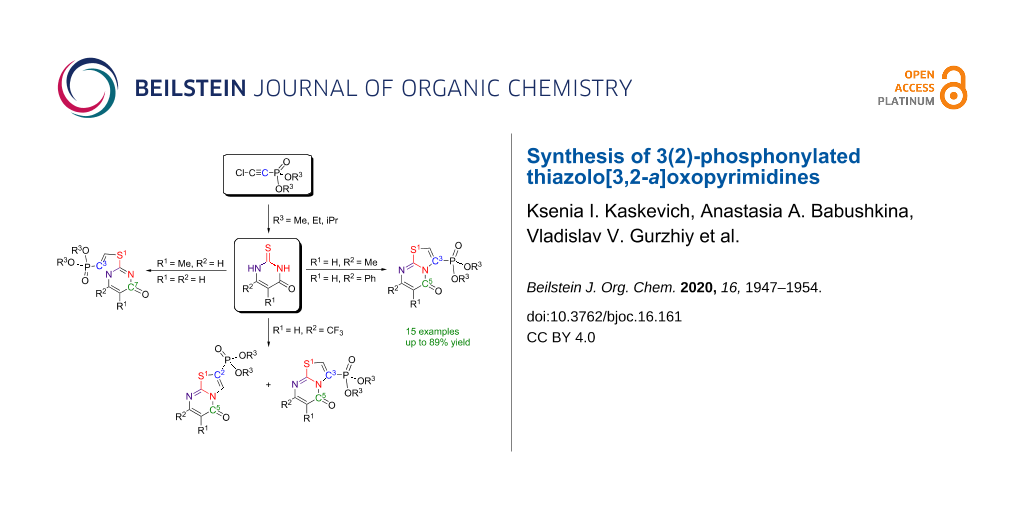
Abstract
A series of 3(2)-phosphonylated thiazolo[3,2-a]oxopyrimidines was synthesized for the first time by the reactions of chloroethynylphosphonates with unsubstituted and 5(6)-substituted 2-thiouracils. The reaction of chloroethynylphosphonates with 6-substituted 2-thiouracils bearing electron-donor groups (CH3, Ph) proceeded with high regioselectivity involving the cyclization through the N3-nitrogen atom to form new 3-phosphonylated thiazolo[3,2-a]-5-oxopyrimidines with good yield. In the case of unsubstituted and 5-methyl-2-thiouracils, cyclization occurred predominantly through the N1 atom and partially via the N3-nitrogen atom to form a mixture of the corresponding thiazolo[3,2-a]-7- and 5-oxopyrimidines. A dramatic change in the reaction regioselectivity was observed in the case of 6-trifluoromethyl-2-thiouracil that afforded 2- and 3-phosphonylated 5-oxothiazolopyrimidines in a 1:1 ratio.

Graphical Abstract
Introduction
Thiazolopyrimidines, whose molecules includes both thiazole and pyrimidine rings, have a structural analogy with the antipsychotic drugs ritanserin and setoperone (Figure 1) [1-3]. To date, a wide spectrum of biological activity of thiazolopyrimidines has been determined: anticancer [4,5], antimicrobial [6,7], anti-inflammatory [8,9], and antiviral [10,11].
![[1860-5397-16-161-1]](/bjoc/content/figures/1860-5397-16-161-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of ritanserin and setoperone drugs.
Figure 1: Structure of ritanserin and setoperone drugs.
The best known methods for the preparation of thiazolopyrimidines are based on condensation reactions. The most commonly used synthesis is the three-component condensation of 2-aminothiazoline, aromatic aldehyde, and ethyl cyanoacetate, which leads to the formation of 5- and 7-oxothiazolopyrimidine-6-carbonitriles (Scheme 1) [12,13].
![[1860-5397-16-161-i1]](/bjoc/content/inline/1860-5397-16-161-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-pot synthesis of 5(7)-oxothiazolopyrimidine-6-carbonitriles.
Scheme 1: One-pot synthesis of 5(7)-oxothiazolopyrimidine-6-carbonitriles.
The synthesis of thiazolopyrimidines through the reaction of 2-aminothiazoles with 1,3-ketoesters in the presence of acids, bases or condensing agents (Scheme 2) has been studied fairly well [6,8,14-16].
![[1860-5397-16-161-i2]](/bjoc/content/inline/1860-5397-16-161-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of thiazolopyrimidine-5-ones through the reaction of 2-aminothiazoles with ethyl acetoacetate.
Scheme 2: Synthesis of thiazolopyrimidine-5-ones through the reaction of 2-aminothiazoles with ethyl acetoace...
The most accessible approach to the synthesis of 5H-thiazolo[3,2-a]pyrimidine-5(7)-ones is the reaction of 2-thiouracil derivatives with α-halo ketones and α-halo acids, involving successive alkylation and condensation steps (Scheme 3) [17-21].
![[1860-5397-16-161-i3]](/bjoc/content/inline/1860-5397-16-161-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2-(benzo[d]thiazol-2-yl)-2-(7-R-5-oxo-5H-thiazolo[3,2-a]pyrimidin-3-yl)acetonitriles.
Scheme 3: Synthesis of 2-(benzo[d]thiazol-2-yl)-2-(7-R-5-oxo-5H-thiazolo[3,2-a]pyrimidin-3-yl)acetonitriles.
A convenient one-step synthesis of thiazolopyrimidine-5-ones by reacting 6-methyl-2-thiouracils with bromoethynylketones has been reported by Shishkin and co-workers (Scheme 4) [20]. The authors for the first time proved the structure of the 5-oxo isomer by single crystal X-ray diffraction analysis.
![[1860-5397-16-161-i4]](/bjoc/content/inline/1860-5397-16-161-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 3-acyl-7-methyl-5H-thiazolo[3,2-a]pyrimidin-5-ones.
