, T.; Ryu, I. Org. Lett. 2009, 11, 3934–3937. doi:10.1021/ol901544q
Return to citation in text: [1] -
Yang, L.; Wang, F.; Chua, P. J.; Lv, Y.; Zhong, L.-J.; Zhong, G. Org. Lett. 2012, 14, 2894–2897. doi:10.1021/ol301175z
Return to citation in text: [1] -
Jian, T.-Y.; Sun, L.-H.; Ye, S. Chem. Commun. 2012, 48, 10907–10909. doi:10.1039/c2cc35273g
Return to citation in text: [1] -
Lv, H.; Chen, X.-Y.; Sun, L.-h.; Ye, S. J. Org. Chem. 2010, 75, 6973–6976. doi:10.1021/jo101318u
Return to citation in text: [1] -
Jian, T.-Y.; Shao, P.-L.; Ye, S. Chem. Commun. 2011, 47, 2381–2383. doi:10.1039/c0cc04839a
Return to citation in text: [1] -
Chen, X.; Fang, X.; Chi, Y. R. Chem. Sci. 2013, 4, 2613–2618. doi:10.1039/c3sc50666e
Return to citation in text: [1] -
Hao, L.; Du, Y.; Lv, H.; Chen, X.; Jiang, H.; Shao, Y.; Chi, Y. R. Org. Lett. 2012, 14, 2154–2157. doi:10.1021/ol300676w
Return to citation in text: [1] -
Lee, A.; Younai, A.; Price, C. K.; Izquierdo, J.; Mishra, R. K.; Scheidt, K. A. J. Am. Chem. Soc. 2014, 136, 10589–10592. doi:10.1021/ja505880r
Return to citation in text: [1] -
Cheng, J.-T.; Chen, X.-Y.; Ye, S. Org. Biomol. Chem. 2015, 13, 1313–1316. doi:10.1039/c4ob02330g
Return to citation in text: [1] -
Lee, A.; Scheidt, K. A. Chem. Commun. 2015, 51, 3407–3410. doi:10.1039/c4cc09590a
Return to citation in text: [1] -
Zhao, X.; Ruhl, K. E.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 12330–12333. doi:10.1002/anie.201206490
Return to citation in text: [1] -
Dong, X.; Yang, W.; Hu, W.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 660–663. doi:10.1002/anie.201409961
Return to citation in text: [1] -
Lin, L.; Yang, Y.; Wang, M.; Lai, L.; Guo, Y.; Wang, R. Chem. Commun. 2015, 51, 8134–8137. doi:10.1039/c5cc01587a
Return to citation in text: [1] -
Wang, X.-N.; Shao, P.-L.; Lv, H.; Ye, S. Org. Lett. 2009, 11, 4029–4031. doi:10.1021/ol901290z
Return to citation in text: [1] -
Mo, J.; Yang, R.; Chen, X.; Tiwari, B.; Chi, Y. R. Org. Lett. 2013, 15, 50–53. doi:10.1021/ol303035r
Return to citation in text: [1] -
Ling, K. B.; Smith, A. D. Chem. Commun. 2011, 47, 373–375. doi:10.1039/c0cc02456b
Return to citation in text: [1] -
Davies, A. T.; Taylor, J. E.; Douglas, J.; Collett, C. J.; Morrill, L. C.; Fallan, C.; Slawin, A. M. Z.; Churchill, G.; Smith, A. D. J. Org. Chem. 2013, 78, 9243–9257. doi:10.1021/jo401433q
Return to citation in text: [1] -
Taylor, J. E.; Daniels, D. S. B.; Smith, A. D. Org. Lett. 2013, 15, 6058–6061. doi:10.1021/ol402955f
Return to citation in text: [1] -
Davies, A. T.; Pickett, P. M.; Slawin, A. M. Z.; Smith, A. D. ACS Catal. 2014, 4, 2696–2700. doi:10.1021/cs500667g
Return to citation in text: [1] -
Attaba, N.; Taylor, J. E.; Slawin, A. M. Z.; Smith, A. D. J. Org. Chem. 2015, 80, 9728–9739. doi:10.1021/acs.joc.5b01820
Return to citation in text: [1] -
Kerr, R. W. F.; Greenhalgh, M. D.; Slawin, A. M. Z.; Arnold, P. L.; Smith, A. D. Tetrahedron: Asymmetry 2017, 28, 125–134. doi:10.1016/j.tetasy.2016.10.012
Return to citation in text: [1] -
Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2011, 50, 5331–5334. doi:10.1002/anie.201101522
Return to citation in text: [1] -
Davies, A. T.; Slawin, A. M. Z.; Smith, A. D. Chem. – Eur. J. 2015, 21, 18944–18948. doi:10.1002/chem.201504256
Return to citation in text: [1] [2] -
McDaniel, D. H.; Brown, H. C. J. Org. Chem. 1958, 23, 420–427. doi:10.1021/jo01097a026
Return to citation in text: [1] -
Crystallographic data for 12 has been deposited with the Cambridge Crystallographic Data Centre (ascension number: CCDC 1987497).
