1Department of Studies in Chemistry, University of Mysore, Manasagangothri, Mysuru, Karnataka 570 006, India
2Department of Chemistry, Memorial University of Newfoundland, St. John’s, Newfoundland and Labrador A1B3X7, Canada
3Aramco Laboratory for Applied Sensing Research, King Abdullah Institute for Nanotechnology, King Saudi University, Riyadh, Saudi Arabia
4Surface, and Interface Sciences, Department, of Physics and Astronomy, College of Science, King Saudi University, Riyadh 11451, Saudi Arabia
5Department of Physics, National Institute of Engineering, Mysuru, Karnataka 570 008, India
Corresponding author email
Associate Editor: J. A. Murphy Beilstein J. Org. Chem.2020,16, 159–167.https://doi.org/10.3762/bjoc.16.18 Received 15 Aug 2019,
Accepted 24 Jan 2020,
Published 03 Feb 2020
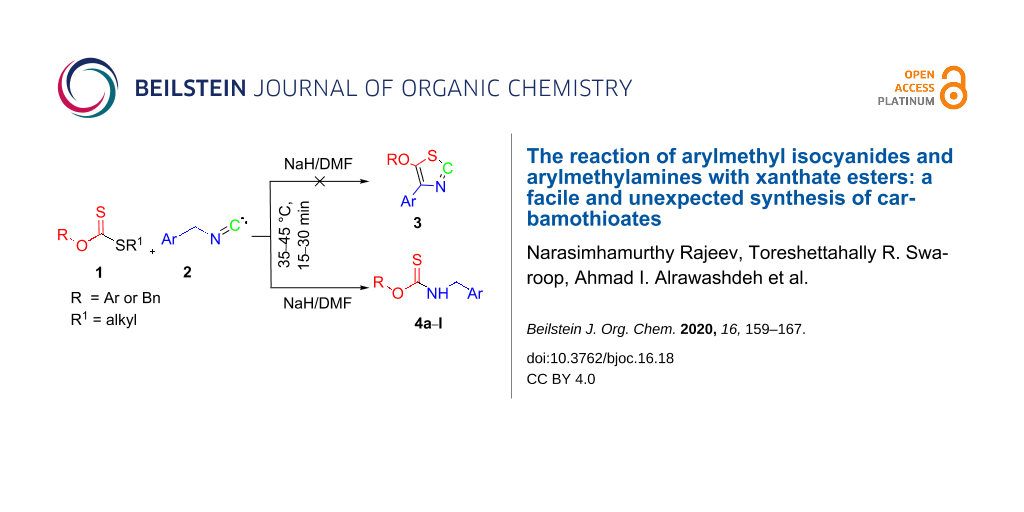
An unexpected formation of carbamothioates by a sodium hydride-mediated reaction of arylmethyl isocyanides with xanthate esters in DMF is reported. The products thus obtained were compared with the carbamothioates obtained by the sodium hydride-mediated condensation of the corresponding benzylamines and xanthate esters in DMF. To account for these unexpected reactions, a mechanism is proposed in which the key steps are supported by quantum chemical calculations.
Carbamothioates (thiocarbamates) have been reported to have antimicrobial [1], antifungal (e.g., tolnaftate and tolciclate) [2], antimycobacterial [3], human leucocyte elastase inhibitory [4], TRPV1 antagonistic [5], and PPARα1γ dual antagonistic [6] properties, and also act as intermediates in the syntheses of HIV-1 integrase ligands [7], insecticides (cartap) [8], and herbicides [9]. They are also used as key intermediates in the generation of carbonyl sulfide/hydrogen sulfide [10], the synthesis of isothiocyanates [11], asymmetric thioureas [12], and thiazolidine/thiaoxazine [13]. Therefore, as a result, numerous synthetic methods for carbamothioates have been reported. These include reactions of chlorothioformates with amines [14], thiocarbonyl benzotriazoles with alcohols [15], copper-catalyzed reactions of α-substituted stannanes with carbamothioates [16], reactions of isothiocyanates with alcohols [6,17], and reactions of xanthate esters with amines [18]. Furthermore, many methods have also been reported for the synthesis of cyclic thiocarbamates, and these include reactions of isothiocyanates with aldehydes in the presence of organocatalysts [19,20], reactions of vicinal diols with potassium thiocyanate [21], iron nanoparticle-catalyzed reactions of 2-naphthol with benzaldehyde and some of its derivatives with thiourea [22], isothiocyanato oxindoles with ketones [23], ammonium isothiocyanates with chalcones [24], and α-isothiocyanato esters with α-keto amides [25]. Among the synthetic methods available for the synthesis of open-chain thiocarbamates, however, many suffer from limitations, such as the use of less stable and sensitive reactants, for example, chlorothioformates [6,14,16,17], toxic stannates [16], and isothiocyanates. In a single patent disclosure, thiocarbamates were reported to have been synthesized from xanthate esters, but the methodology described is limited to only a few examples with aliphatic substituents and furthermore suffers from a tedious isolation protocol [18].