Scheme 4: Synthesis of 3-acyl-7-methyl-5H-thiazolo[3,2-a]pyrimidin-5-ones.
The Pd-catalyzed Sonogashira coupling reaction between 2-thiouracil and propargyl bromide yielded 5H-thiazolo[3,2-a]pyrimidine-5-one (Scheme 5) [20-24].
![[1860-5397-16-161-i5]](/bjoc/content/inline/1860-5397-16-161-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Sonogashira coupling reaction of 6-amino-2-thiouracil with propargyl bromide.
Scheme 5: Sonogashira coupling reaction of 6-amino-2-thiouracil with propargyl bromide.
Despite the wide variety of thiazolopyrimidines reported to date, phosphonylated analogues of compounds of this series are unknown. Of special interest is the design of molecules containing practically significant heteroaromatic rings and a biologically active and hydrolysis-resistant phosphonate group, as it has been reported that the combination of several pharmacophore fragments in one molecule can lead to a synergistic increase in biological activity or an additional variety of the latter [22-28].
Herein, we report the synthesis of a new series of phosphonylated thiazolopyrimidines. In our studies, chloroethynylphosphonate was used as the phosphonylating agent, which allowed the formation of a thiazole ring with simultaneous phosphonylation of the latter. As the second reaction component, available 2-thiouracils were chosen as the most studied objects used for creating thiazolopyrimidine systems. The main objective of the study was to determine the regioselectivity of the reaction. Literature data mainly report the formation of 5-oxopyrimidines by cyclization through the N3 atom of the starting 2-thiouracil [14-24]. The formation of 7-oxopyrimidines by cyclization through the N1 atom has been noted only in a few reports [29-32]. However, reliable data for the identification of the 5- and 7-oxo isomers are not available to date and the determination of the structures of 5- and 7-oxo isomers were mainly based on 1H NMR spectroscopy. The most comprehensive and convincing evidence for the formation of a 5-oxothiazolopyrimidine was provided by Shishkin and co-workers [20], who performed single crystal X-ray diffraction analysis along with 1H and 13C NMR spectral studies. Unfortunately, the majority of reports on the synthesis of thiazolopyrimidines relied on 1H NMR data to prove the structure of the obtained compounds [18,33-35]. There are no systematic data on 13C NMR spectroscopy of thiazolopyrimidines, which could be used as an additional approach to estimate the regioselectivity of the reaction. A single example of the use of 13C NMR spectroscopy for unambiguous establishing the structure of thiazolo[3,2-a]pyrimidines obtained was given by Iranian researchers [36], but only for the 5-oxo isomers. In our opinion, the presence of a phosphorus fragment in a thiazolo[3,2-a]pyrimidine molecule significantly facilitates the determination of the structure by means of 13C, 1H, and 31P NMR spectroscopy methods.
Results and Discussion
Aiming to synthesize a new series of phosphonylated thiazolopyrimidines, we performed reactions of unsubstituted and substituted 2-thiouracils 1a–e with chloroethynylphosphonates 2a–c. We found that the reaction with 6-substituted 2-thiouracils bearing either methyl or phenyl groups occurred regioselectively with a N3 atom ring-closure to afford the 3-phosphonylated thiazolo[3,2-a]-5-oxopyrimidines 3a–f in good yields (Scheme 6). Likely, in this case the attack by the N3 atom was more favorable than by the N1 atom due to the steric effect of the substituent in position 6. The reactions proceeded under mild conditions within 3–5 hours. Anhydrous K2CO3 was used as a base to neutralize HCl formed during the reaction. The need for the use of anhydrous solvents and reagents is caused by the possibility of the formation of byproducts, if any, due to hydrolysis as we have noted earlier in the case of the reactions of chloroethynylphosphonates with nitrogen-containing nucleophiles [37-39].
![[1860-5397-16-161-i6]](/bjoc/content/inline/1860-5397-16-161-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactions of 6-substituted 2-thiouracils 1a,b with chloroethynylphosphonates 2a–c.
Scheme 6: Reactions of 6-substituted 2-thiouracils 1a,b with chloroethynylphosphonates 2a–c.
The assignment of the reaction product to the 5-oxo isomer was made based on 13C NMR spectral analysis: the carbon atoms of the CH=СR fragment (R=CH3, Ph) are represented by a strong signal at δC 101–105 ppm (СН=) and a weak signal at δC 157–158 ppm (=CR). These data coincide with those for 3-phenylthiazolo[3,2-a]-5-oxopyrimidine [40]. In addition, the structures of the phosphonylated thiazolopyrimidines 3a and 3d were unambiguously confirmed by single crystal X-ray diffraction data.
The presence of the CH3 group at the position 5 of the thiouracil ring changes the reaction regioselectivity, as the main direction is cyclization through the N1 nitrogen atom with the formation of 3-phosphonylated thiazolo[3,2-a]-7-oxopyrimidines 4a–c and 5-oxo regioisomers 5a–c in a ≈1:0.1–0.3 ratio with yields of 87–91% (Scheme 7).
![[1860-5397-16-161-i7]](/bjoc/content/inline/1860-5397-16-161-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction of 5-methyl-2-thiouracil (1c) with chloroethynylphosphonates 2a–c.
Scheme 7: Reaction of 5-methyl-2-thiouracil (1c) with chloroethynylphosphonates 2a–c.