Return to citation in text: [1] -
Massey, R. S.; Collett, C. J.; Lindsay, A. G.; Smith, A. D.; O’Donoghue, A. C. J. Am. Chem. Soc. 2012, 134, 20421–20432. doi:10.1021/ja308420c
Return to citation in text: [1] -
Collett, C. J.; Massey, R. S.; Maguire, O. R.; Batsanov, A. S.; O' Donoghue, A. C.; Smith, A. D. Chem. Sci. 2013, 4, 1514–1522. doi:10.1039/c2sc22137c
Return to citation in text: [1] -
Barrios Antúnez, D.-J.; Greenhalgh, M. D.; Brueckner, A. C.; Walden, D. M.; Elías-Rodríguez, P.; Roberts, P.; Young, B. G.; West, T. H.; Slawin, A. M. Z.; Ha-Yeon Cheong, P.; Smith, A. D. Chem. Sci. 2019, 10, 6162–6173. doi:10.1039/c9sc00390h
Return to citation in text: [1] -
The research data underpinning this publication can be accessed at doi:https://doi.org/10.17630/7bc9507c-e817-4da7-988a-02fcd1a6f4fa.
Return to citation in text: [1]
| 1. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 2. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 3. | Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950–8958. doi:10.1002/anie.201202624 |
| 4. | Tomashenko, O. A.; Grushin, V. V. Chem. Rev. 2011, 111, 4475–4521. doi:10.1021/cr1004293 |
| 5. | Dilman, A. D.; Levin, V. V. Eur. J. Org. Chem. 2011, 831–841. doi:10.1002/ejoc.201001558 |
| 6. | Liu, H.; Gu, Z.; Jiang, X. Adv. Synth. Catal. 2013, 355, 617–626. doi:10.1002/adsc.201200764 |
| 14. | Yang, W.; Cui, Y.-M.; Zhou, W.; Li, L.; Yang, K.-F.; Zheng, Z.-J.; Lu, Y.; Xu, L.-W. Synlett 2014, 25, 1461–1465. doi:10.1055/s-0033-1341281 |
| 15. | Vlasserou, I.; Sfetsa, M.; Gerokonstantis, D.-T.; Kokotos, C. G.; Moutevelis-Minakakis, P. Tetrahedron 2018, 74, 2338–2349. doi:10.1016/j.tet.2018.03.054 |
| 16. | Duangdee, N.; Harnying, W.; Rulli, G.; Neudörfl, J.-M.; Gröger, H.; Berkessel, A. J. Am. Chem. Soc. 2012, 134, 11196–11205. doi:10.1021/ja302511t |
| 17. | Hara, N.; Tamura, R.; Funahashi, Y.; Nakamura, S. Org. Lett. 2011, 13, 1662–1665. doi:10.1021/ol2001039 |
| 18. | Kokotos, C. G. J. Org. Chem. 2012, 77, 1131–1135. doi:10.1021/jo2020104 |
| 19. | Qui, L.-H.; Shen, Z.-X.; Shi, C.-Q.; Liu, Y.-H.; Zhang, Y.-W. Chin. J. Chem. 2005, 23, 584–588. doi:10.1002/cjoc.200590584 |
| 47. | Mo, J.; Yang, R.; Chen, X.; Tiwari, B.; Chi, Y. R. Org. Lett. 2013, 15, 50–53. doi:10.1021/ol303035r |
| 13. |
Valero, G.; Companyó, X.; Rios, R. Chem. – Eur. J. 2011, 17, 2018–2037. doi:10.1002/chem.201001546
See for a review. |
| 48. | Ling, K. B.; Smith, A. D. Chem. Commun. 2011, 47, 373–375. doi:10.1039/c0cc02456b |
| 49. | Davies, A. T.; Taylor, J. E.; Douglas, J.; Collett, C. J.; Morrill, L. C.; Fallan, C.; Slawin, A. M. Z.; Churchill, G.; Smith, A. D. J. Org. Chem. 2013, 78, 9243–9257. doi:10.1021/jo401433q |
