

Abstract
Herein we report the synthesis and characterization of a new series of styryl-based push-pull dyes containing a free amino group and their Schiff base derivatives. The dyes include the dicyanomethylene group as an acceptor and different para-substituted alkylamines as donors. Morever as a proton-sensitive group a pyridin-2-yl substituent was attached to the para-position of the phenyl moiety in both series of compounds. The photophysical properties of the dyes were examined in various solvents with different polarities and showed absorption in the visible region and green-red emission with low quantum yields. The absorption and the emission maxima were shifted bathocromically by increasing the solvent’s polarity. However, there was no correlation with the polarity parameters of the solvents. The pH-sensitive properties of all prepared Schiff bases were examined against TBAOH in DMSO, via deprotonation of the OH group in the salicylidene moiety and their reverse protonation was also investigated using TFA. The Schiff bases exhibited a bathochromic shift upon the addition of TBAOH to their solutions in DMSO. Therefore, they showed potential to be utilized as colorimetric and luminescence pH sensors. The second-order nonlinear optical (NLO) responses of the dyes were measured by the electric field-induced second harmonic (EFISH) generation method. The highest μβ values were obtained for the dyes bearing the julolidine donor as 1430 × 10−48 esu (for free amino derivative) and 1950 × 10−48 esu (for Schiff base derivative), respectively. The structural and electronic properties of the dyes as well as their NLO properties were further studied using DFT calculations. The thermal stabilities of all dyes were evaluated by thermogravimetric analysis (TGA). The TGA data showed that all dyes were thermally stable up to 250 °C.
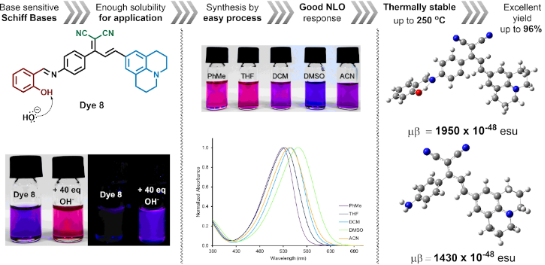
Graphical Abstract
Introduction
Push-pull organic molecules are a class of organic dyes comprising of electron-donating and accepting groups in a donor–π–acceptor (D–π–A) system. The dyes exhibit strong absorption and emission properties in solution and in the solid state [1-3]. One important feature of these molecules is an exceptional polarizability which is a crucial criterion for NLO materials. Nowadays, organic NLO materials bearing strong donor–acceptor groups with a π-bridge have shown extensive usages in signal processing, optical storage, and telecommunication devices [4-10]. The flexibility to change electron-donor and acceptor groups in the molecules allows to tune intramolecular charge transfer (ICT) intensity and it can increase NLO properties [11-14].
Some ions such as proton (H+) and hydroxide (OH−), in aqueous environments, play significant roles in industrial, biological, and environmental processes [15-17]. Their presence in aqueous media directly determines the pH level that affects organisms living in the corresponding area [18-22]. Therefore, pH-sensitive dyes play a critical role in various sensor applications for easy determination of such ions. These dyes show different spectral properties upon protonation/deprotonation processes [23-25]. Fluorescent dyes as chemosensors offer unique merits such as low energy consumption, ease of handling, high selectivity, and notable sensitivity [26,27]. Moreover, some fluorescent dyes demonstrate remarkable ICT characteristics, and could therefore serve as NLO dyes [11-14].
Among dyestuffs classes, the push-pull fluorescent dyes are renowned to own such special behaviors. The push-pull dyes generate higher charge delocalization upon excitation, thus enhance both polarizability and fluorescence emission [12-14,18]. The charge delocalization upon excitation leads to a red-shifted emission which is viable for the detection of various substrates in biological tissues and samples [28-30]. Recently, there was an increasingly growing interest in studies regarding push-pull organic molecules comprising of the dicyanomethylene group as a strong electron-accepting group coupled with various donors connected via a π-conjugation bridge [31-33]. Such dyes offer good NLO characteristics when compared to Disperse Red 1 as well as remarkable thermal stabilities with dissociation temperatures up to 300 °C [31,32].
Schiff bases containing an azomethine group are one of the most widely used organic dyes because of their easy and cheap synthetic accessibility through various methodologies and suitable photophysical properties. In addition, they exhibit a broad range of biological activities such as antimicrobial, antifungal, antiviral, and anticancer activity, to name a few. Moreover, Schiff bases bearing a salicylidene moiety are also used as chemosensors for sensing of specific ions. In addition, some derivatives also show NLO activity [25,34-37].
Herein, we report on the synthesis and full photophysical characterization of a series of new styryl-based organic chromophores containing a free amino group and the corresponding Schiff base derivatives. The photophysical, pH sensitivity, NLO properties, and thermal stabilities of all synthesized dyes were investigated. Density Functional Theory (DFT) calculations were also employed to gain insight into the experimental data.
Results and Discussion
Synthesis
As depicted in Scheme 1, dye 2 was obtained in good yield by reacting malononitrile and 4-aminoacetophenone (compound 1) according to our previous published procedure [31,32].
![[1860-5397-16-189-i1]](/bjoc/content/inline/1860-5397-16-189-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic pathways of dyes 3–7 and Schiff base analogs 8–12.
Scheme 1: Synthetic pathways of dyes 3–7 and Schiff base analogs 8–12.
