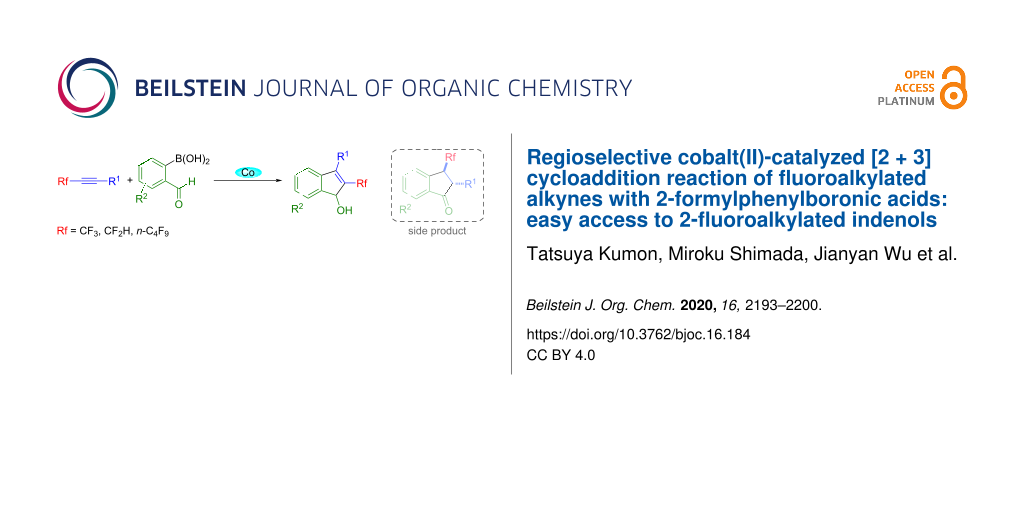
Abstract
[2 + 3] cycloaddition reactions of fluorinated alkynes with 2-formylphenylboronic acids under the influence of Co(acac)2·2H2O in two-component solvents of acetonitrile/2-propanol at reflux temperature for 18 h took place smoothly, affording the corresponding fluoroalkylated indenol derivatives in good yields. This reaction shows excellent regioselectivity, giving 2-fluoroalkylated indenols, together with a very small amount of 3-fluoroalkylated indanones as side products.

Graphical Abstract
Introduction
2,3-Disubstituted indenol derivatives are important compounds possessing high potential due to the insecticidal, myorelaxation, and antiproliferative properties (Figure 1) [1-12]. Thus, enormous attention has been paid to 2- or 3-fluoroalkylated indenol derivatives in the field of medicinal and agrochemical drug design since a fluorine atom can very often bring about an increasing effect on the pharmacological activity owing to the unique nature of the fluorine atom(s) [13-18]. However, reports on a synthetic protocol for 2- or 3-fluoroalkylated indenols are very limited [19-22]. Recently, Yamazaki et al. have reported the reaction of the CF3-containing phthalide B prepared from the 1,2-diester A with the Ruppert–Prakash reagent (TMSCF3) in the presence of a catalytic amount of CsF (Scheme 1a). According to this paper, phthalide B reacted smoothly with phenylthiomethyllithium to give the corresponding CF3-containing indanone C. Then, dehydration of C using p-TsOH⋅H2O, followed by a 1,2-reduction led to 3-fluoroalkylated indenol D. All of these reactions produced the desired products in good yield, however, this protocol is limited to the introduction of a trifluoromethyl group at the 3-position of the indenol. To the best of our knowledge, there are no reports on the practical synthesis of disubstituted 2-fluoroalkylated indenols so far.
![[1860-5397-16-184-1]](/bjoc/content/figures/1860-5397-16-184-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-184-i1]](/bjoc/content/inline/1860-5397-16-184-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2,3-disubstituted indene derivatives.
Scheme 1: Synthesis of 2,3-disubstituted indene derivatives.
Transition-metal-catalyzed carbocyclization reactions of alkynes with benzene derivatives having a leaving group X (X = Br, I, OTs, B(OH)2) have been widely considered as one of the most efficient and convenient protocols for the construction of various 2,3-disubstituted indene derivatives, such as indenols and indenamines (Scheme 1b) [23-25]. There have been numerous studies on the reaction with nonfluorinated alkynes under the influence of various transition metals. Despite their effective advantages, on the other hand, the reports on cycloaddition reactions with fluorine-containing alkynes for the construction of fluoroalkylated indene derivatives are rare [21,26].