The structure of thiazolopyrimidines 4a–c and 5a–с is difficult to establish from the 1H and 13C NMR spectral data. The signals of the vinyl proton of the uracil moiety are represented by quartets (4JHH = 1.4 Hz) in the δH 8.1 ppm region (quartet at δH 6 ppm for 6-methyl-5-oxo isomer). In the 13C NMR spectra, the signals of ethylene carbons of the uracil ring are presented at δC 131 and 122 ppm (δC 101 and 158 ppm for 5-oxopyrimidines 5a–c). These data are in accordance with values of the chemical shifts of carbon atoms and protons of the pyrimidine ring in 3-methylthiazolo[3,2-a]pyrimidine-7-one [29], 3-phenylthiazolo[3,2-a]pyrimidine-7-one [40], and 5-phenylthiazolo[3,2-a]pyrimidine-7-one [16].
A similar reaction outcome was observed when chloroethynylphosphonates 2a–c were reacted with unsubstituted 2-thiouracil (1d). The cyclization reaction also proceeded predominantly through the N1 atom to form the corresponding 3-phosphonylated thiazolo[3,2-a]-7-oxopyrimidines 6a–c together with a small amount of 5-oxo isomers 7a–c (Scheme 8). In addition, it should be noted that the regioselectivity of the reaction was higher when using diisopropyl 2-chloroetynylphosphonate (2c).
![[1860-5397-16-161-i8]](/bjoc/content/inline/1860-5397-16-161-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reaction of 2-thiouracil (1d) with chloroethynylphosphonates 2a–c.
Scheme 8: Reaction of 2-thiouracil (1d) with chloroethynylphosphonates 2a–c.
The assignment of the major reaction product to the 7-oxo isomers was made by help of 13C NMR spectroscopy. In the 13C NMR spectra of the thiazolopyrimidines 6a–c, the O=C–CH=CH fragment is observed by signals of equal intensity at δC 112–113 and 135–136 ppm. However, the unambiguous proof of the structure of thiazolopyrimidine-7-one 6b was obtained by single crystal X-ray diffraction analysis.
It is important to note, that the reaction of dimethyl 2-chloroethynylphosphonate (2a) with 2-thiouracil had some features. The reaction proceeded with the formation of a mixture of 7-oxo and 5-oxo isomers in a ≈1: 0.1–0.3 ratio. However, a decrease in the signal of the 7-oxo isomer was observed in the 31P NMR spectrum upon standing at room temperature for 24 hours. The analysis of the formed precipitate identified product 6aa, representing the monodealkylation product of the dimethylphosphonate group. A similar phenomenon has been reported earlier [41].
In the case of 6-trifluoromethyl-2-thiouracil (1e), a dramatic change in the reaction regioselectivity was observed. The cyclization took place with the formation of a mixture of the corresponding 2- and 3-phosphonylated thiazolopyrimidine-5-ones 8a–c and 9a–c (Scheme 9). As in the case of the 6-substituted 2-thiouracils 1a and 1b, the presence of a trifluoromethyl group in position 6 favored an attack by the N3-nitrogen atom, resulting in the formation of the 5-oxo isomer. The formation of the 2-phosphonylated thiazolopyrimidine-5-one could be explained by a primary attack of the electrophilic carbon atom bonded to the chlorine atom by the N3 nitrogen atom of the uracil fragment followed by cyclization.
![[1860-5397-16-161-i9]](/bjoc/content/inline/1860-5397-16-161-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reaction of 6-trifluoromethyl-2-thiouracil (1e) with chloroethynylphosphonates 2a–c.
Scheme 9: Reaction of 6-trifluoromethyl-2-thiouracil (1e) with chloroethynylphosphonates 2a–c.
The mixture of phosphonylated 5-oxothiazolopyrimidines 8c and 9c was separated by column chromatography and the 1Н, 13С, and 31Р NMR spectral data of the individual thiazolopyrimidine isomers did not allow convincing assignment. In the 1H NMR spectra, the proton of the thiazole ring of both isomers is represented by a doublet signal in the low field region at δH 7.91 (3JHP = 3.2 Hz) and 8.41 ppm (3JHP = 7.5 Hz), whereas the corresponding proton of the uracil ring resonated as a singlet at δH 6.61 and 6.71 ppm, respectively. Note that in the case of the reported 3-(4-bromophenyl)-7-trifluoromethyl-5H-[1,3]thiazolo[3,2-a]pyrimidine-5-one [18] the signal of the uracil proton H6 appeared at δH 6.67 ppm.
The 13C NMR spectra of the isomers 8c and 9c differed only in the values of the carbon signals at the phosphorus atom, i.e., doublets at 119.78 (1JHP = 209.5 Hz) and 130.24 ppm (1JHP = 217.5 Hz). The remaining signals were completely identical. Finally, the X-ray diffraction analysis of the 2-phosphonylated thiazolopyrimidine 9c allowed us to uniquely determine its structure.
In our opinion, the unusual formation of the 2-phosphonylated thiazolopyrimidine can be explained by the electron-withdrawing effect of the trifluoromethyl group in the starting 2-thiouracil. In contrast to 6-methyl- or 6-phenyl-2-thiouracil, where the nucleophilicity is localized on the sulfur atom, the presence of the electron-withdrawing CF3 group in 6-trifluoromethyl-2-thiouracil (1e) enhances the acidity of the N3H hydrogen by direct conjugation to the carbonyl moiety. As a result, 2-thiouracil 1e acts as an ambident nucleophile. Thus, the attack of the carbon atom attached to the chlorine by the N3 nitrogen atom is accompanied by the elimination of hydrogen chloride (Scheme 10). A further 5-endo-dig-type cyclization results in the formation of the 2-phosphonylated 5-oxopyrimidines 9a–c. The formation of the 3-phosphonylated 5-oxopyrimidines 8a–c is due to the implementation of the usual favorable direction with the attack of chloroethynylphosphonate by the sulfur atom [33,42-46].
![[1860-5397-16-161-i10]](/bjoc/content/inline/1860-5397-16-161-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: A plausible mechanism of the reaction between 6-trifluoromethyl-2-thiouracil (1e) and chloroethynylphosphonates.