As a part of our work on the development of new synthetic methods [26-30], we have recently reported the synthesis of thiazoles from xanthate esters [31]. In continuation of this ongoing work, we planned to synthesize 5-alkoxy-4-arylthiazoles 3 by the sodium hydride/DMF-mediated reaction of arylmethyl isocyanides 2 with S-alkyl xanthate esters 1 or O-aryl/O-alkyl dithiocarbonates. Unexpectedly, however, carbamothioates 4a–l were instead obtained in 76–88% yield (Scheme 1). Herein, we report on this intriguing finding and show several examples, including a single crystal X-ray structure of one of the products so obtained. A plausible mechanism to explain the reaction using density functional theory (DFT) analysis is also presented in this article.
Scheme 1:
Synthesis of carbamothioates from xanthate esters and benzyl isocyanides.
Scheme 1:
Synthesis of carbamothioates from xanthate esters and benzyl isocyanides.
At the onset of our study, the reaction between O-benzyl S-methyl dithiocarbonate (1a) and benzyl isocyanide was conducted in the presence of sodium hydride in DMF. The product, obtained in 85% yield after 10 min (Table 1, method A, entry 1), was unexpectedly found to be O-benzyl benzylcarbamothioate (4a). The spectral data indicated that the product existed in cis- and trans-geometrical isomeric forms (rotamers) because of free rotation along the thioamide bond. When the same reaction was conducted in other solvents, such as THF, acetonitrile, dioxane, DMSO, or toluene, in the presence of sodium hydride, none of these reactions afforded the product 4a in a satisfactory yield (Table 1, method A, entries 2–6). Replacement of sodium hydride by DBU did not furnish any product at all (Table 1, method A, entry 7), and a 50% reduction in the quantity of sodium hydride did not affect the yield (Table 1, method A, entry 8). Notably, however, the use of only a catalytic amount of sodium hydride also failed to afford any product.
Table 1:
Optimization data for the synthesis of 4a.
method Aa
entry
solvent
base
time
yield of 4a, %
1
DMF
NaHa
10 min
85
2
THF
NaHa
4 h
45
3
CH3CN
NaHa
3 h
53
4
dioxane
NaHa
4 h
48
5
DMSO
NaHa
2 h
58
6
toluene
NaHa
24 h
10
7
DMF
DBU
10 h
0
8
DMF
NaHc
15 min
83
9
DMF
NaHd
24 h
0
method Bb
entry
solvent
base
time
yield of 4a, %
1
DMF
NaH
1 h
80
2
THF
NaH
6 h
35
3
CH3CN
NaH
6 h
55
4
DMSO
NaH
3 h
58
5
toluene
NaH
12 h
29
6
DMF
NaHc
1 h
74
7
DMF
DBU
24 h
0
aReaction conditions: O-benzyl S-methyl dithiocarbonate (1a, 1.0 mmol), benzyl isocyanide (2a, 1.0 mmol), NaH (2.0 mmol), DMF (2.0 mL), 35–45 °C. bReaction conditions: 1a (1.0 mmol), 5a (1.0 mmol), NaH (2.0 mmol), DMF (2.0 mL), 30–40 °C. cNaH (1.0 mmol) was used. dA catalytic amount of 5 mol % NaH was used.
Using the optimized reaction conditions that were established for 4a, the reactions of 1a with 4-methylbenzyl isocyanide (2b) and 4-fluorobenzyl isocyanide (2c) gave the corresponding products 4b and 4c in 84% and 87% yield, respectively (Figure 1). S-Methyl O-(2-methylbenzyl) dithiocarbonate (1b) reacted with benzyl isocyanide (2a) to give O-(2-methylbenzyl) benzylcarbamothioate (4d) in 81% yield. O-(3-Methoxybenzyl) S-methyl dithiocarbonate (1c) reacted with 4-fluorobenzyl isocyanide (2c) or 4-chlorobenzyl isocyanide (2d) to give the corresponding carbamothioates 4e and 4f in 83% and 79% yield, respectively. The generality of the reaction was further probed by reacting O-(4-bromobenzyl) S-methyl dithiocarbonate (1d) with benzyl isocyanide (2a) and 4-methylbenzyl isocyanide (2b), which afforded the corresponding carbamothioates 4g and 4h in 80% and 76% yield, respectively. Interestingly, with O-butyl S-methyl dithiocarbonate (1e), the xanthate ester synthesized from n-butanol, the corresponding O-butyl (4-fluorobenzyl)carbamothioate (4i) and O-butyl (4-chlorobenzyl)carbamothioate (4j), were produced when reacted with 4-fluorobenzyl isocyanide (2c) and 4-chlorobenzyl isocyanide (2d) in similar yields of 86% and 84%, respectively. Finally, S-methyl O-(3-methylcyclohexyl) dithiocarbonate (1f) also afforded the corresponding carbamothioates 4k and 4l in 82% and 88% yields, with benzyl isocyanide (2a) and 4-fluorobenzyl isocyanide (2c), respectively. The use of a weaker base, such as DBU, failed to form any product (Table 1, methods A and B, entry 7).