Recently, our group has reported the first practical synthesis of fluoroalkylated indenol derivatives using various fluoroalkylated alkynes with 2-iodoaryl ketones via cobalt-catalyzed carbocyclizations (Scheme 1c) [21]. Although our previous work was practical to produce interesting fluoroalkylated indenol derivatives, some drawbacks still remain unsolved. Initially, the reaction showed a low regioselectivity, leading to a mixture of 3-fluoroalkylated and 2-fluoroalkylated indenols, which was difficult to separate. Secondly, a trimer of fluoroalkylated alkynes was obtained as a side product since the cobalt(I) species produced by the cobalt(II)/Zn system worked as a suitable catalyst, leading to the corresponding trimer of the fluoroalkylated alkyne, as reported by our group [27]. Finally, only 2-iodoaryl ketones (R3 = Me, Cy, Ph) were applicable in this catalytic reaction, whereas the cycloaddition using 2-iodobenzaldehyde (R3 = H) did not work at all. Therefore, the development of practical protocols for [2 + 3] cycloaddition reactions with a broader substrate scope for the synthesis of fluoroalkylated indenols is still required. Herein we present a synthesis of 2-fluoroalkylated indenols via [2 + 3] cycloadditions of various fluorinated alkynes with 2-formylphenylboronic acids in the presence of cobalt(II) species as a catalyst to suppress the trimerization products (Scheme 1d).
Results and Discussion
Initially, we carried out the screening of the reaction conditions for the cobalt-catalyzed [2 + 3] cycloaddition using fluoroalkylated alkyne 1a and 2-formylphenylboronic acid (2A) [28]. The results are summarized in Table 1.
Table 1: Screening for the reaction conditions of the cobalt-catalyzed [2 + 3] cycloaddition using the fluoroalkylated alkyne 1a and 2-formylphenylboronic acid (2A).
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-184-i6.svg?max-width=637&scale=1.0)
|
||||||
| entry | solvent | catalyst | ligand | yielda/% | ratioa | yielda/% |
| 3aA + 4aA | 3aA/4aA | 5aA | ||||
| 1 | CH3CN | Co(acac)2·2H2O | dppe | 76 | 66:34 | 12 |
| 2 | DCE | Co(acac)2·2H2O | dppe | 8 | 49:51 | 0 |
| 3 | iPrOH | Co(acac)2·2H2O | dppe | 6 | >99:1 | 3 |
| 4 | 1,4-dioxane | Co(acac)2·2H2O | dppe | 14 | 64:36 | 1 |
| 5 | CH3CN/iPrOH 3:1, v/v | Co(acac)2·2H2O | dppe | 48 | >99:1 | 48 |
| 6 | CH3CN/iPrOH 3:1, v/v | CoCl2 | dppe | 81 | 66:34 | 16 |
| 7 | CH3CN/iPrOH 3:1, v/v | Co(OAc)2·4H2O | dppe | 24 | >99:1 | 24 |
| 8 | CH3CN/iPrOH 3:1, v/v | Co(acac)3 | dppe | 15 | 70:30 | 11 |
| 9 | CH3CN/iPrOH 3:1, v/v | Co(OH)2 | dppe | 8 | 39:61 | 3 |
| 10 | CH3CN/iPrOH 3:1, v/v | Co(acac)2·2H2O | dppp | 59 | 98:2 | 3 |
| 11 | CH3CN/iPrOH 3:1, v/v | Co(acac)2·2H2O | dppb | trace | >99:1 | 2 |
| 12b | CH3CN/iPrOH 3:1, v/v | Co(acac)2·2H2O | dppp | 75 | >99:1 | 6 |
| 13c | CH3CN/iPrOH 3:1, v/v | Co(acac)2·2H2O | dppp | 49 | 73:27 | – |
aDetermined by 19F NMR spectroscopy. bCarried out at the reflux temperature. c2-Acetylphenylboronic acid was used.