Scheme 10: A plausible mechanism of the reaction between 6-trifluoromethyl-2-thiouracil (1e) and chloroethynyl...
Conclusion
In conclusion, a series of phosphonylated thiazolo[3,2-a]oxopyrimidines was first synthesized by reacting unsubstituted and substituted 2-thiouracils with chloroethynylphosphonates. The main regularities of this reaction were revealed. In the case of 6-substituted 2-thiouracil the primary attack by the most favorable nucleophilic site C=S takes place with further cyclization through the N3 atom of 2-thiouracil to form 5-oxopyrimidines. When using unsubstituted and 5-substituted 2-thiouracils, cyclization occurs predominantly through the N1 atom of the uracil ring, leading to the formation of 7-oxopyrimidines.
Supporting Information
| Supporting Information File 1: General experimental procedure, characterization data, and copies of NMR spectra. | ||
| Format: PDF | Size: 7.9 MB | Download |
References
-
Awad, S. M.; Youns, M. M.; Ahmed, N. M. Pharmacophore 2018, 9, 37–49.
Return to citation in text: [1] -
Cui, P.; Li, X.; Zhu, M.; Wang, B.; Liu, J.; Chen, H. Bioorg. Med. Chem. Lett. 2017, 27, 2234–2237. doi:10.1016/j.bmcl.2016.11.060
Return to citation in text: [1] -
Ahmed, N. M.; Mohamed, M. S. J. Adv. Pharm. Res. 2017, 1, 75–88.
Return to citation in text: [1] -
Shin, N.; Li, Y.-L.; Mei, S.; Wang, K. H.; Hall, L.; Katiyar, K.; Wang, Q.; Yang, G.; Rumberger, B.; Leffet, L.; He, X.; Rupar, M.; Bowman, K.; Favata, M.; Li, J.; Liu, M.; Li, Y.; Covington, M.; Koblish, H.; Soloviev, M.; Shuey, D.; Burn, T.; Diamond, S.; Fridman, J.; Combs, A.; Yao, W.; Yeleswaram, S.; Hollis, G.; Vaddi, K.; Huber, R.; Newton, R.; Scherle, P. J. Pharmacol. Exp. Ther. 2018, 364, 120–130. doi:10.1124/jpet.117.244947
Return to citation in text: [1] -
Abdo, N. Y. M. Acta Chim. Slov. 2015, 62, 168–180.
Return to citation in text: [1] -
Khobragade, C. N.; Bodade, R. G.; Konda, S. G.; Dawane, B. S.; Manwar, A. V. Eur. J. Med. Chem. 2010, 45, 1635–1638. doi:10.1016/j.ejmech.2009.12.040
Return to citation in text: [1] [2] -
Mohamed, M. M.; Khalil, A. K.; Abbass, E. M.; El-Naggar, A. M. Synth. Commun. 2017, 47, 1441–1457. doi:10.1080/00397911.2017.1332223
Return to citation in text: [1] -
El-Borai, M. A.; Rizk, H. F.; Ibrahim, S. A.; El-Sayed, H. F. J. Heterocycl. Chem. 2017, 54, 1031–1041. doi:10.1002/jhet.2671
Return to citation in text: [1] [2] -
Banothu, J.; Khanapur, M.; Basavoju, S.; Bavantula, R.; Narra, M.; Abbagani, S. RSC Adv. 2014, 4, 22866–22874. doi:10.1039/c4ra02514h
Return to citation in text: [1] -
Ravendra Babu, K.; Koteswara Rao, V.; Nanda Kumar, Y.; Polireddy, K.; Venkata Subbaiah, K.; Bhaskar, M.; Lokanatha, V.; Naga Raju, C. Antiviral Res. 2012, 95, 118–127. doi:10.1016/j.antiviral.2012.05.010
Return to citation in text: [1] -
Mohamed, S. F.; Flefel, E. M.; Amr, A. E.-G. E.; Abd El-Shafy, D. N. Eur. J. Med. Chem. 2010, 45, 1494–1501. doi:10.1016/j.ejmech.2009.12.057
Return to citation in text: [1] -
Jeanneau-Nicolle, E.; Benoit-Guyod, M.; Namil, A.; Leclerc, G. Eur. J. Med. Chem. 1992, 27, 115–120. doi:10.1016/0223-5234(92)90099-m
Return to citation in text: [1] -
Jeanneau-Nicolle, E.; Benoit-Guyod, M.; Leclerc, G. Synth. Commun. 1991, 21, 1443–1454. doi:10.1080/00397919108016417
Return to citation in text: [1] -
Awadallah, F. M. Sci. Pharm. 2008, 76, 415–438. doi:10.3797/scipharm.0804-20
Return to citation in text: [1] [2] -
Veretennikov, E. A.; Pavlov, A. V. Russ. J. Org. Chem. 2013, 49, 575–579. doi:10.1134/s1070428013040143
Return to citation in text: [1] [2] -
Yaragatti, N. B.; Kulkarni, M. V.; Ghate, M. D.; Hebbar, S. S.; Hegde, G. R. J. Sulfur Chem. 2010, 31, 123–133. doi:10.1080/17415990903569544
Return to citation in text: [1] [2] [3] -
