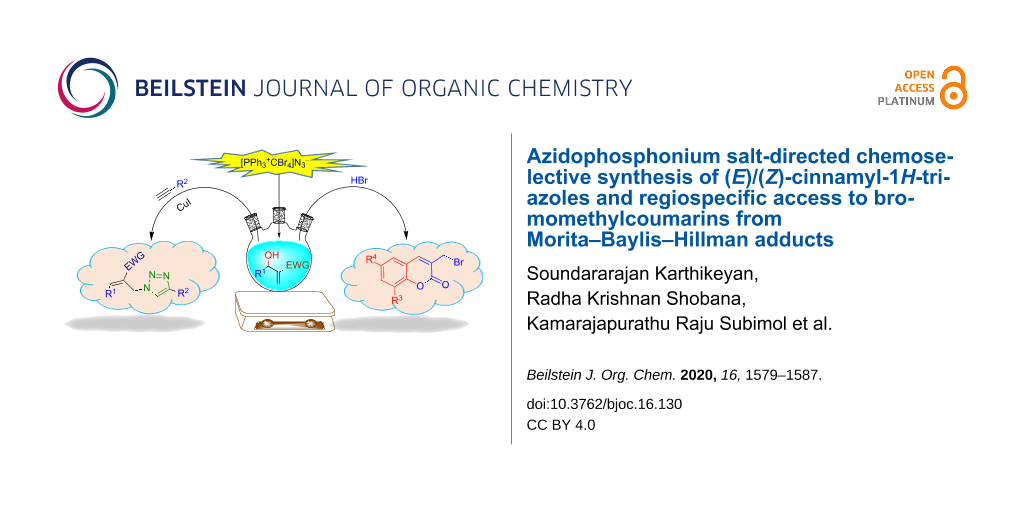
Abstract
The direct transformation of Morita–Baylis–Hillman (MBH) adducts into molecules of interest is a crucial process wherein allylic hydroxy-protected or halogenated MBH adducts are commonly preferred. Herein, we report an azidophosphonium salt (AzPS)-catalysed straight forward protocol for synthesising structurally demanding (E)/(Z)-cinnamyl-1H-1,2,3-triazoles and halomethylcoumarins from MBH adducts. The novel methodology, efficient catalyst, and direct utilization of MBH adducts under mild reaction conditions qualify the reported procedures as powerful synthetic tools.

Graphical Abstract
Introduction
The presence of versatile functional groups in close proximity classifies Morita–Baylis–Hillman adducts as privileged key scaffolds for synthetic organic chemists. Accordingly, MBH adducts have been explored as strategic intermediates for the synthesis of interesting molecules, such as carbamates of unsaturated β-amino acids [1], β-phenylsylfenyl-α-cyanohydrocinnamaldhydes [2], 2-alkylcarbonyl-1-indanols [3], dihydropyrazoles [4], tetrahydroacridines [5], γ-lactams [6], quinolin-5-ones [7], spirobisglutarimides [8], indolizines [9], and spiro carbocyclic frameworks [10]. However, most of the reported synthetic transformations utilize either allylic hydroxy-protected or allyl halide-substituted MBH adducts [11-23].
Among the known synthetic transformations using functionalized MBH adducts, cycloaddition reactions are challenging and attractive for synthetic organic chemists. In this context, acetate-functionalized Morita–Baylis–Hillman adducts have been extensively utilized over other precursors. For example, heterocycles such as, pyrroles (e.g., IV) [24], keto pyrroles (e.g., V) [25], pyridines (e.g., VI) [26], pyrrolotriazoles (e.g., VII) [27], and triazolobenzoxazonines (e.g., VIII) [28] result from MBH acetates (Scheme 1). From these synthetic elaborations, three successive steps are universally utilized: (i) acetylation, (ii) azidation, and (iii) cycloaddition to produce IV–VIII. In spite of the broad scope and synthetic utility, it is evident that the multistep synthetic methodology is the only existing module for cycloaddition reactions.
![[1860-5397-16-130-i1]](/bjoc/content/inline/1860-5397-16-130-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Literature-reported cycloaddition reactions of MBH acetates involving azides and alkynes [24-28].
Scheme 1: Literature-reported cycloaddition reactions of MBH acetates involving azides and alkynes [24-28].
Our research group is focused on developing one-pot synthetic transformations for complex molecules [29-31]. Two individual research groups have reported the multistep pathway to access the cinnamyl-1H-1,2,3-triazole derivatives IX from acetates of MBH adducts (Scheme 2) [32,33]. The other preferable moiety for triazole transformations is the allyl halide of MBH adducts, however, the vicinity of its (E)- and (Z)-isomers restricts their use as a favourable starting moiety [34]. After a careful bibliographic investigation, it became evident that there were no one-pot protocols for direct transformations of MBH adducts to cinnamyl triazoles. The outcome of developing a one-pot synthetic strategy will be worthwhile for pharmacologically important triazoles, such as isavuconazole, tazobactam, and ravuconazole [35].
![[1860-5397-16-130-i2]](/bjoc/content/inline/1860-5397-16-130-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic methodologies for triazolations of MBH adducts. a) Literature-reported indirect triazolation of MBH adducts [32,33]. b) This work: phosphonium salt-catalysed triazolation of MBH adducts.
Scheme 2: Synthetic methodologies for triazolations of MBH adducts. a) Literature-reported indirect triazolat...