Figure 1:
Substrate scope for the synthesis of carbamothioates. Reaction conditions for methods A and B: sodium hydride (2.0 mmol), 1a–f (1.0 mmol), 2 or 5 (1.0 mmol), DMF (2.0 mL). aIsolated yield from method A. bIsolated yield from method B.
Figure 1:
Substrate scope for the synthesis of carbamothioates. Reaction conditions for methods A and B: sodi...
For the purpose of comparison, the condensation reaction of O-benzyl S-methyl dithiocarbonate (1a) with benzylamine 5a in the presence of sodium hydride as the base of choice was evaluated using different solvents, DMF, THF, acetonitrile, DMSO, and toluene (Table 1, method B, entries 1–5). DMF was found to be the best solvent, yielding O-benzyl benzylcarbamothioate (4a) in 80% yield after 1 h (Table 1, method B, entry 1). A decreased amount of base reduced the yield slightly (Table 1, method B, entry 6). The versatility of the synthetic methodology was further investigated by reacting O-benzyl S-methyl dithiocarbonate (1a) with 4-methylbenzylamine (5b) and 4-fluorobenzylamine (5c), which respectively yielded O-benzyl (4-methylbenzyl)carbamothioate (4b) and O-benzyl (4-fluorobenzyl)carbamothioate (4c) in 82% and 77% yield (Figure 1).
S-Methyl O-(2-methylbenzyl) dithiocarbonate (1b) reacted smoothly with benzylamine (5a) to give O-(2-methylbenzyl) benzylcarbamothioate (4d) in 81% yield. The xanthate ester O-(3-methoxybenzyl) S-methyl carbonodithioate (1c) underwent condensation with 4-fluorobenzylamine (5c) and 4-chlorobenzylamine (5d) to afford the corresponding O-(3-methoxybenzyl) (4-fluorobenzyl)carbamothioate and O-(3-methoxybenzyl) (4-chlorobenzyl)carbamothioate 4e and 4f in 75% and 83% yield, respectively. The xanthate ester O-(4-bromobenzyl) S-methyl dithiocarbonate (1d) also reacted successfully with benzylamine (5a) and 4-methylbenzylamine (5b) to furnish the corresponding carbamothioates 4g and 4h in 82% and 86% yield, respectively. O-Butyl S-methyl dithiocarbonate (1e), the xanthate ester derived from the aliphatic alcohol n-butanol, also gave the corresponding O-butyl (4-fluorobenzyl)carbamothioate and O-butyl (4-chlorobenzyl)carbamothioate 4i and 4j in 74% and 79% yield, respectively, from the reactions with 4-fluorobenzylamine (5c) and 4-chlorobenzylamine (5d). Finally, the cycloalkyl xanthate ester S-methyl O-(3-methylcyclohexyl) carbonodithioate (1f) also underwent a condensation with benzylamine (5a) and 4-fluorobenzylamine (5c) to give carbamothioates 4k and 4l in 81 and 71% yield, respectively. The NMR spectra of the carbamothioate products obtained indicated that, apart from 4e and 4f, all existed as rotamers and that the ratios of rotamers, where present, were the same whether derived from either method A or B. Alajarin et al. [32] noted a similar doubling of 1H and 13C NMR signals due to rotamers in one of their O-benzyl N-thiocarbamates. The structure of one of the carbamothioates, 4c, was confirmed by a single crystal X-ray diffraction study (Figure 2 as well as Tables S1 and S2, Supporting Information File 1, CCDC reference number: 1831389) [33]. A DFT modeling study was then conducted at the B3LYP/6-311++G(d,p) level of theory, with solvent corrections for chloroform, for two rotamers, namely 4cA and 4cB (generically represented as 4A and 4B in Figure 3). These structures were generated based on the X-ray structure of 4c and afforded a computed Gibbs free energy difference of −1.769 kJ mol−1 in favor of 4cB. The resulting calculated equilibrium constant of 2.042, corresponding to a 67.1/32.9 ratio of the rotamers (4cB/4cA), was in good agreement with the experimentally by 1H NMR (CDCl3) observed ratio of 65/35. Significantly, the single crystal of 4c, which afforded the crystal structure shown in Figure 3, corresponds to rotamer 4cB. A limited variable-temperature 1H NMR study was conducted by heating a solution of 4c in DMSO-d6 from ambient temperature up to 60 °C, but no changes were observed in the ratio of the rotamers.