Abdel-fattah, A.-S. M.; Negm, A. M.; Gaafar, A. E. M. Phosphorus, Sulfur Silicon Relat. Elem. 1992, 72, 145–156. doi:10.1080/10426509208031548
Return to citation in text: [1] [2] -
Frolova, T. V.; Kim, D. G.; Sharutin, V. V.; Shal’kova, E. N. Russ. J. Org. Chem. 2016, 52, 96–98. doi:10.1134/s1070428016010188
Return to citation in text: [1] [2] [3] [4] -
Mohamed, K. S.; Abdulaziz, N. M.; Fadda, A. A. J. Heterocycl. Chem. 2013, 50, 645–649. doi:10.1002/jhet.1672
Return to citation in text: [1] [2] -
Elokhina, V. N.; Nakhmanovich, A. S.; Stepanova, Z. V.; Lopyrev, V. A.; Bannikova, O. B.; Struchkov, Y. T.; Shishkin, O. V. Russ. Chem. Bull. 1996, 45, 2189–2191. doi:10.1007/bf01430737
Return to citation in text: [1] [2] [3] [4] [5] -
El-Emary, T. I.; Abdel-Mohsen, S. A. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 2459–2474. doi:10.1080/10426500600754695
Return to citation in text: [1] [2] [3] -
Lukáč, M.; Hocková, D.; Keough, D. T.; Guddat, L. W.; Janeba, Z. Tetrahedron 2017, 73, 692–702. doi:10.1016/j.tet.2016.12.046
Return to citation in text: [1] [2] [3] -
Abdou, W. M.; Barghash, R. F.; Khidre, R. E. Monatsh. Chem. 2013, 144, 1233–1242. doi:10.1007/s00706-013-0950-6
Return to citation in text: [1] [2] [3] -
Modranka, J.; Jakubowski, R.; Janecki, T. Synlett 2014, 25, 105–109. doi:10.1055/s-0033-1340071
Return to citation in text: [1] [2] [3] -
Abdou, W. M.; Shaddy, A. A.; Kamel, A. A. Chem. Pap. 2017, 71, 1961–1973. doi:10.1007/s11696-017-0190-z
Return to citation in text: [1] -
Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 884–895. doi:10.1002/ardp.201200142
Return to citation in text: [1] -
Radi, S.; Lazrek, H. B. J. Chem. Res., Miniprint 2002, 264–266. doi:10.3184/030823402103172149
Return to citation in text: [1] -
Jansa, P.; Hradil, O.; Baszczyňski, O.; Dračínský, M.; Klepetářová, B.; Holý, A.; Balzarini, J.; Janeba, Z. Tetrahedron 2012, 68, 865–871. doi:10.1016/j.tet.2011.11.040
Return to citation in text: [1] -
Andrew, H. F.; Bradsher, C. K. J. Heterocycl. Chem. 1967, 4, 577–581. doi:10.1002/jhet.5570040419
Return to citation in text: [1] [2] -
Attaby, F. A.; Eldin, S. M. Z. Naturforsch., B: J. Chem. Sci. 1999, 54, 788–798. doi:10.1515/znb-1999-0614
Return to citation in text: [1] -
Weng, Y. Y.; Ying, L. M.; Chen, Q. X.; Su, W. K. Chin. Chem. Lett. 2012, 23, 911–914. doi:10.1016/j.cclet.2012.05.025
Return to citation in text: [1] -
Keivanloo, A.; Bakherad, M.; Rajaei, M. Phosphorus, Sulfur Silicon Relat. Elem. 2014, 189, 1656–1663. doi:10.1080/10426507.2014.885972
Return to citation in text: [1] -
Khudina, O. G.; Ivanova, A. E.; Burgart, Y. V.; Pervova, M. G.; Shatunova, T. V.; Borisevich, S. S.; Khursan, S. L.; Saloutin, V. I. Russ. J. Org. Chem. 2019, 55, 782–791. doi:10.1134/s1070428019060071
Return to citation in text: [1] [2] -
Althagafi, I.; El-Metwaly, N.; Farghaly, T. A. Molecules 2019, 24, 1741. doi:10.3390/molecules24091741
Return to citation in text: [1] -
Ramadan, S. K.; El-Helw, E. A. E.; Sallam, H. A. Heterocycl. Commun. 2019, 25, 107–115. doi:10.1515/hc-2019-0008
Return to citation in text: [1] -
Morsy, H. A.; Moustafa, A. H. J. Iran. Chem. Soc. 2020, 17, 119–125. doi:10.1007/s13738-019-01760-w
Return to citation in text: [1] -
Krylov, A. S.; Kaskevich, K. I.; Erkhitueva, E. B.; Svintsitskaya, N. I.; Dogadina, A. V. Tetrahedron Lett. 2018, 59, 4326–4329. doi:10.1016/j.tetlet.2018.10.052
Return to citation in text: [1] -
Erkhitueva, E. B.; Panikorovskii, T. L.; Svintsitskaya, N. I.; Trifonov, R. E.; Dogadina, A. V. Synlett 2018, 29, 933–937. doi:10.1055/s-0036-1591919
Return to citation in text: [1] -
Krylov, A. S.; Petrosian, A. A.; Piterskaya, J. L.; Svintsitskaya, N. I.; Dogadina, A. V. Beilstein J. Org. Chem. 2019, 15, 1563–1568. doi:10.3762/bjoc.15.159
Return to citation in text: [1] -