Results and Discussion
Initially, phosphonium salts were barely utilised or exploited in synthetic transformations. Later, in 2014, several organic transformations employed quaternary phosphonium salts as favourable catalysts [36]. Their synthetic utility was not only confined to catalysis, but they were also used as intermediates for the synthesis of 1H-indazoles [37], as promoters for stereoselective rearrangements [38], and as temporary protectors of O,P-acetals [39], which branded them as promising motifs. The above reports and the Lewis acid character of quaternary phosphonium salts (QPS) [40-48] qualifies them as reliable catalysts for the proposed methodology. The most elaborate process in the proposed methodology is the protection and elimination of the allylic hydroxy group. We believe that this crucial strategy could be primarily resolved by a quaternary phosphonium salt. After the initial screening of various quaternary phosphonium salts, the azidophosphonium salt [Ph3P+CBr3]N3−, reported by Blanco and co-workers, was opted to accomplish our goal [49-51]. The AzPS surprisingly synchronised with the functional and structural requirements of the proposed work. The azidophosphonium salt was generated and purified according to a modified literature procedure [49].
The one-pot model reaction was investigated using the MBH adduct 1a (1 equiv) and propargyl alcohol (2a, 1.2 equiv) in presence of the AzPS [Ph3P+CBr3]N3− (see Supporting Information File 1 for the substituent patterns of the compounds 1a–o). In this precedent reaction, the adduct 1a and propargyl alcohol (2a) in THF were treated with the AzPS (1 equiv) and CuI (3 mol %) at room temperature. To our expectations, the reaction afforded the (E)-cinnamyl-1H-1,2,3-triazole in a low yield of 24% (Table 1, entry 1). Thereby, we anticipated that an increase in the proportion of the AzPS would substantially increase the yield of 3a (Table 1, entries 2 and 3), but unexpectedly, the reaction demonstrated an unsatisfactory yield. Thereafter, on attempting the reaction with an improved ratio of CuI (5 mol %) and AzPS (2 equiv), the expected product 3a was obtained in a moderate yield (71%, Table 1, entry 4). However, a further increase in the AzPS ascertained a gradual decrease in the yield of 3a (Table 1, entries 5 and 6). The outcome of this analysis might have been due to the formation of large amounts of the byproduct triphenylphosphine oxide, which impeded the purification process and decreased the yield of 3a. Alternative Cu(I) catalysts, CuCl and CuBr, were also used at 5 mol % with the AzPS (2 equiv), however, the combination showed no potential increase in the yield of 3a (Table 1, entries 7 and 8). Comprehensive investigations on the proposed methodology revealed 2 equiv of the AzPS and 5 mol % of CuI as the optimized catalytic combination. Further, the optimized reaction was screened in presence of various solvents (Table 1, entries 9–13), and the outcome revealed acetonitrile as the most preferable solvent, yielding 3a in 83% yield (Table 1, entry 11). Interestingly, the dilution of the reaction mixture did not alter the efficiency of this reaction.
Table 1: Optimization of the triazolation of the MBH adduct 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-130-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | equiv of AzPS | Cu(I) salt (mol %) | solvent | yield (%) |
| 1 | 1 | CuI (3) | THF | 24 |
| 2 | 1.5 | CuI (3) | THF | 33 |
| 3 | 2 | CuI (3) | THF | 42 |
| 4 | 2 | CuI (5) | THF | 71 |
| 5 | 3 | CuI (5) | THF | 45 |
| 6 | 4 | CuI (5) | THF | 36 |
| 7 | 2 | CuCl (5) | THF | 47 |
| 8 | 2 | CuBr (5) | THF | 54 |
| 9 | 2 | CuI (5) | EtOAc | 66 |
| 10 | 2 | CuI (5) | acetone | 69 |
| 11 | 2 | CuI (5) | CH3CN | 83 |
| 12 | 2 | CuI (5) | DMF | 64 |
| 13 | 2 | CuI (5) | DMSO | 69 |
The substrate scope of the optimized reaction and its limitations were further extended to structurally distinct MBH adducts (Scheme 3). The MBH adducts derived from methoxy and ethoxy acrylate stereochemically afforded the (E)-cinnamyl-1,4-disubstituted 1,2,3-triazole derivatives 3a–d/g–k/m–q in a yield of 70–88%. Distinctively, the cyano acrylate-substituted MBH adduct stereoselectively afforded the (Z)-cinnamyl-1,4-disubstituted 1,2,3-triazole derivatives 3e/f/l in a yield of 82–92%. Irrespective of the acetylene moiety, the MBH adducts derived from acrylonitrile comparatively afforded cinnamyl-1,4-disubstituted 1,2,3-triazoles at an improved yield compared to that of the methyl and ethyl counterparts. Notably, the MBH adducts derived from the para-bromo-, para-chloro-, and para-nitrobenzaldehydes favourably assisted the formation of the corresponding (E)-cinnamyl-1,4-disubstituted 1,2,3-triazole derivatives 3g–m in a yield of 72–87%. Alternatively, the ortho- and meta-substituted aryl-MBH adducts were incompatible with the optimized reaction conditions, and this was presumably due to the apparent steric hindrance. Similarly, the MBH adducts derived from aliphatic aldehydes, salicylaldehydes, and methyl- or methoxy-substituted benzaldehydes were also inert under the optimized reaction conditions. Therefore, it is evident that the electronic variation of the substituents on the aromatic moiety of the MBH adducts played a crucial role in determining the outcome of the optimized reactions. We further extended the scope of this transformation to five-membered heterocyclic MBH adducts. To our delight, except pyrroles, the proposed methodology was amenable to MBH adducts of furan and thiophene (3n–q, 70–80%).