Figure 2:
ORTEP diagram of O-benzyl (4-fluorobenzyl)carbamothioate (4c).
Figure 2:
ORTEP diagram of O-benzyl (4-fluorobenzyl)carbamothioate (4c).
We initially hypothesized that the isocyanides could have undergone a reductive cleavage to give the corresponding benzylamines, which might have reacted with 1 to give 4. A control experiment was therefore conducted with only the isocyanide under standard reaction conditions. Only unchanged isocyanide was found under these conditions, thus ruling out this initial hypothesis.
Computational studies on the proposed reaction mechanism
Several possible reaction mechanisms were considered to account for the unexpected products obtained. Ultimately, we employed quantum chemical calculations to shed light on the most probable reaction pathway for the observed products, as shown in Scheme 2. For simplicity, the reaction of benzyl isocyanide (2a) with O-benzyl S-methyl dithiocarbonate (1a) was chosen for the calculations, forming the intermediates Int1–3 via the most probable transition states TS1–3, respectively, which, after hydrolysis, formed the observed product 4a. To simplify the quantum chemical calculations, the reactions shown in Scheme 2 involve a hydride as the nucleophile or base [34], although it is possible that dimethylamide, formed from the reaction of sodium hydride with DMF [35], could be the initiating nucleophile/base. All computations were carried out with Gaussian 09 [36]. The HF/6-31G(d) level of theory in the gas phase was only used to locate the transition state geometries. An intrinsic reaction coordinate (IRC) analysis was conducted for each transition state studied in this work to confirm that the transition states were associated with the respective minima. The final IRC structures were further optimized (Figure S14, Supporting Information File 1). The geometries of all reactants, transition states, and intermediates were then fully optimized at the B3LYP/6-311++G(d,p) level of theory in the DMF solvent phase using the polarized continuum model (PCM). Vibrational frequencies for all of the optimized structures were calculated to ensure the presence of a single imaginary frequency for each transition state, and the absence of imaginary frequencies for reactants, intermediates, and products and also to obtain thermal corrections for energies at 298.15 K. The optimized geometries of reactants, transition states, intermediates, and the product of the proposed reaction mechanism are shown in Figure 4. The relative energies are shown in Figure 5 and are summarized in Table S3, Supporting Information File 1. However, it should be noted that quantum chemical calculations for the hydrolysis steps subsequent to the formation of Int3 (i.e., steps A–C leading to the hydrolysis products in Scheme 2) were not conducted.
Scheme 2:
Proposed general reaction mechanism for the formation of carbamothioates (e.g., 4a) from xanthate esters (e.g., 1a) and benzyl isocyanides (e.g., 2a). The counterion in all steps is presumed to be Na+ from NaH.
Scheme 2:
Proposed general reaction mechanism for the formation of carbamothioates (e.g., 4a) from xanthate e...
The proposed mechanism (Scheme 2 and Figure 4) involves several steps, the most significant one being the formation of the anion Int1 by hydride addition to the terminal carbon atom of the isocyanide group in 2a via transition state TS1. This anion undergoes nucleophilic addition to the thiocarbonyl moiety of the xanthate 1a to generate the intermediate Int2 via transition state TS2. In the next step, elimination of the thiomethyl group via transition state TS3 forms the third intermediate Int3, consisting of a carbene and a thiolate anion. The steps leading to the observed product carbamothioate 4a occur from the final quenching hydrolysis of Int3, which occurs via several energetically favorable steps (e.g., A–C), as has been reported for other carbene hydrolyses [37-39]. As can be seen from Figure 5, the highest activation energy barrier is 42.2 kJ/mol.
Figure 4:
Optimized geometries of the reactants, transition states, intermediates, and products of the proposed reaction mechanism, as shown schematically in Scheme 2, determined at the B3LYP/6-311++G(d,p) level of theory in DMF (using a PCM).
Figure 4:
Optimized geometries of the reactants, transition states, intermediates, and products of the propos...
Figure 5:
Relative energies of the reactants, transition states (TS1–TS3), and intermediates (Int1–Int3) of the proposed reaction mechanism, computed at the B3LYP/6-311++G(d,p) level of theory in DMF as the solvent.
Figure 5:
Relative energies of the reactants, transition states (TS1–TS3), and intermediates (Int1–Int3) of t...