Xu, H.; Zhang, Y.; Huang, J.; Chen, W. Org. Lett. 2010, 12, 3704–3707. doi:10.1021/ol101563f
Return to citation in text: [1] [2] -
Erkhitueva, E. B.; Dogadina, A. V.; Khramchikhin, A. V.; Ionin, B. I. Tetrahedron Lett. 2012, 53, 4304–4308. doi:10.1016/j.tetlet.2012.05.157
Return to citation in text: [1] -
Mehta, S.; Larock, R. C. J. Org. Chem. 2010, 75, 1652–1658. doi:10.1021/jo902639f
Return to citation in text: [1] -
Cho, C.-H.; Neuenswander, B.; Larock, R. C. J. Comb. Chem. 2010, 12, 278–285. doi:10.1021/cc900172u
Return to citation in text: [1] -
Kolli, S. K.; Nakhi, A.; Medishetti, R.; Yellanki, S.; Kulkarni, P.; Ramesh Raju, R.; Pal, M. Bioorg. Med. Chem. Lett. 2014, 24, 4460–4465. doi:10.1016/j.bmcl.2014.07.096
Return to citation in text: [1] -
Sanz, R.; Guilarte, V.; Hernando, E.; Sanjuán, A. M. J. Org. Chem. 2010, 75, 7443–7446. doi:10.1021/jo101436f
Return to citation in text: [1] -
Kashiki, T.; Shinamura, S.; Kohara, M.; Miyazaki, E.; Takimiya, K.; Ikeda, M.; Kuwabara, H. Org. Lett. 2009, 11, 2473–2475. doi:10.1021/ol900809w
Return to citation in text: [1]
| 29. | Andrew, H. F.; Bradsher, C. K. J. Heterocycl. Chem. 1967, 4, 577–581. doi:10.1002/jhet.5570040419 |
| 37. | Krylov, A. S.; Kaskevich, K. I.; Erkhitueva, E. B.; Svintsitskaya, N. I.; Dogadina, A. V. Tetrahedron Lett. 2018, 59, 4326–4329. doi:10.1016/j.tetlet.2018.10.052 |
| 38. | Erkhitueva, E. B.; Panikorovskii, T. L.; Svintsitskaya, N. I.; Trifonov, R. E.; Dogadina, A. V. Synlett 2018, 29, 933–937. doi:10.1055/s-0036-1591919 |
| 39. | Krylov, A. S.; Petrosian, A. A.; Piterskaya, J. L.; Svintsitskaya, N. I.; Dogadina, A. V. Beilstein J. Org. Chem. 2019, 15, 1563–1568. doi:10.3762/bjoc.15.159 |
| 40. | Xu, H.; Zhang, Y.; Huang, J.; Chen, W. Org. Lett. 2010, 12, 3704–3707. doi:10.1021/ol101563f |
| 1. | Awad, S. M.; Youns, M. M.; Ahmed, N. M. Pharmacophore 2018, 9, 37–49. |
| 2. | Cui, P.; Li, X.; Zhu, M.; Wang, B.; Liu, J.; Chen, H. Bioorg. Med. Chem. Lett. 2017, 27, 2234–2237. doi:10.1016/j.bmcl.2016.11.060 |
| 3. | Ahmed, N. M.; Mohamed, M. S. J. Adv. Pharm. Res. 2017, 1, 75–88. |
| 10. | Ravendra Babu, K.; Koteswara Rao, V.; Nanda Kumar, Y.; Polireddy, K.; Venkata Subbaiah, K.; Bhaskar, M.; Lokanatha, V.; Naga Raju, C. Antiviral Res. 2012, 95, 118–127. doi:10.1016/j.antiviral.2012.05.010 |
| 11. | Mohamed, S. F.; Flefel, E. M.; Amr, A. E.-G. E.; Abd El-Shafy, D. N. Eur. J. Med. Chem. 2010, 45, 1494–1501. doi:10.1016/j.ejmech.2009.12.057 |
| 18. | Frolova, T. V.; Kim, D. G.; Sharutin, V. V.; Shal’kova, E. N. Russ. J. Org. Chem. 2016, 52, 96–98. doi:10.1134/s1070428016010188 |
| 33. | Khudina, O. G.; Ivanova, A. E.; Burgart, Y. V.; Pervova, M. G.; Shatunova, T. V.; Borisevich, S. S.; Khursan, S. L.; Saloutin, V. I. Russ. J. Org. Chem. 2019, 55, 782–791. doi:10.1134/s1070428019060071 |
| 34. | Althagafi, I.; El-Metwaly, N.; Farghaly, T. A. Molecules 2019, 24, 1741. doi:10.3390/molecules24091741 |
| 35. | Ramadan, S. K.; El-Helw, E. A. E.; Sallam, H. A. Heterocycl. Commun. 2019, 25, 107–115. doi:10.1515/hc-2019-0008 |
| 8. | El-Borai, M. A.; Rizk, H. F.; Ibrahim, S. A.; El-Sayed, H. F. J. Heterocycl. Chem. 2017, 54, 1031–1041. doi:10.1002/jhet.2671 |
| 9. | Banothu, J.; Khanapur, M.; Basavoju, S.; Bavantula, R.; Narra, M.; Abbagani, S. RSC Adv. 2014, 4, 22866–22874. doi:10.1039/c4ra02514h |
| 36. | Morsy, H. A.; Moustafa, A. H. J. Iran. Chem. Soc. 2020, 17, 119–125. doi:10.1007/s13738-019-01760-w |
| 6. | Khobragade, C. N.; Bodade, R. G.; Konda, S. G.; Dawane, B. S.; Manwar, A. V. Eur. J. Med. Chem. 2010, 45, 1635–1638. doi:10.1016/j.ejmech.2009.12.040 |
| 7. | Mohamed, M. M.; Khalil, A. K.; Abbass, E. M.; El-Naggar, A. M. Synth. Commun. 2017, 47, 1441–1457. doi:10.1080/00397911.2017.1332223 |
| 29. | Andrew, H. F.; Bradsher, C. K. J. Heterocycl. Chem. 1967, 4, 577–581. doi:10.1002/jhet.5570040419 |