The cycloaddition of the fluoroalkylated alkyne 1a with 2.0 equiv of 2-formylphenylboronic acid (2A) in the presence of 10 mol % each of Co(acac)2·2H2O and 1,2- bis(diphenylphosphino)ethane (dppe) in CH3CN at 80 °C for 18 h proceeded to afford the corresponding cyclic products 3aA and 4aA in 76% yield as a regioisomeric 66:34 mixture (Table 1, entry 1). Intriguingly, a small amount of the undesired trans-3-fluoroalkylated indanone 5aA was obtained as a side product, whereas the cis-3-fluoroalkylated and cis/trans-2-fluoroalkylated indanones were not observed. As shown in Table 1, entries 2–4, changing the solvent from CH3CN to DCE, iPrOH, or 1,4-dioxane caused an appreciable decrease in the yield. It should be noted that the use of a mixed solvent of CH3CN and iPrOH significantly improved the isomeric ratio of 3aA and 4aA (>99:1), although 48% of the indanone 5aA was also obtained (Table 1, entry 5). The cycloaddition reaction in the presence of CoCl2 instead of Co(acac)2·2H2O produced the desired 3aA and 4aA in 81% yield, with a lower selectivity of 66:34 (Table 1, entry 6), while the other cobalt catalysts, such as Co(OAc)2·4H2O, Co(acac)3, and Co(OH)2 resulted in the sluggish formation of the fluoroalkylated indenols or indanone (Table 1, entries 7–9). When 1,3-bis(diphenylphosphino)propane (dppp) was used as a phosphine ligand, the desired indenol 3aA was obtained in a moderate yield and with high regioselectivity of 98:2, together with a very small amount of the fluoroalkylated indanone 5aA (Table 1, entry 10). On the other hand, 1,4-bis(diphenylphosphino)butane (dppb) was not a suitable ligand for this reaction, leading only to trace amount of the indanone 5aA (Table 1, entry 11). Finally, the desired cyclization product was produced in a good yield at the reflux temperature while maintaining the selectivity (>99:1, Table 1, entry 12). Unfortunately, 2-acetylphenylboronic acid instead of 2-formylphenylboronic acid (2A) showed a lower reactivity and a poor regioselectivity (73:27, Table 1, entry 13) [29].
With the optimized reaction conditions (Table 1, entry 12), we explored the substrate scope for the [2 + 3] cycloaddition of various fluoroalkylated alkynes 1 and 2-formylphenylboronic acids 2. The results are summarized in Scheme 2.
![[1860-5397-16-184-i2]](/bjoc/content/inline/1860-5397-16-184-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cobalt-catalyzed [2 + 3] cycloaddition reaction of the fluorinated alkynes 1 with various 2-formylphenylboronic acids 2. The yields were determined by 19F NMR spectroscopy. The values in parentheses are the isolated yield of 3. aAtropisomers of 3fA and 5fA were detected. b3.0 equiv of 2 were used. cThe reaction was carried out using 3.0 equiv of 2 in the presence of 20 mol % each of Co(acac)2·2H2O and dppp at 110 °C in a sealed tube.
Scheme 2: Cobalt-catalyzed [2 + 3] cycloaddition reaction of the fluorinated alkynes 1 with various 2-formylp...
The substrates 1, having an electron-donating substituent on the benzene ring of the fluoroalkylated alkyne, such as t-Bu or MeO, reacted efficiently, leading to the desired fluoroalkylated indenols 3bA and 3cA in 74% and 68% yield, respectively. However, the substrate containing the electron-withdrawing group CO2Et on the benzene ring of 1 showed a lower reactivity in this reaction (see 3dA). The cycloaddition reaction using a fluoroalkylated alkyne having a 4-biphenyl group as R1 took place smoothly, giving the corresponding indenol 3eA in a good yield. Though bulkier groups as R1, such as 1-naphthyl or 3-chlorophenyl, reduced the reactivity (see 3fA and 3gA), an excess loading of the boronic acid improved the yield appreciably. For the difluoromethylated alkyne 1h, the desired indenol 3hA was obtained in 62% yield under optimal conditions. However, the nonafluorobutylated alkyne 1i showed a lower reactivity, giving the corresponding indenol 3iA in 22% yield. Therefore, when the reaction was carried out using 3.0 equiv of the boronic acid 2A and 20 mol % each of Co(acac)2·2H2O and dppp at 110 °C, the desired 2-fluoroalkylated indenol 3iA was obtained in 43% yield.
Subsequently, we investigated the [2 + 3] cycloaddition reaction of the fluoroalkylated alkyne 1a (R1 = 4-ClC6H4) with variously substituted 2-formylphenylboronic acids 2. Electron-deficient formylphenylboronic acids possessing a fluorine or chlorine atom on the benzene ring gave the corresponding indenols 3aB and 3aC in 41% and 32% yield, respectively, which could not be improved even when an excess loading of the reagents, prolonging the reaction time, and a higher reaction temperature were applied. However, the exposure of the fluoroalkylated alkynes 1a to electron-rich substrates, e.g., 2D and 2E, lead to the indenols in approximately 30% yield, and the use of an excessive amount of boronic acid, Co(acac)2·2H2O, or dppp improved the yields of the desired indenols, forming 3aD in 51% and 3aE in 41% yield. Changing the substituent position on the aromatic ring of the phenylboronic acid from the 4- to the 5-position did not affect the result, giving the indenol 3aF in 52% yield. However, a boronic acid with a fluorine atom at the 3-position did not lead to a satisfactory result, with the cyclic product 3aG being formed in only 22% yield.