![[1860-5397-16-130-i3]](/bjoc/content/inline/1860-5397-16-130-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the one-pot cascade reaction of the unprotected Morita–Baylis–Hillman adducts 3a–q.
Scheme 3: Scope of the one-pot cascade reaction of the unprotected Morita–Baylis–Hillman adducts 3a–q.
The mechanistic pathway for the triazolation proceeded via a nucleophilic attack on the AzPS by the allylic alcohol of the MBH adduct Ia. Subsequently, the azide ion undergoes a nucleophilic attack on the allylic carbon atom of the oxyphosphonium intermediate IIa and generates the 2-azidoalkene IIIa. Interestingly, the consecutive nucleophilic attack by the azido ion smoothly initiates the allylic rearrangement and thereby facilitates the removal of the crucial phosphonium oxide. The outcome of this process is the structurally relevant azido moiety IIIa, which then undergoes a 1,3-dipolar cycloaddition with the copper acetylide IVa to furnish the 1,4-disubstituted 1,2,3-triazoles Va (Figure 1).
![[1860-5397-16-130-1]](/bjoc/content/figures/1860-5397-16-130-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Proposed mechanism for the synthesis of 1,4-disubstituted triazoles.
Figure 1: Proposed mechanism for the synthesis of 1,4-disubstituted triazoles.
At this stage, we sought to analyse the outcome of the proposed reaction following a sequential addition of the reagents utilised for the synthesis of AzPS. Therefore, a preliminary investigation was attempted using the MBH adduct 1a (1 equiv) and propargyl alcohol (2a,1.2 equiv) in the presence of CuI (5 mol %), triphenylphosphine (1 equiv), bromomethane (1.1 equiv), and sodium azide (2 equiv). Unexpectedly, the reaction yielded (Z)-methyl-2-(bromomethyl)-3-phenylacrylate (58%) over the expected triazole. Similarly, the MBH adduct derived from furan, 1i, and phenylacetylene (2b) also yielded (Z)-methyl 2-(bromomethyl)-3-(furan-2-yl)acrylate (42%) rather than the expected triazole (Scheme 4). Thereby, it was clearly evident that the addition of the individual reagents prevented the formation of complicated triazoles.
![[1860-5397-16-130-i4]](/bjoc/content/inline/1860-5397-16-130-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Comparative analysis of the sequential one-pot reaction.
Scheme 4: Comparative analysis of the sequential one-pot reaction.
Interestingly, the MBH adducts derived from salicylaldehydes were inert to triazolations, surprisingly affords bromomethylcoumarin in the presence of AzPS and HBr. The reaction was optimized using salicylaldehyde (1 equiv) in the presence of AzPS (2 equiv) and HBr (2.0 equiv). The reaction afforded 6-(bromomethyl)coumarin (4a) in a yield of 78% (Table 2, entry 3). The synthetic utility of the reaction was further extended to ortho-vanillin and para-bromobenzaldehyde to afford the corresponding halomethylcoumarins (4b/c). However, this regiospecific transformation was restricted only to the MBH adducts derived from salicyladehydes and tert-butyl acrylate [52,53]. Among the reported methodologies on synthesis of halomethylcoumarins [54,55], the present methodology was attractive due to its good yield and the simple reaction conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-130-i6.svg?max-width=637&scale=1.0)
As shown in Figure 2, the mechanistic pathway for 4a–c progressed via treating the MBH adduct (1m) with AzPS and HBr. The outcome of this process was the phosphonium-protected MBH moiety Ib and hydrazoic acid. The counter ion bromine facilitated the nucleophilic attack at the vinylic centre of Ib and the spontaneous removal of triphenylphosphine oxide to yield IIb. A consecutive intramolecular nucleophilic attack of the hydroxy moiety at the carbonyl carbon of IIIb further drove the cyclisation to afford the bromomethylcoumarin 4a.
![[1860-5397-16-130-2]](/bjoc/content/figures/1860-5397-16-130-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Proposed mechanism for the synthesis of 3-(bromomethyl)coumarins.
Figure 2: Proposed mechanism for the synthesis of 3-(bromomethyl)coumarins.
Conclusion
In summary, we reported the first protocol on the quaternary phosphonium salt-mediated direct synthesis of cinnamyltriazoles and 3-(bromomethyl)coumarins from Morita–Baylis–Hillman adducts. In contrast to the contending reports on the synthesis of 1,2,3-triazoles and halomethylcoumarins from MBH adducts, our studies report moderate reaction conditions with an improved yield. The above investigation provides a useful synthetic tool for synthetic organic chemists. The synthesis of biologically important triazoles using the reported methodology is underway in our laboratory.
Experimental
General information
Chemicals were purchased from Sigma-Aldrich, Spectrochem (P) Ltd., Central Drug House (P) Ltd., and Rankem, India. All chemicals were used without further purification. The solvents were purified using standard procedures. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer using CDCl3 and DMSO-d6 as the solvent. Tetramethylsilane (TMS) was used as an internal standard. Chemical shifts are given in δ relative to TMS. High-resolution mass spectra were recorded on an Agilent Technologies 6540 UHD accurate mass Q-TOF LC–MS spectrometer. Melting points are uncorrected. The compounds were purified using column chromatography on silica gel (100–200 mesh) using hexane/ethyl acetate and chloroform/methanol as eluent.