We had previously considered an alternative mechanism in which a benzylic proton is instead removed by the base [40]. For the previous mechanism, which we have now recalculated at the B3LYP/6-311++G(d,p) level of theory in DMF (using a PCM, see Scheme S1 and Figures S15 and S16, Supporting Information File 1), an activation energy barrier of 20.6 kJ/mol was obtained for the formation of the resulting benzylic α-carbanion and H2. This benzylic α-carbanion could subsequently undergo a nucleophilic addition to the thiocarbonyl group via a transition state, which was analogous to TS2 depicted in Scheme 2 above. However, in this step, instead of the nitrogen anion, the carbanion was the nucleophile. Subsequently, elimination of the thiomethyl or thiolate anion, either by a stepwise or by a concerted five-membered ring transition state, followed by several subsequent steps could lead to the observed products. However, the mechanism involving all of those steps required an improbably higher overall activation energy of 173.9 kJ/mol (Figure S16, Supporting Information File 1) for the products observed. This alternative mechanism is therefore unlikely, considering the mild conditions employed (35–45 °C), as compared to the proposed reaction mechanism shown in Scheme 2 and Figure 4, which had an overall activation energy of only 42.2 kJ/mol (Figure 5).
Due to the subsequent dilute aqueous/DMF quenching conditions, we were unable to detect the hydrolysis products by HRMS. Furthermore, using the same reaction conditions, which were employed to the S-methyl dithiocarbonates 1a–f, but using S-ethyl or S-benzyl dithiocarbonates, none of the corresponding expected reaction products were obtained. As well, reductive quenching with aqueous NaCNBH3 failed to trap Int3 and only afforded the same reaction products.
Conclusion
There are several reports, which have discussed interesting reactions or reactivity of isocyanides. Among these are those which showed that the isocyanide carbon atom can act as either a nucleophile or an electrophile. To account for the reactions reported herein, the isocyanide carbon atom acted as an electrophile in the reaction with a hydride (or a dimethylamide anion stemming from DMF). A facile general protocol was described for the unexpected formation of carbamothioates 4a–l by the reaction of the corresponding isocyanides 2a–d with S-methyl xanthate esters (or S-methyl dithiocarbonates) 1a–f in the presence of sodium hydride in DMF (method A). The short reaction time and simple work-up procedure were noteworthy features of this protocol. As well, these carbamothioates 4a–l were also synthesized by the condensation of xanthate esters 1a–f with benzylamines 5a–d in the presence of sodium hydride in DMF (method B) for comparison. The reaction times required using method A were shorter than those required by method B. In most cases, rotamers of the final products were detected in the NMR spectra, and a representative DFT computational analysis conducted with 4c (this compound also yielded a crystal structure) was in agreement with the ratio of the two rotamers that were observed in the corresponding NMR spectra. A mechanism was proposed that could be supported by quantum chemical calculations. Of course, other alternative mechanisms that can be envisioned include one in which the thiolate anions, which were generated, could also either a) regenerate dimethylamide anions from the surrounding DMF solvent; and/or b) possibly add to the isocyanide carbon atom to generate a nucleophilic nitrogen atom akin to the step that led to the analogous TS1. However, the fact that catalytic amounts of NaH were not sufficient to afford the observed product (Table 1, method A, entry 9) and that the reactions using method A required at least an equimolar amount of NaH (Table 1, method A, entry 8, cf. entry 1) but 2 equivalents in the other solvents suggested that the latter scenarios (a and b) were less likely and that the methylthiolate was perhaps countered by the sodium cation from sodium hydride. Further work on this and other isocyanide-mediated cyclization reactions are currently in progress in our laboratory.
Supporting Information
Supporting Information File 1:
Experimental procedures, analytical data, copies of 1H and 13C NMR spectra of all studied compounds, and computational details.
All authors are grateful to the Institution of Excellence, University of Mysore, Mysuru for spectral analyses. Compute Canada and Dr. Oliver Stueker are gratefully acknowledged for ongoing support with the computational work.
Funding
M.P.S. thanks UGC-SAP DRS III (grant no. UGC F-540/10/DRS-III (SAP-I) dated 14-09-2016). N.R. thanks UGC, New Delhi for providing an RGNF. T.R.S. thanks SICI for a postdoctoral fellowship. S.R. and A.A. are grateful to the Deanship of Scientific Research, King Saudi University for funding through the Vice Deanship of Scientific Research Chairs.