Substituted indenones and indanones widely exist in nature, and these skeletons are important classes of organic compounds due to diverse biological and pharmacological activities [30-32]. Therefore, we also accomplished the synthesis of 2-fluoroalkylated indenone and indanone by simple reactions (Scheme 3). An allylic oxidation was carried out with 10 equiv of manganese dioxide in dichloromethane as the solvent at 0 °C for 30 minutes, leading to the corresponding 2-fluoroalkylated indenone 6 in 98% yield. Subsequently, a hydrogenolysis with 1 mol % of Pd/C under a hydrogen atmosphere in methanol at room temperature for 15 h produced the desired 2-fluoroalkylated indanone 7 as the trans-isomer in 69% yield [33-35].
![[1860-5397-16-184-i3]](/bjoc/content/inline/1860-5397-16-184-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the fluoroalkylated indenone 6 and the indanone 7 from the indenol 3aA. The yields were determined by 19F NMR spectroscopy. The values in parentheses are the isolated yields.
Scheme 3: Synthesis of the fluoroalkylated indenone 6 and the indanone 7 from the indenol 3aA. The yields wer...
The stereochemical assignment of 5aA and 7 was carried out based on NMR techniques, as shown in Scheme 4 [36]. A strong correlation between the carbonyl carbon atom and Ha was obtained, whereas the cross-peak between the carbonyl carbon atom and Hb was not observed, strongly indicating that the indanone 5aA possessed a CF3 group at the 3-position. On the other hand, the cross-peak between the carbonyl carbon atom and Hc of the indanone 7 was obtained, but there was no cross-peak between the carbonyl carbon atom and Hd, meaning that the indanone 7 has a CF3 group at the 2-position. This result indicated that the indenol 3aA, which is the precursor of the 2-fluoroalkylated indanone 7, also possess a trifluoromethyl group at the 2-position. Each Ha–Hb coupling of 5aA and each Hc–Hd coupling of 7 appeared as a doublet, with J = 4.0 Hz and 4.6 Hz, respectively. These NMR results suggested that the fluoroalkylated indanones 5aA and 7 were trans-isomers, and previous results showed that the J value of the nonfluoroalkylated trans-2,3-disubstituted indanone 8 is 5.0 Hz, whereas the J value of the cis-isomer 9 is 8.0 Hz [37].
![[1860-5397-16-184-i4]](/bjoc/content/inline/1860-5397-16-184-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Stereochemical assignment of 5aA and 7 based on NMR techniques. The cross-peaks were observed through HMBC measurements.
Scheme 4: Stereochemical assignment of 5aA and 7 based on NMR techniques. The cross-peaks were observed throu...
The proposed catalytic cycles for the cobalt-catalyzed [2 + 3] cycloaddition process leading to 2-fluoroalkylated indenols and 3-fluoroalkylated indanones are shown in Scheme 5 [28,38]. Thus, the reaction presumably proceeds as follows: (1) transmetalation of the cobalt catalyst with 2-formylphenylboronic acids (2) gives the arylcobalt species Int-1, (2) insertion of the alkyne 1 into the [Co]–Ar bond (see Int-2a) [39], (3) migration insertion into the formyl moiety to afford the corresponding cobalt alkoxide Int-3a, (4) protonation of Int-3a with HX (X = acac, OiPr, or OH), giving rise to the desired 2-fluoroalkylated indenol 3.
![[1860-5397-16-184-i5]](/bjoc/content/inline/1860-5397-16-184-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The formation of the 3-fluoroalkylated indanones as a side product may be explained based on the previous literature [20,40]. The reaction of Int-1 with the alkynes 1 also gives Int-2b as a regioisomeric intermediate of Int-2a, leading to the 3-fluoroalkylated cobalt alkoxide Int-3b. Subsequently, the proton shift of the cobalt alkoxide Int-3b provides the allylcobalt species Int-4b because the acidity of the proton Ha of Int-3b is the same as, or higher than the hydroxy group of Int-4b due to the electron-withdrawing effect of the fluoroalkyl group. It should be noted that this process is probably accelerated by 2-propanol or H2O, which is explained by the results that the most 3-fluoroalkylated indenol 4aA was converted into the indanone 5aA in the reaction, using 2-propanol as the solvent and a cobalt hydrate as a catalyst, such as Co(acac)2·2H2O and Co(OAc)2·4H2O, respectively (Table 1, entries 3, 5, and 7).