Typical procedure for quaternary phosphonium salts
As described in [49]. Typically, triphenylphosphine, bromomethane, and sodium azide at a molar ratio of 1.1:1.1:5 were utilized for synthesising the quaternary phosphonium salt. Initially, triphenylphosphine and sodium azide were stirred at 0 °C in dimethylformamide (5 mL) for 30 minutes. To the mixture, bromomethane in DMF was added slowly to avoid a sudden increase in temperature. The reaction was slowly warmed to room temperature and stirred for another 30 minutes. Finally, the reaction was quenched by the addition of diethyl ether. The filtration of insoluble inorganic salts resulted in a transparent liquid, which, upon concentration by evaporation, provided a crude oily residue. The residue was dissolved in ethyl acetate, washed with brine, and dried over sodium sulphate to yield a clear oily residue of the quaternary phosphonium salt.
Typical procedure for 1a–o
As described in [52]. A mixture of benzaldehyde (1.1 g, 1.14 mL, 0.01 mol), methyl acrylate (2.05 g, 2.15 mL, 0.023 mol) and DABCO (0.87 g, 0.0077 mol) in chloroform (5 mL) was stirred at room temperature for 7 d. The reaction mixture was quenched with 10% aqueous hydrochloric acid (50 mL) and washed repeatedly with water. The chloroform extract was then dried, concentrated, and purified by column chromatography (hexane/EtOAc 8:2, v/v) to afford 1a as colourless oil (1.64 gm, 85%).
Typical procedure for 3a–q
A solution of AzPS (2 equiv) in acetonitrile (5 mL) was added to a solution of the Morita–Baylis–Hillman adduct 1a (1 equiv) in acetonitrile (3 mL). The reaction mixture was then stirred for an hour, and 1.2 equiv of propargyl alcohol (2a) and CuI (5 mol %) were added. The reaction mixture was stirred for another 4 hours, followed by TLC analysis. After the completion of the reaction, the solution was concentrated, diluted, and extracted with EtOAc. The combined extracts were washed with brine, filtered through a celite bed, and dried over anhydrous Na2SO4. Thereafter, the solvent was removed, and the isolated crude oily product was purified over silica gel (CHCl3/MeOH) to obtain 3a as a white solid.
Typical procedure for 4a–c
To a mixture of the Morita–Baylis–Hillman adduct (1 equiv) and AzPS (2 equiv) in acetonitrile (3 mL), HBr (2 equiv) was added carefully at room temperature. After 2 hours, the reaction mixture was quenched with water (20 mL) and then extracted with ethyl acetate. The organic layer was washed with brine and dried over anhydrous MgSO4. The removal of the solvent in vacuo afforded the crude product, which was purified over silica gel (using hexane/EtOAc) to acquire 4a as colorless crystals.
Supporting Information
| Supporting Information File 1: Compound characterization data and NMR spectra. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Mamaghani, M.; Badrian, A. Tetrahedron Lett. 2004, 45, 1547–1550. doi:10.1016/j.tetlet.2003.12.046
Return to citation in text: [1] -
Yadav, L. D. S.; Srivastava, V. P.; Patel, R. Tetrahedron Lett. 2008, 49, 3142–3146. doi:10.1016/j.tetlet.2008.03.016
Return to citation in text: [1] -
Tu, S.; Xu, L.-H.; Yu, C.-R. Synth. Commun. 2008, 38, 2662–2671. doi:10.1080/00397910601038731
Return to citation in text: [1] -
Gowrisankar, S.; Kim, K.-H.; Kim, J.-N. Bull. Korean Chem. Soc. 2008, 29, 2537–2539. doi:10.5012/bkcs.2008.29.12.2537
Return to citation in text: [1] -
Basavaiah, D.; Rao, J. S.; Reddy, R. J. J. Org. Chem. 2004, 69, 7379–7382. doi:10.1021/jo0489871
Return to citation in text: [1] -
Basavaiah, D.; Srivardhana Rao, J. Tetrahedron Lett. 2004, 45, 1621–1625. doi:10.1016/j.tetlet.2003.12.133
Return to citation in text: [1] -