References
Krátký, M.; Volková, M.; Novotná, E.; Trejtnar, F.; Stolaříková, J.; Vinšová, J. Bioorg. Med. Chem.2014,22, 4073–4082. doi:10.1016/j.bmc.2014.05.064
Return to citation in text:
[1]
Ryder, N. S.; Frank, I.; Dupont, M. C. Antimicrob. Agents Chemother.1986,29, 858–860. doi:10.1128/aac.29.5.858
Return to citation in text:
[1]
Hu, J.-F.; Schetz, J. A.; Kelly, M.; Peng, J.-N.; Ang, K. K. H.; Flotow, H.; Leong, C. Y.; Ng, S. B.; Buss, A. D.; Wilkins, S. P.; Hamann, M. T. J. Nat. Prod.2002,65, 476–480. doi:10.1021/np010471e
Return to citation in text:
[1]
Li-Pan, Z. S.; Joshi, H. V.; Digenis, G. A. J. Enzyme Inhib.1999,15, 63–77. doi:10.1080/14756369909030341
Return to citation in text:
[1]
Lee, J.; Jin, M.-K.; Kang, S.-U.; Kim, S. Y.; Lee, J.; Shin, M.; Hwang, J.; Cho, S.; Choi, Y.-S.; Choi, H.-K.; Kim, S.-E.; Suh, Y.-G.; Lee, Y.-S.; Kim, Y.-H.; Ha, H.-J.; Toth, A.; Pearce, L. V.; Tran, R.; Szabo, T.; Welter, J. D.; Lundberg, D. J.; Wang, Y.; Lazar, J.; Pavlyukovets, V. A.; Morgan, M. A.; Blumberg, P. M. Bioorg. Med. Chem. Lett.2005,15, 4143–4150. doi:10.1016/j.bmcl.2005.06.006
Return to citation in text:
[1]
Kim, N.-J.; Lee, K.-O.; Koo, B.-W.; Li, F.; Yoo, J.-K.; Park, H.-J.; Min, K.-H.; Lim, J. I.; Kim, M. K.; Kim, J.-K.; Suh, Y.-G. Bioorg. Med. Chem. Lett.2007,17, 3595–3598. doi:10.1016/j.bmcl.2007.04.057
Return to citation in text:
[1]
[2]
[3]
Zhang, X.; Neamati, N.; Lee, Y. K.; Orr, A.; Brown, R. D.; Whitaker, N.; Pommier, Y.; Burke, T. R., Jr. Bioorg. Med. Chem.2001,9, 1649–1657. doi:10.1016/s0968-0896(01)00075-x
Return to citation in text:
[1]
Hartley, D.; Kidd, H., Eds. The Agrochemicals Handbook, 2nd ed.; Taylor & Francis: Abingdon-on-Thames, U.K., 1987; pp 1614 ff.
Return to citation in text:
[1]
Steiger, A. K.; Yang, Y.; Royzen, M.; Pluth, M. D. Chem. Commun.2017,53, 1378–1380. doi:10.1039/c6cc09547j
Return to citation in text:
[1]
Li, Z.; Zhang, J.; Shi, S.; Zheng, C.; Zhuang, Y.; Yin, Y.; Sun, X. A process for preparing isothiocyanates. Chin. Patent CN 104860856 A, May 14, 2015.
Return to citation in text:
[1]
Li, Z.; Shi, S.; Sun, X.; Chen, X. Green method for synthesizing asymmetric thiourea. Chin. Patent CN 103435431 A, Aug 30, 2013.
Return to citation in text:
[1]
Meng, X.; Kim, S. Org. Biomol. Chem.2011,9, 4429–4431. doi:10.1039/c1ob05512g
Return to citation in text:
[1]
Katritzky, A. R.; Witek, R. M.; Rodriguez-Garcia, V.; Mohapatra, P. P.; Rogers, J. W.; Cusido, J.; Abdel-Fattah, A. A. A.; Steel, P. J. J. Org. Chem.2005,70, 7866–7881. doi:10.1021/jo050670t
Return to citation in text:
[1]
Falck, J. R.; Bhatt, R. K.; Ye, J. J. Am. Chem. Soc.1995,117, 5973–5982. doi:10.1021/ja00127a010
Return to citation in text:
[1]
[2]
[3]
Plutín, A. M.; Suárez, M.; Ochoa, E.; Machado, T.; Mocelo, R.; Concellón, J. M.; Rodríguez-Solla, H. Tetrahedron2005,61, 5812–5817. doi:10.1016/j.tet.2005.04.018
Return to citation in text:
[1]
[2]
Harris, G. H.; Fischback, B. C. Process for the manufacture of dialkyl thionocarbamates. U.S. Patent US2,691,635, Oct 12, 1954.