The compound Int-4b produced the allylcobalt species Int-5b with a stabilized C–[Co] bond due to the electron-withdrawing ability of the fluoroalkyl group. The Int-5b species undergoes protonation at the carbon bonded to the fluoroalkyl group, giving the enol Int-6b. Finally, the enol Int-6b produces the 3-fluoroalkylated indanone 5 via keto–enol tautomerism.
Conclusion
In conclusion, we developed a practical and efficient synthetic protocol for 2-fluoroalkylated indenol derivatives via a cobalt-catalyzed [2 + 3] cycloaddition reaction using fluorinated alkynes and 2-formylphenylboronic acids. It was revealed that the reaction using dppp as a ligand showed a high regioselectivity, leading to 2-fluoroalkylated indenols in good yield. Moreover, the side product was a small amount of 3-fluoroalkylated indanones, which was easily separated from the 2-fluoroalkylated indenols due to the lower polarity of indanones compared to indenols. Finally, 2-fluoroalkylated indenol was simply converted into the corresponding 2-fluoroalkylated indenone and indanone in good to excellent yield.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data (1H, 13C, 19F NMR, IR, and HRMS), copies of 1H, 13C, and 19F NMR spectra. | ||
| Format: PDF | Size: 6.9 MB | Download |
References
-
Ishiguro, Y.; Okamoto, K.; Ojima, F.; Sonoda, Y. Chem. Lett. 1993, 22, 1139–1140. doi:10.1246/cl.1993.1139
Return to citation in text: [1] -
Clark, W. M.; Tickner-Eldridge, A. M.; Huang, G. K.; Pridgen, L. N.; Olsen, M. A.; Mills, R. J.; Lantos, I.; Baine, N. H. J. Am. Chem. Soc. 1998, 120, 4550–4551. doi:10.1021/ja973882j
Return to citation in text: [1] -
Jiang, Z.-H.; Hwang, G.-S.; Xi, Z.; Goldberg, I. H. J. Am. Chem. Soc. 2002, 124, 3216–3217. doi:10.1021/ja0176292
Return to citation in text: [1] -
Upadhayaya, R. S.; Shinde, P. D.; Kadam, S. A.; Bawane, A. N.; Sayyed, A. Y.; Kardile, R. A.; Gitay, P. N.; Lahore, S. V.; Dixit, S. S.; Földesi, A.; Chattopadhyaya, J. Eur. J. Med. Chem. 2011, 46, 1306–1324. doi:10.1016/j.ejmech.2011.01.053
Return to citation in text: [1] -
Liebeskind, L. S.; Gasdaska, J. R.; McCallum, J. S.; Tremont, S. J. J. Org. Chem. 1989, 54, 669–677. doi:10.1021/jo00264a030
Return to citation in text: [1] -
Robinson, N. P.; Main, L.; Nicholson, B. K. J. Organomet. Chem. 1989, 364, C37–C39. doi:10.1016/0022-328x(89)87157-8
Return to citation in text: [1] -
Quan, L. G.; Gevorgyan, V.; Yamamoto, Y. J. Am. Chem. Soc. 1999, 121, 3545–3546. doi:10.1021/ja983645w
Return to citation in text: [1] -
Chang, K.-J.; Rayabarapu, D. K.; Cheng, C.-H. J. Org. Chem. 2004, 69, 4781–4787. doi:10.1021/jo049506g
Return to citation in text: [1] -
Singh, P. P.; Reddy, P. B.; Sawant, S. D.; Koul, S.; Taneja, S. C.; Kumar, H. M. S. Tetrahedron Lett. 2006, 47, 7241–7243. doi:10.1016/j.tetlet.2006.07.126
Return to citation in text: [1] -
Chinnagolla, R. K.; Jeganmohan, M. Eur. J. Org. Chem. 2012, 417–423. doi:10.1002/ejoc.201101364
Return to citation in text: [1] -
Zhang, C.; Li, H.; Pei, C.; Qiu, L.; Hu, W.; Bao, X.; Xu, X. ACS Catal. 2019, 9, 2440–2447. doi:10.1021/acscatal.8b04144
Return to citation in text: [1] -
Gu, C.-X.; Chen, W.-W.; Xu, M.-H. J. Org. Chem. 2020, 85, 3887–3893. doi:10.1021/acs.joc.9b02958
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992–1012. doi:10.1016/j.jfluchem.2006.05.006
Return to citation in text: [1] -
Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011
Return to citation in text: [1] -
Allen, A. D.; Fujio, M.; Mohammed, N.; Tidwell, T. T.; Tsuji, Y. J. Org. Chem. 1997, 62, 246–252. doi:10.1021/jo961387k
Return to citation in text: [1] -