Zhong, W.; Lin, F.; Chen, R.; Su, W. Synthesis 2008, 2561–2568. doi:10.1055/s-2008-1078601
Return to citation in text: [1] -
Basavaiah, D.; Reddy, R. J. Org. Biomol. Chem. 2008, 6, 1034–1039. doi:10.1039/b717843c
Return to citation in text: [1] -
Basavaiah, D.; Devendar, B.; Lenin, D. V.; Satyanarayana, T. Synlett 2009, 411–416. doi:10.1055/s-0028-1087533
Return to citation in text: [1] -
Basavaiah, D.; Roy, S. Org. Lett. 2008, 10, 1819–1822. doi:10.1021/ol800424v
Return to citation in text: [1] -
Weichert, A.; Hoffmann, H. M. R. J. Org. Chem. 1991, 56, 4098–4112. doi:10.1021/jo00013a007
Return to citation in text: [1] -
Basavaiah, D.; Suguna Hyma, R.; Kumaragurubaran, N. Tetrahedron 2000, 56, 5905–5907. doi:10.1016/s0040-4020(00)00478-6
Return to citation in text: [1] -
Muthiah, C.; Kumar, K. S.; Vittal, J. J.; Kumara Swamy, K. C. Synlett 2002, 1787–1790. doi:10.1055/s-2002-34899
Return to citation in text: [1] -
Mase, N.; Wake, S.; Watanabe, Y.; Toru, T. Tetrahedron Lett. 1998, 39, 5553–5556. doi:10.1016/s0040-4039(98)01118-6
Return to citation in text: [1] -
Drewes, S. E.; Emslie, N. D.; Karodia, N.; Loizou, G. Synth. Commun. 1990, 20, 1437–1443. doi:10.1080/00397919008052859
Return to citation in text: [1] -
Chung, Y. M.; Gong, J. H.; Kim, T. H.; Kim, J. N. Tetrahedron Lett. 2001, 42, 9023–9026. doi:10.1016/s0040-4039(01)01971-2
Return to citation in text: [1] -
Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 295–299. doi:10.1055/s-2000-6257
Return to citation in text: [1] -
Ciclosi, M.; Fava, C.; Galeazzi, R.; Orena, M.; Sepulveda-Arques, J. Tetrahedron Lett. 2002, 43, 2199–2202. doi:10.1016/s0040-4039(02)00233-2
Return to citation in text: [1] -
Wang, M.-X.; Wu, Y. Org. Biomol. Chem. 2003, 1, 535–540. doi:10.1039/b209791e
Return to citation in text: [1] -
Keck, G. E.; Welch, D. S. Org. Lett. 2002, 4, 3687–3690. doi:10.1021/ol026638s
Return to citation in text: [1] -
Marino, J. P.; Nguyen, H. N. J. Org. Chem. 2002, 67, 6291–6296. doi:10.1021/jo0110146
Return to citation in text: [1] -
Ó Dálaigh, C.; Connon, S. J. J. Org. Chem. 2007, 72, 7066–7069. doi:10.1021/jo071223b
Return to citation in text: [1] -
Liu, J.; Li, Q.; Cao, Z.-M.; Jin, Y.; Lin, J.; Yan, S.-J. J. Org. Chem. 2019, 84, 1797–1807. doi:10.1021/acs.joc.8b02594
Return to citation in text: [1] -
Reddy, C. R.; Reddy, M. D.; Srikanth, B. Org. Biomol. Chem. 2012, 10, 4280–4288. doi:10.1039/c2ob25272d
Return to citation in text: [1] [2] -
Reddy, C. R.; Panda, S. A.; Ramaraju, A. J. Org. Chem. 2017, 82, 944–949. doi:10.1021/acs.joc.6b02468
Return to citation in text: [1] [2] -
Raji Reddy, C.; Panda, S. A.; Reddy, M. D. Org. Lett. 2015, 17, 896–899. doi:10.1021/ol503752k
Return to citation in text: [1] [2] -
Park, S. P.; Ahn, S.-H.; Lee, K.-J. Tetrahedron 2010, 66, 3490–3498. doi:10.1016/j.tet.2010.03.017
Return to citation in text: [1] [2] -
Basavaiah, D.; Reddy, B. S.; Lingam, H. Tetrahedron 2013, 69, 10060–10067. doi:10.1016/j.tet.2013.09.056
Return to citation in text: [1] [2] -
Kumar, K. K.; Kumar, R. M.; Subramanian, V.; Das, T. M. Carbohydr. Res. 2010, 345, 2297–2304. doi:10.1016/j.carres.2010.07.037
Return to citation in text: [1] -
Karthik Kumar, K.; Prabu Seenivasan, S.; Kumar, V.; Mohan Das, T. Carbohydr. Res. 2011, 346, 2084–2090. doi:10.1016/j.carres.2011.06.028
Return to citation in text: [1] -
Karthik Kumar, K.; Mohan Das, T. Carbohydr. Res. 2011, 346, 728–732. doi:10.1016/j.carres.2011.02.008
Return to citation in text: [1] -
Chandrasekhar, S.; Basu, D.; Rambabu, C. Tetrahedron Lett. 2006, 47, 3059–3063. doi:10.1016/j.tetlet.2006.03.037
Return to citation in text: [1] [2] -
Sreedhar, B.; Reddy, P. S.; Kumar, N. S. Tetrahedron Lett. 2006, 47, 3055–3058. doi:10.1016/j.tetlet.2006.03.007