Return to citation in text:
[1]
[2]
Han, W.-Y.; Zhao, J.-Q.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem.2013,78, 10541–10547. doi:10.1021/jo401779x
Return to citation in text:
[1]
Chen, W.-B.; Han, W.-Y.; Han, Y.-Y.; Zhang, X.-M.; Yuan, W.-C. Tetrahedron2013,69, 5281–5286. doi:10.1016/j.tet.2013.05.002
Return to citation in text:
[1]
Baranov, V. V.; Kravchenko, A. N.; Nelyubina, Y. V. Mendeleev Commun.2014,24, 105–107. doi:10.1016/j.mencom.2014.03.014
Return to citation in text:
[1]
Basavegowda, N.; Somai Magar, K. B.; Mishra, K.; Lee, Y. R. New J. Chem.2014,38, 5415–5420. doi:10.1039/c4nj01155d
Return to citation in text:
[1]
Lingaraju, G. S.; Swaroop, T. R.; Vinayaka, A. C.; Sharath Kumar, K. S.; Sadashiva, M. P.; Rangappa, K. S. Synthesis2012,44, 1373–1379. doi:10.1055/s-0031-1290762
Return to citation in text:
[1]
Swaroop, T. R.; Roopashree, R.; Ila, H.; Rangappa, K. S. Tetrahedron Lett.2013,54, 147–150. doi:10.1016/j.tetlet.2012.10.110
Return to citation in text:
[1]
Swaroop, T. R.; Ila, H.; Rangappa, K. S. Tetrahedron Lett.2013,54, 5288–5292. doi:10.1016/j.tetlet.2013.07.079
Return to citation in text:
[1]
Vinayaka, A. C.; Swaroop, T. R.; Chikkade, P. K.; Rangappa, K. S.; Sadashiva, M. P. RSC Adv.2016,6, 11528–11535. doi:10.1039/c5ra21421a
Return to citation in text:
[1]
Vinay Kumar, K. S.; Swaroop, T. R.; Rajeev, N.; Vinayaka, A. C.; Lingaraju, G. S.; Rangappa, K. S.; Sadashiva, M. P. Synlett2016,27, 1363–1366. doi:10.1055/s-0035-1561391
Return to citation in text:
[1]
Rajeev, N.; Swaroop, T. R.; Anil, S. M.; Bommegowda, Y. K.; Rangappa, K. S.; Sadashiva, M. P. Synlett2017,28, 2281–2284. doi:10.1055/s-0036-1590811
Return to citation in text:
[1]
Alajarin, M.; Marin-Luna, M.; Ortin, M.-M.; Sanchez-Andrada, P.; Vidal, A. Tetrahedron2009,65, 2579–2590. doi:10.1016/j.tet.2009.01.064
Return to citation in text:
[1]
CCDC 1831389 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/getstructures.
Return to citation in text:
[1]
Spielmann, J.; Harder, S. Chem. – Eur. J.2007,13, 8928–8938. doi:10.1002/chem.200701028
Return to citation in text:
[1]
Filali, E.; Lloyd-Jones, G. C.; Sale, D. A. Synlett2009, 205–208. doi:10.1055/s-0028-1087668
Return to citation in text:
[1]
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text:
[1]
Hollóczki, O.; Terleczky, P.; Szieberth, D.; Mourgas, G.; Gudat, D.; Nyulászi, L. J. Am. Chem. Soc.2011,133, 780–789. doi:10.1021/ja103578y
Return to citation in text:
[1]
Bernasconi, C. F.; Perez, G. S. J. Am. Chem. Soc.2000,122, 12441–12446. doi:10.1021/ja002371l
Return to citation in text:
[1]
Nenajdenko, V. G., Ed. Isocyanide Chemistry: Applications in Synthesis and Material Science; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527652532
Return to citation in text:
[1]
Rajeev, N.; Swaroop, T. R.; Anil, S. M.; Kiran, K. R.; Chandru, C.; Georghiou, P. E.; Alrawashde, A. I.; Rahman, S.; Alodhayb, A.; Rangappa, K. S.; Sadashiva, M. P. Beilstein Arch.2019, No. 201926. doi:10.3762/bxiv.2019.26.v1
Return to citation in text:
[1]
References 26-30
26.
Lingaraju, G. S.; Swaroop, T. R.; Vinayaka, A. C.; Sharath Kumar, K. S.; Sadashiva, M. P.; Rangappa, K. S. Synthesis2012,44, 1373–1379. doi:10.1055/s-0031-1290762
Vinayaka, A. C.; Swaroop, T. R.; Chikkade, P. K.; Rangappa, K. S.; Sadashiva, M. P. RSC Adv.2016,6, 11528–11535. doi:10.1039/c5ra21421a
30.