Mitobe, K.; Terashima, K.; Kawasaki-Takasuka, T.; Agou, T.; Kubota, T.; Yamazaki, T. Eur. J. Org. Chem. 2018, 6944–6951. doi:10.1002/ejoc.201801384
Return to citation in text: [1] [2] -
Kumon, T.; Yoshida, K.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. Tetrahedron 2019, 75, 3713–3721. doi:10.1016/j.tet.2019.05.042
Return to citation in text: [1] [2] [3] -
Ghavtadze, N.; Fröhlich, R.; Bergander, K.; Würthwein, E.-U. Synthesis 2008, 3397–3406. doi:10.1055/s-0028-1083188
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2014, 114, 1783–1826. doi:10.1021/cr400133p
Return to citation in text: [1] -
Gandeepan, P.; Cheng, C.-H. Acc. Chem. Res. 2015, 48, 1194–1206. doi:10.1021/ar500463r
Return to citation in text: [1] -
Boyarskiy, V. P.; Ryabukhin, D. S.; Bokach, N. A.; Vasilyev, A. V. Chem. Rev. 2016, 116, 5894–5986. doi:10.1021/acs.chemrev.5b00514
Return to citation in text: [1] -
Jeon, S. L.; Kim, J. K.; Son, J. B.; Kim, B. T.; Jeong, I. H. Tetrahedron Lett. 2006, 47, 9107–9111. doi:10.1016/j.tetlet.2006.10.088
Return to citation in text: [1] -
Kumon, T.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 213, 11–17. doi:10.1016/j.jfluchem.2018.06.004
Return to citation in text: [1] -
Ueda, M.; Ueno, T.; Suyama, Y.; Ryu, I. Tetrahedron Lett. 2017, 58, 2972–2974. doi:10.1016/j.tetlet.2017.06.049
Return to citation in text: [1] [2] -
The lower regioselectivity when using 2-acetylphenylboronic acid instead of 2-formylphenylboronic (2A) still remains unclear. However, a similar tendency for the regioselectivity has been observed in the cobalt-catalyzed [2 + 3] cycloaddition using an unsymmetrical internal alkyne for the construction of indenol derivatives, see [8].
Return to citation in text: [1] -
Ernst-Russell, M. A.; Chai, C. L. L.; Wardlaw, J. H.; Elix, J. A. J. Nat. Prod. 2000, 63, 129–131. doi:10.1021/np9903245
Return to citation in text: [1] -
Morrell, A.; Placzek, M.; Parmley, S.; Grella, B.; Antony, S.; Pommier, Y.; Cushman, M. J. Med. Chem. 2007, 50, 4388–4404. doi:10.1021/jm070307+
Return to citation in text: [1] -
Turek, M.; Szczęsna, D.; Koprowski, M.; Bałczewski, P. Beilstein J. Org. Chem. 2017, 13, 451–494. doi:10.3762/bjoc.13.48
Return to citation in text: [1] -
It is well-known that α-trifluoromethylated carbonyl compounds have a strongly acidic α-hydrogen atom due to the highly electron-withdrawing trifluoromethyl and carbonyl groups and are easily enolized. In the present study, it is therefore highly possible that the cis-isomer generated through the catalytic hydrogenation of the indenone 6 isomerized to the much more thermally stable trans-isomer 7 via enolization.
Return to citation in text: [1] -
Cooke, E.; Paradellis, T. C.; Edward, J. T. Can. J. Chem. 1982, 60, 29–34. doi:10.1139/v82-006
Return to citation in text: [1] -
Barth, W.; Paquette, L. A. J. Org. Chem. 1985, 50, 2438–2443. doi:10.1021/jo00214a008
Return to citation in text: [1] -
Generally, the signal of the 2-fluoroalkylated indenols 3 appeared at −56 ppm and that of the 3-fluoroalkylated indenols 4 appeared at −59 ppm.
Return to citation in text: [1] -
Minatti, A.; Zheng, X.; Buchwald, S. L. J. Org. Chem. 2007, 72, 9253–9258. doi:10.1021/jo701741y
Return to citation in text: [1] -
Lin, P.-S.; Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 11296–11299. doi:10.1002/chem.200801858
Return to citation in text: [1] -
Since a fluoroalkyl group has a very strong electron-withdrawing ability, the Rf–Cα–[Co] bond may be stronger than the Cβ–[Co] bond, which stabilizes the transition state of the insertion step. Therefore, Int-2a was produced as the major regioisomeric intermediate.