Return to citation in text: [1] [2] -
Singh, V.; Hutait, S.; Batra, S. Eur. J. Org. Chem. 2009, 3454–3466. doi:10.1002/ejoc.200900336
Return to citation in text: [1] -
Dheer, D.; Singh, V.; Shankar, R. Bioorg. Chem. 2017, 71, 30–54. doi:10.1016/j.bioorg.2017.01.010
Return to citation in text: [1] -
Chen, L.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2019, 21, 5733–5736. doi:10.1021/acs.orglett.9b02108
Return to citation in text: [1] -
Paul, S.; Panda, S.; Manna, D. Tetrahedron Lett. 2014, 55, 2480–2483. doi:10.1016/j.tetlet.2014.03.001
Return to citation in text: [1] -
Ji, S.-Y.; Sun, Y.-M.; Zhang, H.; Hou, Q.-G.; Zhao, C.-Q. Tetrahedron Lett. 2014, 55, 5742–5744. doi:10.1016/j.tetlet.2014.08.118
Return to citation in text: [1] -
Yahata, K.; Minami, M.; Watanabe, K.; Fujioka, H. Org. Lett. 2014, 16, 3680–3683. doi:10.1021/ol501463p
Return to citation in text: [1] -
Werner, T. Adv. Synth. Catal. 2009, 351, 1469–1481. doi:10.1002/adsc.200900211
Return to citation in text: [1] -
Terada, M.; Kouchi, M. Tetrahedron 2006, 62, 401–409. doi:10.1016/j.tet.2005.09.075
Return to citation in text: [1] -
Mukaiyama, T.; Kashiwagi, K.; Matsui, S. Chem. Lett. 1989, 18, 1397–1400. doi:10.1246/cl.1989.1397
Return to citation in text: [1] -
Amurrio, I.; Córdoba, R.; Csákÿ, A. G.; Plumet, J. Tetrahedron 2004, 60, 10521–10524. doi:10.1016/j.tet.2004.07.101
Return to citation in text: [1] -
Wang, X.; Tian, S.-K. Tetrahedron Lett. 2007, 48, 6010–6013. doi:10.1016/j.tetlet.2007.06.132
Return to citation in text: [1] -
Cairns, A. G.; McQuaker, S. J.; Murphy, M. P.; Hartley, R. C. Targeting mitochondria with small molecules: The preparation of MitoB and MitoP as exomarkers of mitochondrial hydrogen peroxide. In Mitochondrial Medicine: Volume II, Manipulating Mitochondrial Function; Weissig, V.; Edeas, M., Eds.; Methods in Molecular Biology, Vol. 1265; Humana Press: Totowa, NJ, USA, 2015; pp 25–50. doi:10.1007/978-1-4939-2288-8_3
Return to citation in text: [1] -
Fraser, K. J.; MacFarlane, D. R. Aust. J. Chem. 2009, 62, 309. doi:10.1071/ch08558
Return to citation in text: [1] -
Enders, D.; Nguyen, T. V. Org. Biomol. Chem. 2012, 10, 5327. doi:10.1039/c2ob25823d
Return to citation in text: [1] -
Golandaj, A.; Ahmad, A.; Ramjugernath, D. Adv. Synth. Catal. 2017, 359, 3676–3706. doi:10.1002/adsc.201700795
Return to citation in text: [1] -
Jiménez Blanco, J.; García Fernández, J.; Gadelle, A.; Defaye, J. Carbohydr. Res. 1997, 303, 367–372. doi:10.1016/s0008-6215(97)00176-6
Return to citation in text: [1] [2] [3] -
Buder, W.; Schmidt, A. Chem. Ber. 1973, 106, 3812–3816. doi:10.1002/cber.19731061207
Return to citation in text: [1] -
Trippett, S.; Smith, D. J. H. Phosphines and phosphonium salts. In Organophosphorus Chemistry; Trippett, S., Ed.; Royal Society of Chemistry: Cambridge, U.K., 1975; Vol. 6, pp 1–26. doi:10.1039/9781847554291
Return to citation in text: [1] -
Kaye, P. T.; Musa, M. A. Synthesis 2002, 2701–2706. doi:10.1055/s-2002-35984
Return to citation in text: [1] [2] -
Kaye, P. T.; Musa, M. A. Synth. Commun. 2003, 33, 1755–1770. doi:10.1081/scc-120018937
Return to citation in text: [1] -
Hong, W. P.; Lee, K.-J. Synthesis 2005, 33–38. doi:10.1055/s-2004-834916
Return to citation in text: [1] -
Kaye, P. T.; Musa, M. A.; Nocanda, X. W.; Robinson, R. S. Org. Biomol. Chem. 2003, 1, 1133–1138. doi:10.1039/b300360d
Return to citation in text: [1]
| 49. | Jiménez Blanco, J.; García Fernández, J.; Gadelle, A.; Defaye, J. Carbohydr. Res. 1997, 303, 367–372. doi:10.1016/s0008-6215(97)00176-6 |
| 50. | Buder, W.; Schmidt, A. Chem. Ber. 1973, 106, 3812–3816. doi:10.1002/cber.19731061207 |