| 30. | Attaby, F. A.; Eldin, S. M. Z. Naturforsch., B: J. Chem. Sci. 1999, 54, 788–798. doi:10.1515/znb-1999-0614 |
| 31. | Weng, Y. Y.; Ying, L. M.; Chen, Q. X.; Su, W. K. Chin. Chem. Lett. 2012, 23, 911–914. doi:10.1016/j.cclet.2012.05.025 |
| 32. | Keivanloo, A.; Bakherad, M.; Rajaei, M. Phosphorus, Sulfur Silicon Relat. Elem. 2014, 189, 1656–1663. doi:10.1080/10426507.2014.885972 |
| 33. | Khudina, O. G.; Ivanova, A. E.; Burgart, Y. V.; Pervova, M. G.; Shatunova, T. V.; Borisevich, S. S.; Khursan, S. L.; Saloutin, V. I. Russ. J. Org. Chem. 2019, 55, 782–791. doi:10.1134/s1070428019060071 |
| 42. | Mehta, S.; Larock, R. C. J. Org. Chem. 2010, 75, 1652–1658. doi:10.1021/jo902639f |
| 43. | Cho, C.-H.; Neuenswander, B.; Larock, R. C. J. Comb. Chem. 2010, 12, 278–285. doi:10.1021/cc900172u |
| 44. | Kolli, S. K.; Nakhi, A.; Medishetti, R.; Yellanki, S.; Kulkarni, P.; Ramesh Raju, R.; Pal, M. Bioorg. Med. Chem. Lett. 2014, 24, 4460–4465. doi:10.1016/j.bmcl.2014.07.096 |
| 45. | Sanz, R.; Guilarte, V.; Hernando, E.; Sanjuán, A. M. J. Org. Chem. 2010, 75, 7443–7446. doi:10.1021/jo101436f |
| 46. | Kashiki, T.; Shinamura, S.; Kohara, M.; Miyazaki, E.; Takimiya, K.; Ikeda, M.; Kuwabara, H. Org. Lett. 2009, 11, 2473–2475. doi:10.1021/ol900809w |
| 4. | Shin, N.; Li, Y.-L.; Mei, S.; Wang, K. H.; Hall, L.; Katiyar, K.; Wang, Q.; Yang, G.; Rumberger, B.; Leffet, L.; He, X.; Rupar, M.; Bowman, K.; Favata, M.; Li, J.; Liu, M.; Li, Y.; Covington, M.; Koblish, H.; Soloviev, M.; Shuey, D.; Burn, T.; Diamond, S.; Fridman, J.; Combs, A.; Yao, W.; Yeleswaram, S.; Hollis, G.; Vaddi, K.; Huber, R.; Newton, R.; Scherle, P. J. Pharmacol. Exp. Ther. 2018, 364, 120–130. doi:10.1124/jpet.117.244947 |
| 5. | Abdo, N. Y. M. Acta Chim. Slov. 2015, 62, 168–180. |
| 20. | Elokhina, V. N.; Nakhmanovich, A. S.; Stepanova, Z. V.; Lopyrev, V. A.; Bannikova, O. B.; Struchkov, Y. T.; Shishkin, O. V. Russ. Chem. Bull. 1996, 45, 2189–2191. doi:10.1007/bf01430737 |
| 20. | Elokhina, V. N.; Nakhmanovich, A. S.; Stepanova, Z. V.; Lopyrev, V. A.; Bannikova, O. B.; Struchkov, Y. T.; Shishkin, O. V. Russ. Chem. Bull. 1996, 45, 2189–2191. doi:10.1007/bf01430737 |
| 22. | Lukáč, M.; Hocková, D.; Keough, D. T.; Guddat, L. W.; Janeba, Z. Tetrahedron 2017, 73, 692–702. doi:10.1016/j.tet.2016.12.046 |
| 23. | Abdou, W. M.; Barghash, R. F.; Khidre, R. E. Monatsh. Chem. 2013, 144, 1233–1242. doi:10.1007/s00706-013-0950-6 |
| 24. | Modranka, J.; Jakubowski, R.; Janecki, T. Synlett 2014, 25, 105–109. doi:10.1055/s-0033-1340071 |
| 25. | Abdou, W. M.; Shaddy, A. A.; Kamel, A. A. Chem. Pap. 2017, 71, 1961–1973. doi:10.1007/s11696-017-0190-z |
| 26. | Abdou, W. M.; Barghash, R. F.; Bekheit, M. S. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 884–895. doi:10.1002/ardp.201200142 |
| 27. | Radi, S.; Lazrek, H. B. J. Chem. Res., Miniprint 2002, 264–266. doi:10.3184/030823402103172149 |
| 28. | Jansa, P.; Hradil, O.; Baszczyňski, O.; Dračínský, M.; Klepetářová, B.; Holý, A.; Balzarini, J.; Janeba, Z. Tetrahedron 2012, 68, 865–871. doi:10.1016/j.tet.2011.11.040 |
| 41. | Erkhitueva, E. B.; Dogadina, A. V.; Khramchikhin, A. V.; Ionin, B. I. Tetrahedron Lett. 2012, 53, 4304–4308. doi:10.1016/j.tetlet.2012.05.157 |
| 17. | Abdel-fattah, A.-S. M.; Negm, A. M.; Gaafar, A. E. M. Phosphorus, Sulfur Silicon Relat. Elem. 1992, 72, 145–156. doi:10.1080/10426509208031548 |
| 18. | Frolova, T. V.; Kim, D. G.; Sharutin, V. V.; Shal’kova, E. N. Russ. J. Org. Chem. 2016, 52, 96–98. doi:10.1134/s1070428016010188 |
| 19. | Mohamed, K. S.; Abdulaziz, N. M.; Fadda, A. A. J. Heterocycl. Chem. 2013, 50, 645–649. doi:10.1002/jhet.1672 |
| 20. | Elokhina, V. N.; Nakhmanovich, A. S.; Stepanova, Z. V.; Lopyrev, V. A.; Bannikova, O. B.; Struchkov, Y. T.; Shishkin, O. V. Russ. Chem. Bull. 1996, 45, 2189–2191. doi:10.1007/bf01430737 |