Return to citation in text: [1] -
Hamada, Y.; Kawasaki-Takasuka, T.; Yamazaki, T. Beilstein J. Org. Chem. 2017, 13, 1507–1512. doi:10.3762/bjoc.13.149
Return to citation in text: [1]
| 8. | Chang, K.-J.; Rayabarapu, D. K.; Cheng, C.-H. J. Org. Chem. 2004, 69, 4781–4787. doi:10.1021/jo049506g |
| 1. | Ishiguro, Y.; Okamoto, K.; Ojima, F.; Sonoda, Y. Chem. Lett. 1993, 22, 1139–1140. doi:10.1246/cl.1993.1139 |
| 2. | Clark, W. M.; Tickner-Eldridge, A. M.; Huang, G. K.; Pridgen, L. N.; Olsen, M. A.; Mills, R. J.; Lantos, I.; Baine, N. H. J. Am. Chem. Soc. 1998, 120, 4550–4551. doi:10.1021/ja973882j |
| 3. | Jiang, Z.-H.; Hwang, G.-S.; Xi, Z.; Goldberg, I. H. J. Am. Chem. Soc. 2002, 124, 3216–3217. doi:10.1021/ja0176292 |
| 4. | Upadhayaya, R. S.; Shinde, P. D.; Kadam, S. A.; Bawane, A. N.; Sayyed, A. Y.; Kardile, R. A.; Gitay, P. N.; Lahore, S. V.; Dixit, S. S.; Földesi, A.; Chattopadhyaya, J. Eur. J. Med. Chem. 2011, 46, 1306–1324. doi:10.1016/j.ejmech.2011.01.053 |
| 5. | Liebeskind, L. S.; Gasdaska, J. R.; McCallum, J. S.; Tremont, S. J. J. Org. Chem. 1989, 54, 669–677. doi:10.1021/jo00264a030 |
| 6. | Robinson, N. P.; Main, L.; Nicholson, B. K. J. Organomet. Chem. 1989, 364, C37–C39. doi:10.1016/0022-328x(89)87157-8 |
| 7. | Quan, L. G.; Gevorgyan, V.; Yamamoto, Y. J. Am. Chem. Soc. 1999, 121, 3545–3546. doi:10.1021/ja983645w |
| 8. | Chang, K.-J.; Rayabarapu, D. K.; Cheng, C.-H. J. Org. Chem. 2004, 69, 4781–4787. doi:10.1021/jo049506g |
| 9. | Singh, P. P.; Reddy, P. B.; Sawant, S. D.; Koul, S.; Taneja, S. C.; Kumar, H. M. S. Tetrahedron Lett. 2006, 47, 7241–7243. doi:10.1016/j.tetlet.2006.07.126 |
| 10. | Chinnagolla, R. K.; Jeganmohan, M. Eur. J. Org. Chem. 2012, 417–423. doi:10.1002/ejoc.201101364 |
| 11. | Zhang, C.; Li, H.; Pei, C.; Qiu, L.; Hu, W.; Bao, X.; Xu, X. ACS Catal. 2019, 9, 2440–2447. doi:10.1021/acscatal.8b04144 |
| 12. | Gu, C.-X.; Chen, W.-W.; Xu, M.-H. J. Org. Chem. 2020, 85, 3887–3893. doi:10.1021/acs.joc.9b02958 |
| 21. | Kumon, T.; Yoshida, K.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. Tetrahedron 2019, 75, 3713–3721. doi:10.1016/j.tet.2019.05.042 |
| 26. | Jeon, S. L.; Kim, J. K.; Son, J. B.; Kim, B. T.; Jeong, I. H. Tetrahedron Lett. 2006, 47, 9107–9111. doi:10.1016/j.tetlet.2006.10.088 |
| 39. | Since a fluoroalkyl group has a very strong electron-withdrawing ability, the Rf–Cα–[Co] bond may be stronger than the Cβ–[Co] bond, which stabilizes the transition state of the insertion step. Therefore, Int-2a was produced as the major regioisomeric intermediate. |
| 23. | Chinchilla, R.; Nájera, C. Chem. Rev. 2014, 114, 1783–1826. doi:10.1021/cr400133p |
| 24. | Gandeepan, P.; Cheng, C.-H. Acc. Chem. Res. 2015, 48, 1194–1206. doi:10.1021/ar500463r |
| 25. | Boyarskiy, V. P.; Ryabukhin, D. S.; Bokach, N. A.; Vasilyev, A. V. Chem. Rev. 2016, 116, 5894–5986. doi:10.1021/acs.chemrev.5b00514 |
| 20. | Mitobe, K.; Terashima, K.; Kawasaki-Takasuka, T.; Agou, T.; Kubota, T.; Yamazaki, T. Eur. J. Org. Chem. 2018, 6944–6951. doi:10.1002/ejoc.201801384 |
| 40. | Hamada, Y.; Kawasaki-Takasuka, T.; Yamazaki, T. Beilstein J. Org. Chem. 2017, 13, 1507–1512. doi:10.3762/bjoc.13.149 |
| 19. | Allen, A. D.; Fujio, M.; Mohammed, N.; Tidwell, T. T.; Tsuji, Y. J. Org. Chem. 1997, 62, 246–252. doi:10.1021/jo961387k |