| 51. | Trippett, S.; Smith, D. J. H. Phosphines and phosphonium salts. In Organophosphorus Chemistry; Trippett, S., Ed.; Royal Society of Chemistry: Cambridge, U.K., 1975; Vol. 6, pp 1–26. doi:10.1039/9781847554291 |
| 49. | Jiménez Blanco, J.; García Fernández, J.; Gadelle, A.; Defaye, J. Carbohydr. Res. 1997, 303, 367–372. doi:10.1016/s0008-6215(97)00176-6 |
| 52. | Kaye, P. T.; Musa, M. A. Synthesis 2002, 2701–2706. doi:10.1055/s-2002-35984 |
| 53. | Kaye, P. T.; Musa, M. A. Synth. Commun. 2003, 33, 1755–1770. doi:10.1081/scc-120018937 |
| 1. | Mamaghani, M.; Badrian, A. Tetrahedron Lett. 2004, 45, 1547–1550. doi:10.1016/j.tetlet.2003.12.046 |
| 5. | Basavaiah, D.; Rao, J. S.; Reddy, R. J. J. Org. Chem. 2004, 69, 7379–7382. doi:10.1021/jo0489871 |
| 27. | Park, S. P.; Ahn, S.-H.; Lee, K.-J. Tetrahedron 2010, 66, 3490–3498. doi:10.1016/j.tet.2010.03.017 |
| 4. | Gowrisankar, S.; Kim, K.-H.; Kim, J.-N. Bull. Korean Chem. Soc. 2008, 29, 2537–2539. doi:10.5012/bkcs.2008.29.12.2537 |
| 28. | Basavaiah, D.; Reddy, B. S.; Lingam, H. Tetrahedron 2013, 69, 10060–10067. doi:10.1016/j.tet.2013.09.056 |
| 3. | Tu, S.; Xu, L.-H.; Yu, C.-R. Synth. Commun. 2008, 38, 2662–2671. doi:10.1080/00397910601038731 |
| 25. | Reddy, C. R.; Panda, S. A.; Ramaraju, A. J. Org. Chem. 2017, 82, 944–949. doi:10.1021/acs.joc.6b02468 |
| 2. | Yadav, L. D. S.; Srivastava, V. P.; Patel, R. Tetrahedron Lett. 2008, 49, 3142–3146. doi:10.1016/j.tetlet.2008.03.016 |
| 26. | Raji Reddy, C.; Panda, S. A.; Reddy, M. D. Org. Lett. 2015, 17, 896–899. doi:10.1021/ol503752k |
| 9. | Basavaiah, D.; Devendar, B.; Lenin, D. V.; Satyanarayana, T. Synlett 2009, 411–416. doi:10.1055/s-0028-1087533 |
| 11. | Weichert, A.; Hoffmann, H. M. R. J. Org. Chem. 1991, 56, 4098–4112. doi:10.1021/jo00013a007 |
| 12. | Basavaiah, D.; Suguna Hyma, R.; Kumaragurubaran, N. Tetrahedron 2000, 56, 5905–5907. doi:10.1016/s0040-4020(00)00478-6 |
| 13. | Muthiah, C.; Kumar, K. S.; Vittal, J. J.; Kumara Swamy, K. C. Synlett 2002, 1787–1790. doi:10.1055/s-2002-34899 |
| 14. | Mase, N.; Wake, S.; Watanabe, Y.; Toru, T. Tetrahedron Lett. 1998, 39, 5553–5556. doi:10.1016/s0040-4039(98)01118-6 |
| 15. | Drewes, S. E.; Emslie, N. D.; Karodia, N.; Loizou, G. Synth. Commun. 1990, 20, 1437–1443. doi:10.1080/00397919008052859 |
| 16. | Chung, Y. M.; Gong, J. H.; Kim, T. H.; Kim, J. N. Tetrahedron Lett. 2001, 42, 9023–9026. doi:10.1016/s0040-4039(01)01971-2 |
| 17. | Chamakh, A.; M'hirsi, M.; Villiéras, J.; Lebreton, J.; Amri, H. Synthesis 2000, 295–299. doi:10.1055/s-2000-6257 |
| 18. | Ciclosi, M.; Fava, C.; Galeazzi, R.; Orena, M.; Sepulveda-Arques, J. Tetrahedron Lett. 2002, 43, 2199–2202. doi:10.1016/s0040-4039(02)00233-2 |
| 19. | Wang, M.-X.; Wu, Y. Org. Biomol. Chem. 2003, 1, 535–540. doi:10.1039/b209791e |
| 20. | Keck, G. E.; Welch, D. S. Org. Lett. 2002, 4, 3687–3690. doi:10.1021/ol026638s |
| 21. | Marino, J. P.; Nguyen, H. N. J. Org. Chem. 2002, 67, 6291–6296. doi:10.1021/jo0110146 |
| 22. | Ó Dálaigh, C.; Connon, S. J. J. Org. Chem. 2007, 72, 7066–7069. doi:10.1021/jo071223b |
| 23. | Liu, J.; Li, Q.; Cao, Z.-M.; Jin, Y.; Lin, J.; Yan, S.-J. J. Org. Chem. 2019, 84, 1797–1807. doi:10.1021/acs.joc.8b02594 |
| 8. | Basavaiah, D.; Reddy, R. J. Org. Biomol. Chem. 2008, 6, 1034–1039. doi:10.1039/b717843c |
| 24. | Reddy, C. R.; Reddy, M. D.; Srikanth, B. Org. Biomol. Chem. 2012, 10, 4280–4288. doi:10.1039/c2ob25272d |
| 7. | Zhong, W.; Lin, F.; Chen, R.; Su, W. Synthesis 2008, 2561–2568. doi:10.1055/s-2008-1078601 |
| 54. | Hong, W. P.; Lee, K.-J. Synthesis 2005, 33–38. doi:10.1055/s-2004-834916 |
| 55. | Kaye, P. T.; Musa, M. A.; Nocanda, X. W.; Robinson, R. S. Org. Biomol. Chem. 2003, 1, 1133–1138. doi:10.1039/b300360d |