| 50. | Taylor, J. E.; Daniels, D. S. B.; Smith, A. D. Org. Lett. 2013, 15, 6058–6061. doi:10.1021/ol402955f |
| 51. | Davies, A. T.; Pickett, P. M.; Slawin, A. M. Z.; Smith, A. D. ACS Catal. 2014, 4, 2696–2700. doi:10.1021/cs500667g |
| 52. | Attaba, N.; Taylor, J. E.; Slawin, A. M. Z.; Smith, A. D. J. Org. Chem. 2015, 80, 9728–9739. doi:10.1021/acs.joc.5b01820 |
| 53. | Kerr, R. W. F.; Greenhalgh, M. D.; Slawin, A. M. Z.; Arnold, P. L.; Smith, A. D. Tetrahedron: Asymmetry 2017, 28, 125–134. doi:10.1016/j.tetasy.2016.10.012 |
| 10. | Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a |
| 11. | Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 |
| 12. | Ma, J.-A.; Cahard, D. Chem. Rev. 2004, 104, 6119–6146. doi:10.1021/cr030143e |
| 43. | Zhao, X.; Ruhl, K. E.; Rovis, T. Angew. Chem., Int. Ed. 2012, 51, 12330–12333. doi:10.1002/anie.201206490 |
| 44. | Dong, X.; Yang, W.; Hu, W.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 660–663. doi:10.1002/anie.201409961 |
| 45. | Lin, L.; Yang, Y.; Wang, M.; Lai, L.; Guo, Y.; Wang, R. Chem. Commun. 2015, 51, 8134–8137. doi:10.1039/c5cc01587a |
| 7. | Hagmann, W. K. J. Med. Chem. 2008, 51, 4359–4369. doi:10.1021/jm800219f |
| 8. | Sani, M.; Volonterio, A.; Zanda, M. ChemMedChem 2007, 2, 1693–1700. doi:10.1002/cmdc.200700156 |
| 9. | Léger, S.; Bayly, C. I.; Black, W. C.; Desmarais, S.; Falgueyret, J.-P.; Massé, F.; Percival, M. D.; Truchon, J.-F. Bioorg. Med. Chem. Lett. 2007, 17, 4328–4332. doi:10.1016/j.bmcl.2007.05.024 |
| 46. | Wang, X.-N.; Shao, P.-L.; Lv, H.; Ye, S. Org. Lett. 2009, 11, 4029–4031. doi:10.1021/ol901290z |
| 29. | Zhang, Y.-R.; He, L.; Wu, X.; Shao, P.-L.; Ye, S. Org. Lett. 2008, 10, 277–280. doi:10.1021/ol702759b |
| 30. | Duguet, N.; Campbell, C. D.; Slawin, A. M. Z.; Smith, A. D. Org. Biomol. Chem. 2008, 6, 1108–1113. doi:10.1039/b800857b |
| 36. | Lv, H.; Chen, X.-Y.; Sun, L.-h.; Ye, S. J. Org. Chem. 2010, 75, 6973–6976. doi:10.1021/jo101318u |
| 37. | Jian, T.-Y.; Shao, P.-L.; Ye, S. Chem. Commun. 2011, 47, 2381–2383. doi:10.1039/c0cc04839a |
| 38. | Chen, X.; Fang, X.; Chi, Y. R. Chem. Sci. 2013, 4, 2613–2618. doi:10.1039/c3sc50666e |
| 27. | Vora, H. U.; Wheeler, P.; Rovis, T. Adv. Synth. Catal. 2012, 354, 1617–1639. doi:10.1002/adsc.201200031 |
| 28. | Douglas, J.; Churchill, G.; Smith, A. D. Synthesis 2012, 44, 2295–2309. doi:10.1055/s-0031-1289788 |
| 39. | Hao, L.; Du, Y.; Lv, H.; Chen, X.; Jiang, H.; Shao, Y.; Chi, Y. R. Org. Lett. 2012, 14, 2154–2157. doi:10.1021/ol300676w |
| 40. | Lee, A.; Younai, A.; Price, C. K.; Izquierdo, J.; Mishra, R. K.; Scheidt, K. A. J. Am. Chem. Soc. 2014, 136, 10589–10592. doi:10.1021/ja505880r |
| 41. | Cheng, J.-T.; Chen, X.-Y.; Ye, S. Org. Biomol. Chem. 2015, 13, 1313–1316. doi:10.1039/c4ob02330g |