| 20. | Mitobe, K.; Terashima, K.; Kawasaki-Takasuka, T.; Agou, T.; Kubota, T.; Yamazaki, T. Eur. J. Org. Chem. 2018, 6944–6951. doi:10.1002/ejoc.201801384 |
| 21. | Kumon, T.; Yoshida, K.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. Tetrahedron 2019, 75, 3713–3721. doi:10.1016/j.tet.2019.05.042 |
| 22. | Ghavtadze, N.; Fröhlich, R.; Bergander, K.; Würthwein, E.-U. Synthesis 2008, 3397–3406. doi:10.1055/s-0028-1083188 |
| 37. | Minatti, A.; Zheng, X.; Buchwald, S. L. J. Org. Chem. 2007, 72, 9253–9258. doi:10.1021/jo701741y |
| 13. | Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788 |
| 14. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 15. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 16. | Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007 |
| 17. | Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992–1012. doi:10.1016/j.jfluchem.2006.05.006 |
| 18. | Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011 |
| 28. | Ueda, M.; Ueno, T.; Suyama, Y.; Ryu, I. Tetrahedron Lett. 2017, 58, 2972–2974. doi:10.1016/j.tetlet.2017.06.049 |
| 38. | Lin, P.-S.; Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 11296–11299. doi:10.1002/chem.200801858 |
| 29. | The lower regioselectivity when using 2-acetylphenylboronic acid instead of 2-formylphenylboronic (2A) still remains unclear. However, a similar tendency for the regioselectivity has been observed in the cobalt-catalyzed [2 + 3] cycloaddition using an unsymmetrical internal alkyne for the construction of indenol derivatives, see [8]. |
| 33. | It is well-known that α-trifluoromethylated carbonyl compounds have a strongly acidic α-hydrogen atom due to the highly electron-withdrawing trifluoromethyl and carbonyl groups and are easily enolized. In the present study, it is therefore highly possible that the cis-isomer generated through the catalytic hydrogenation of the indenone 6 isomerized to the much more thermally stable trans-isomer 7 via enolization. |
| 34. | Cooke, E.; Paradellis, T. C.; Edward, J. T. Can. J. Chem. 1982, 60, 29–34. doi:10.1139/v82-006 |
| 35. | Barth, W.; Paquette, L. A. J. Org. Chem. 1985, 50, 2438–2443. doi:10.1021/jo00214a008 |
| 28. | Ueda, M.; Ueno, T.; Suyama, Y.; Ryu, I. Tetrahedron Lett. 2017, 58, 2972–2974. doi:10.1016/j.tetlet.2017.06.049 |
| 36. | Generally, the signal of the 2-fluoroalkylated indenols 3 appeared at −56 ppm and that of the 3-fluoroalkylated indenols 4 appeared at −59 ppm. |
| 27. | Kumon, T.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. J. Fluorine Chem. 2018, 213, 11–17. doi:10.1016/j.jfluchem.2018.06.004 |
| 21. | Kumon, T.; Yoshida, K.; Yamada, S.; Agou, T.; Kubota, T.; Konno, T. Tetrahedron 2019, 75, 3713–3721. doi:10.1016/j.tet.2019.05.042 |
| 30. | Ernst-Russell, M. A.; Chai, C. L. L.; Wardlaw, J. H.; Elix, J. A. J. Nat. Prod. 2000, 63, 129–131. doi:10.1021/np9903245 |
| 31. | Morrell, A.; Placzek, M.; Parmley, S.; Grella, B.; Antony, S.; Pommier, Y.; Cushman, M. J. Med. Chem. 2007, 50, 4388–4404. doi:10.1021/jm070307+ |
| 32. | Turek, M.; Szczęsna, D.; Koprowski, M.; Bałczewski, P. Beilstein J. Org. Chem. 2017, 13, 451–494. doi:10.3762/bjoc.13.48 |
© 2020 Kumon et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)