| 6. | Basavaiah, D.; Srivardhana Rao, J. Tetrahedron Lett. 2004, 45, 1621–1625. doi:10.1016/j.tetlet.2003.12.133 |
| 49. | Jiménez Blanco, J.; García Fernández, J.; Gadelle, A.; Defaye, J. Carbohydr. Res. 1997, 303, 367–372. doi:10.1016/s0008-6215(97)00176-6 |
| 32. | Chandrasekhar, S.; Basu, D.; Rambabu, C. Tetrahedron Lett. 2006, 47, 3059–3063. doi:10.1016/j.tetlet.2006.03.037 |
| 33. | Sreedhar, B.; Reddy, P. S.; Kumar, N. S. Tetrahedron Lett. 2006, 47, 3055–3058. doi:10.1016/j.tetlet.2006.03.007 |
| 24. | Reddy, C. R.; Reddy, M. D.; Srikanth, B. Org. Biomol. Chem. 2012, 10, 4280–4288. doi:10.1039/c2ob25272d |
| 25. | Reddy, C. R.; Panda, S. A.; Ramaraju, A. J. Org. Chem. 2017, 82, 944–949. doi:10.1021/acs.joc.6b02468 |
| 26. | Raji Reddy, C.; Panda, S. A.; Reddy, M. D. Org. Lett. 2015, 17, 896–899. doi:10.1021/ol503752k |
| 27. | Park, S. P.; Ahn, S.-H.; Lee, K.-J. Tetrahedron 2010, 66, 3490–3498. doi:10.1016/j.tet.2010.03.017 |
| 28. | Basavaiah, D.; Reddy, B. S.; Lingam, H. Tetrahedron 2013, 69, 10060–10067. doi:10.1016/j.tet.2013.09.056 |
| 29. | Kumar, K. K.; Kumar, R. M.; Subramanian, V.; Das, T. M. Carbohydr. Res. 2010, 345, 2297–2304. doi:10.1016/j.carres.2010.07.037 |
| 30. | Karthik Kumar, K.; Prabu Seenivasan, S.; Kumar, V.; Mohan Das, T. Carbohydr. Res. 2011, 346, 2084–2090. doi:10.1016/j.carres.2011.06.028 |
| 31. | Karthik Kumar, K.; Mohan Das, T. Carbohydr. Res. 2011, 346, 728–732. doi:10.1016/j.carres.2011.02.008 |
| 39. | Yahata, K.; Minami, M.; Watanabe, K.; Fujioka, H. Org. Lett. 2014, 16, 3680–3683. doi:10.1021/ol501463p |
| 40. | Werner, T. Adv. Synth. Catal. 2009, 351, 1469–1481. doi:10.1002/adsc.200900211 |
| 41. | Terada, M.; Kouchi, M. Tetrahedron 2006, 62, 401–409. doi:10.1016/j.tet.2005.09.075 |
| 42. | Mukaiyama, T.; Kashiwagi, K.; Matsui, S. Chem. Lett. 1989, 18, 1397–1400. doi:10.1246/cl.1989.1397 |
| 43. | Amurrio, I.; Córdoba, R.; Csákÿ, A. G.; Plumet, J. Tetrahedron 2004, 60, 10521–10524. doi:10.1016/j.tet.2004.07.101 |
| 44. | Wang, X.; Tian, S.-K. Tetrahedron Lett. 2007, 48, 6010–6013. doi:10.1016/j.tetlet.2007.06.132 |
| 45. | Cairns, A. G.; McQuaker, S. J.; Murphy, M. P.; Hartley, R. C. Targeting mitochondria with small molecules: The preparation of MitoB and MitoP as exomarkers of mitochondrial hydrogen peroxide. In Mitochondrial Medicine: Volume II, Manipulating Mitochondrial Function; Weissig, V.; Edeas, M., Eds.; Methods in Molecular Biology, Vol. 1265; Humana Press: Totowa, NJ, USA, 2015; pp 25–50. doi:10.1007/978-1-4939-2288-8_3 |
| 46. | Fraser, K. J.; MacFarlane, D. R. Aust. J. Chem. 2009, 62, 309. doi:10.1071/ch08558 |
| 47. | Enders, D.; Nguyen, T. V. Org. Biomol. Chem. 2012, 10, 5327. doi:10.1039/c2ob25823d |
| 48. | Golandaj, A.; Ahmad, A.; Ramjugernath, D. Adv. Synth. Catal. 2017, 359, 3676–3706. doi:10.1002/adsc.201700795 |
| 37. | Paul, S.; Panda, S.; Manna, D. Tetrahedron Lett. 2014, 55, 2480–2483. doi:10.1016/j.tetlet.2014.03.001 |
| 38. | Ji, S.-Y.; Sun, Y.-M.; Zhang, H.; Hou, Q.-G.; Zhao, C.-Q. Tetrahedron Lett. 2014, 55, 5742–5744. doi:10.1016/j.tetlet.2014.08.118 |
| 32. | Chandrasekhar, S.; Basu, D.; Rambabu, C. Tetrahedron Lett. 2006, 47, 3059–3063. doi:10.1016/j.tetlet.2006.03.037 |
| 33. | Sreedhar, B.; Reddy, P. S.; Kumar, N. S. Tetrahedron Lett. 2006, 47, 3055–3058. doi:10.1016/j.tetlet.2006.03.007 |
| 36. | Chen, L.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2019, 21, 5733–5736. doi:10.1021/acs.orglett.9b02108 |
| 34. | Singh, V.; Hutait, S.; Batra, S. Eur. J. Org. Chem. 2009, 3454–3466. doi:10.1002/ejoc.200900336 |
| 35. | Dheer, D.; Singh, V.; Shankar, R. Bioorg. Chem. 2017, 71, 30–54. doi:10.1016/j.bioorg.2017.01.010 |
© 2020 Karthikeyan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)