| 42. | Lee, A.; Scheidt, K. A. Chem. Commun. 2015, 51, 3407–3410. doi:10.1039/c4cc09590a |
| 21. | Moore, J. L.; Rovis, T. Top. Curr. Chem. 2010, 291, 77–144. |
| 22. | Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380 |
| 23. | Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z |
| 24. | Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384 |
| 25. | Ryan, S. J.; Candish, L.; Lupton, D. W. Chem. Soc. Rev. 2013, 42, 4906–4917. doi:10.1039/c3cs35522e |
| 26. | Flanigan, D. M.; Romanov-Michailidis, F.; White, N. A.; Rovis, T. Chem. Rev. 2015, 115, 9307–9387. doi:10.1021/acs.chemrev.5b00060 |
| 20. | Thomson, C. J.; Barber, D. M.; Dixon, D. J. Angew. Chem., Int. Ed. 2020, 59, 5359–5364. doi:10.1002/anie.201916129 |
| 31. | He, M.; Uc, G. J.; Bode, J. W. J. Am. Chem. Soc. 2006, 128, 15088–15089. doi:10.1021/ja066380r |
| 32. | He, M.; Beahm, B. J.; Bode, J. W. Org. Lett. 2008, 10, 3817–3820. doi:10.1021/ol801502h |
| 33. | Kobayashi, S.; Kinoshita, T.; Uehara, H.; Sudo, T.; Ryu, I. Org. Lett. 2009, 11, 3934–3937. doi:10.1021/ol901544q |
| 34. | Yang, L.; Wang, F.; Chua, P. J.; Lv, Y.; Zhong, L.-J.; Zhong, G. Org. Lett. 2012, 14, 2894–2897. doi:10.1021/ol301175z |
| 35. | Jian, T.-Y.; Sun, L.-H.; Ye, S. Chem. Commun. 2012, 48, 10907–10909. doi:10.1039/c2cc35273g |
| 55. | Davies, A. T.; Slawin, A. M. Z.; Smith, A. D. Chem. – Eur. J. 2015, 21, 18944–18948. doi:10.1002/chem.201504256 |
| 54. | Uyanik, M.; Suzuki, D.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2011, 50, 5331–5334. doi:10.1002/anie.201101522 |
| 55. | Davies, A. T.; Slawin, A. M. Z.; Smith, A. D. Chem. – Eur. J. 2015, 21, 18944–18948. doi:10.1002/chem.201504256 |
| 60. | Barrios Antúnez, D.-J.; Greenhalgh, M. D.; Brueckner, A. C.; Walden, D. M.; Elías-Rodríguez, P.; Roberts, P.; Young, B. G.; West, T. H.; Slawin, A. M. Z.; Ha-Yeon Cheong, P.; Smith, A. D. Chem. Sci. 2019, 10, 6162–6173. doi:10.1039/c9sc00390h |
| 61. | The research data underpinning this publication can be accessed at doi:https://doi.org/10.17630/7bc9507c-e817-4da7-988a-02fcd1a6f4fa. |
| 58. | Massey, R. S.; Collett, C. J.; Lindsay, A. G.; Smith, A. D.; O’Donoghue, A. C. J. Am. Chem. Soc. 2012, 134, 20421–20432. doi:10.1021/ja308420c |
| 59. | Collett, C. J.; Massey, R. S.; Maguire, O. R.; Batsanov, A. S.; O' Donoghue, A. C.; Smith, A. D. Chem. Sci. 2013, 4, 1514–1522. doi:10.1039/c2sc22137c |
| 56. | McDaniel, D. H.; Brown, H. C. J. Org. Chem. 1958, 23, 420–427. doi:10.1021/jo01097a026 |
| 57. | Crystallographic data for 12 has been deposited with the Cambridge Crystallographic Data Centre (ascension number: CCDC 1987497). |
© 2020 Davies et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)