| 21. | El-Emary, T. I.; Abdel-Mohsen, S. A. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 2459–2474. doi:10.1080/10426500600754695 |
| 14. | Awadallah, F. M. Sci. Pharm. 2008, 76, 415–438. doi:10.3797/scipharm.0804-20 |
| 15. | Veretennikov, E. A.; Pavlov, A. V. Russ. J. Org. Chem. 2013, 49, 575–579. doi:10.1134/s1070428013040143 |
| 16. | Yaragatti, N. B.; Kulkarni, M. V.; Ghate, M. D.; Hebbar, S. S.; Hegde, G. R. J. Sulfur Chem. 2010, 31, 123–133. doi:10.1080/17415990903569544 |
| 17. | Abdel-fattah, A.-S. M.; Negm, A. M.; Gaafar, A. E. M. Phosphorus, Sulfur Silicon Relat. Elem. 1992, 72, 145–156. doi:10.1080/10426509208031548 |
| 18. | Frolova, T. V.; Kim, D. G.; Sharutin, V. V.; Shal’kova, E. N. Russ. J. Org. Chem. 2016, 52, 96–98. doi:10.1134/s1070428016010188 |
| 19. | Mohamed, K. S.; Abdulaziz, N. M.; Fadda, A. A. J. Heterocycl. Chem. 2013, 50, 645–649. doi:10.1002/jhet.1672 |
| 20. | Elokhina, V. N.; Nakhmanovich, A. S.; Stepanova, Z. V.; Lopyrev, V. A.; Bannikova, O. B.; Struchkov, Y. T.; Shishkin, O. V. Russ. Chem. Bull. 1996, 45, 2189–2191. doi:10.1007/bf01430737 |
| 21. | El-Emary, T. I.; Abdel-Mohsen, S. A. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 2459–2474. doi:10.1080/10426500600754695 |
| 22. | Lukáč, M.; Hocková, D.; Keough, D. T.; Guddat, L. W.; Janeba, Z. Tetrahedron 2017, 73, 692–702. doi:10.1016/j.tet.2016.12.046 |
| 23. | Abdou, W. M.; Barghash, R. F.; Khidre, R. E. Monatsh. Chem. 2013, 144, 1233–1242. doi:10.1007/s00706-013-0950-6 |
| 24. | Modranka, J.; Jakubowski, R.; Janecki, T. Synlett 2014, 25, 105–109. doi:10.1055/s-0033-1340071 |
| 18. | Frolova, T. V.; Kim, D. G.; Sharutin, V. V.; Shal’kova, E. N. Russ. J. Org. Chem. 2016, 52, 96–98. doi:10.1134/s1070428016010188 |
| 6. | Khobragade, C. N.; Bodade, R. G.; Konda, S. G.; Dawane, B. S.; Manwar, A. V. Eur. J. Med. Chem. 2010, 45, 1635–1638. doi:10.1016/j.ejmech.2009.12.040 |
| 8. | El-Borai, M. A.; Rizk, H. F.; Ibrahim, S. A.; El-Sayed, H. F. J. Heterocycl. Chem. 2017, 54, 1031–1041. doi:10.1002/jhet.2671 |
| 14. | Awadallah, F. M. Sci. Pharm. 2008, 76, 415–438. doi:10.3797/scipharm.0804-20 |
| 15. | Veretennikov, E. A.; Pavlov, A. V. Russ. J. Org. Chem. 2013, 49, 575–579. doi:10.1134/s1070428013040143 |
| 16. | Yaragatti, N. B.; Kulkarni, M. V.; Ghate, M. D.; Hebbar, S. S.; Hegde, G. R. J. Sulfur Chem. 2010, 31, 123–133. doi:10.1080/17415990903569544 |
| 40. | Xu, H.; Zhang, Y.; Huang, J.; Chen, W. Org. Lett. 2010, 12, 3704–3707. doi:10.1021/ol101563f |
| 12. | Jeanneau-Nicolle, E.; Benoit-Guyod, M.; Namil, A.; Leclerc, G. Eur. J. Med. Chem. 1992, 27, 115–120. doi:10.1016/0223-5234(92)90099-m |
| 13. | Jeanneau-Nicolle, E.; Benoit-Guyod, M.; Leclerc, G. Synth. Commun. 1991, 21, 1443–1454. doi:10.1080/00397919108016417 |
| 20. | Elokhina, V. N.; Nakhmanovich, A. S.; Stepanova, Z. V.; Lopyrev, V. A.; Bannikova, O. B.; Struchkov, Y. T.; Shishkin, O. V. Russ. Chem. Bull. 1996, 45, 2189–2191. doi:10.1007/bf01430737 |
| 21. | El-Emary, T. I.; Abdel-Mohsen, S. A. Phosphorus, Sulfur Silicon Relat. Elem. 2006, 181, 2459–2474. doi:10.1080/10426500600754695 |
| 22. | Lukáč, M.; Hocková, D.; Keough, D. T.; Guddat, L. W.; Janeba, Z. Tetrahedron 2017, 73, 692–702. doi:10.1016/j.tet.2016.12.046 |
| 23. | Abdou, W. M.; Barghash, R. F.; Khidre, R. E. Monatsh. Chem. 2013, 144, 1233–1242. doi:10.1007/s00706-013-0950-6 |
| 24. | Modranka, J.; Jakubowski, R.; Janecki, T. Synlett 2014, 25, 105–109. doi:10.1055/s-0033-1340071 |
| 16. | Yaragatti, N. B.; Kulkarni, M. V.; Ghate, M. D.; Hebbar, S. S.; Hegde, G. R. J. Sulfur Chem. 2010, 31, 123–133. doi:10.1080/17415990903569544 |
© 2020 Kaskevich et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)