Vinay Kumar, K. S.; Swaroop, T. R.; Rajeev, N.; Vinayaka, A. C.; Lingaraju, G. S.; Rangappa, K. S.; Sadashiva, M. P. Synlett2016,27, 1363–1366. doi:10.1055/s-0035-1561391
Lee, J.; Jin, M.-K.; Kang, S.-U.; Kim, S. Y.; Lee, J.; Shin, M.; Hwang, J.; Cho, S.; Choi, Y.-S.; Choi, H.-K.; Kim, S.-E.; Suh, Y.-G.; Lee, Y.-S.; Kim, Y.-H.; Ha, H.-J.; Toth, A.; Pearce, L. V.; Tran, R.; Szabo, T.; Welter, J. D.; Lundberg, D. J.; Wang, Y.; Lazar, J.; Pavlyukovets, V. A.; Morgan, M. A.; Blumberg, P. M. Bioorg. Med. Chem. Lett.2005,15, 4143–4150. doi:10.1016/j.bmcl.2005.06.006
Katritzky, A. R.; Witek, R. M.; Rodriguez-Garcia, V.; Mohapatra, P. P.; Rogers, J. W.; Cusido, J.; Abdel-Fattah, A. A. A.; Steel, P. J. J. Org. Chem.2005,70, 7866–7881. doi:10.1021/jo050670t
Hu, J.-F.; Schetz, J. A.; Kelly, M.; Peng, J.-N.; Ang, K. K. H.; Flotow, H.; Leong, C. Y.; Ng, S. B.; Buss, A. D.; Wilkins, S. P.; Hamann, M. T. J. Nat. Prod.2002,65, 476–480. doi:10.1021/np010471e
Hollóczki, O.; Terleczky, P.; Szieberth, D.; Mourgas, G.; Gudat, D.; Nyulászi, L. J. Am. Chem. Soc.2011,133, 780–789. doi:10.1021/ja103578y
38.
Bernasconi, C. F.; Perez, G. S. J. Am. Chem. Soc.2000,122, 12441–12446. doi:10.1021/ja002371l
39.
Nenajdenko, V. G., Ed. Isocyanide Chemistry: Applications in Synthesis and Material Science; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527652532
Rajeev, N.; Swaroop, T. R.; Anil, S. M.; Kiran, K. R.; Chandru, C.; Georghiou, P. E.; Alrawashde, A. I.; Rahman, S.; Alodhayb, A.; Rangappa, K. S.; Sadashiva, M. P. Beilstein Arch.2019, No. 201926. doi:10.3762/bxiv.2019.26.v1
Zhang, X.; Neamati, N.; Lee, Y. K.; Orr, A.; Brown, R. D.; Whitaker, N.; Pommier, Y.; Burke, T. R., Jr. Bioorg. Med. Chem.2001,9, 1649–1657. doi:10.1016/s0968-0896(01)00075-x
CCDC 1831389 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/getstructures.
Kim, N.-J.; Lee, K.-O.; Koo, B.-W.; Li, F.; Yoo, J.-K.; Park, H.-J.; Min, K.-H.; Lim, J. I.; Kim, M. K.; Kim, J.-K.; Suh, Y.-G. Bioorg. Med. Chem. Lett.2007,17, 3595–3598. doi:10.1016/j.bmcl.2007.04.057
Kim, N.-J.; Lee, K.-O.; Koo, B.-W.; Li, F.; Yoo, J.-K.; Park, H.-J.; Min, K.-H.; Lim, J. I.; Kim, M. K.; Kim, J.-K.; Suh, Y.-G. Bioorg. Med. Chem. Lett.2007,17, 3595–3598. doi:10.1016/j.bmcl.2007.04.057
17.
Plutín, A. M.; Suárez, M.; Ochoa, E.; Machado, T.; Mocelo, R.; Concellón, J. M.; Rodríguez-Solla, H. Tetrahedron2005,61, 5812–5817. doi:10.1016/j.tet.2005.04.018
Kim, N.-J.; Lee, K.-O.; Koo, B.-W.; Li, F.; Yoo, J.-K.; Park, H.-J.; Min, K.-H.; Lim, J. I.; Kim, M. K.; Kim, J.-K.; Suh, Y.-G. Bioorg. Med. Chem. Lett.2007,17, 3595–3598. doi:10.1016/j.bmcl.2007.04.057
Falck, J. R.; Bhatt, R. K.; Ye, J. J. Am. Chem. Soc.1995,117, 5973–5982. doi:10.1021/ja00127a010
17.
Plutín, A. M.; Suárez, M.; Ochoa, E.; Machado, T.; Mocelo, R.; Concellón, J. M.; Rodríguez-Solla, H. Tetrahedron2005,61, 5812–5817. doi:10.1016/j.tet.2005.04.018



![[1860-5397-16-18-i1]](/bjoc/content/inline/1860-5397-16-18-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-18-i3.svg?max-width=637&scale=1.0)
![[1860-5397-16-18-1]](/bjoc/content/figures/1860-5397-16-18-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-18-2]](/bjoc/content/figures/1860-5397-16-18-2.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-18-3]](/bjoc/content/figures/1860-5397-16-18-3.jpg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-18-i2]](/bjoc/content/inline/1860-5397-16-18-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-18-4]](/bjoc/content/figures/1860-5397-16-18-4.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-18-5]](/bjoc/content/figures/1860-5397-16-18-5.png?scale=2.0&max-width=1024&background=FFFFFF)