The styryl-based organic chromophores 3–7 having a free amino and strong donor-acceptor groups and a pyridin-2-yl moiety were synthesized by the base-catalyzed condensation of 2 with different aldehyde derivatives in ethanol, as depicted in Scheme 1. Further reactions of the dyes 3–7 with salicylaldehyde under basic conditions resulted in the formation of an azomethine bridge on each amine end yielding dyes 8–12 as the final products. All dyes were obtained in moderate to excellent yields (58–96%) and generally without the need for chromatographic purification. The structures of all the new synthesized dyes were confirmed by FTIR, 1H, 13C NMR, and HRMS analyses (Figures S1–S40 in Supporting Information File 1). Moreover, the stereochemistry of the double bonds was determined on the basis of the coupling constants of the vinylic hydrogens in the 1H NMR spectra (J ≈ 15–16 Hz) and revealed that the dyes are stable as E-stereoisomers [31].
Optimized geometries
The optimized geometries of dyes 3–7 and 8-12 were obtained by performing DFT calculations at the B3LYP/6-31+G(d,p) level of theory. The structures are illustrated in Figure 1 and Figures S41 and S42 (Supporting Information File 1). As shown in Figure 1, the different substituents were planar to the dicyanomethylene group. A twist was observed between the dicyanomethylene and aminophenyl groups (salicylidenyl moiety for 8–12) with an angle of about 50°. In addition, the dyes 8–12 exhibited strong intramolecular O–H···N hydrogen bonds with lengths in the range of 2.63–2.64 Å (Figure 1, Figures S41 and S42 in Supporting Information File 1).
![[1860-5397-16-189-1]](/bjoc/content/figures/1860-5397-16-189-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The optimized geometry of dyes 3 and 8.
Figure 1: The optimized geometry of dyes 3 and 8.
Absorption and emission properties
To assess the effect of the solvent on the absorption and emission spectra of 3–7 and 8–12 (c = 10 μM for absorption, and c = 1 μM for emission), various solvents with different polarity were employed at room temperature (Figure 2, Table 1, and Table S1 in Supporting Information File 1). The dyes showed generally good solubility in organic solvents. However, the dyes 8–12 had a low solubility in MeOH. Therefore, the photophysical properties of the dyes 8–12 could not be investigated in this solvent. The Dimroth–Reichardt polarity parameters (ET30, Dimroth–Reichardt polarity parameter in kcal mol−1 of MeOH = 55.4, ACN = 45.6, DMSO = 45.1, DCM = 40.7, THF = 37.4, and PhMe = 33.9) were used in the study [38]. In addition, the absorption spectra of all dyes were obtained by TD-DFT calculations using the PCM model. The absorption maxima (λabsmax), oscillator strengths (f), and relevant transitions and their contributions (w) are given in Table 1 and Table S1 in Supporting Information File 1). The shifts of the absorption maxima of the dyes were little dependent on the solvent polarity and there was no obvious correlation with increasing polarity parameters. There were only small differences observed for the absorption maxima values for both series of compounds. As representative examples, the spectra of 3 and 8 are provided in Figure 2 and Figure 3. The absorption spectra of 3 and 8 in all solvents tested displayed absorption maxima in the range of 512–543 and 529–562 nm, respectively. Of note, the largest bathochromic shifts of the absorption maxima for both series of dyes were observed in DMSO compared to the other solvents applied. Switching the solvent from PhMe to DMSO, bathochromic shifts of 31 nm for 3, 22 nm for 4, 19 nm for 5, and 23 nm for 6 were observed. Similar behaviors were observed for the absorption spectra of compounds 8–12 (Table S1, Supporting Information File 1). The TD-DFT calculation results obtained for the values of absorption wavelengths for the dyes were in the order 3 > 4 > 5 > 6 > 7 and 8 > 9 > 10 > 11 > 12 in DMSO, which was consistent with the experimental data (Table 1 and Table S1 in Supporting Information File 1). A solvent effect on the absorption spectra was observed for compound 4 resulting in a bathochromic shift of about 22 nm from PhMe to DMSO, whereas there was only a little effect on the shift for dye 3 (6 nm). In case of the Schiff bases 8–11, similar bathochromic shifts were observed. In addition, experimentally, there was no solvatochromic behavior observed for dyes 7 and 12 as was predicted by theoretical calculations.
![[1860-5397-16-189-2]](/bjoc/content/figures/1860-5397-16-189-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Absorption spectra of dyes 3 (a, left) and 8 (b, right). Inset: Color of dyes 3 and 8 in the given solvents of different polarities in ambient light, c = 10 μM.
Figure 2: Absorption spectra of dyes 3 (a, left) and 8 (b, right). Inset: Color of dyes 3 and 8 in the given ...
Table 1: Photophysical properties of dyes 3–7 in various solvents with different polarity and the calculated absorption spectra data.
| Experimental | Calculated | |||||||||
| solventa |
λabsmax
(nm) |
λemmax
(nm) |
Stokes shift
(nm) |
Stokes
shift (cm−1) |
ΦFb |
ε
(mM−1cm−1) |
λabsmax
(nm) |
f | transitions, w (%) | |
| 3 | MeOH, (55.4c) | 528 | 630 | 102 | 3074 | <0.01 | 42.6 | 506 | 1.1731 | HOMO→LUMO, 96.2 |
|
ACN
(45.6c) |
527 | 630 | 103 | 3125 | <0.01 | 51.7 | 507 | 1.1786 | HOMO→LUMO, 96.2 | |
|
DMSO
(45.1c) |
543 | 644 | 101 | 2879 | <0.01 | 49.8 | 511 | 1.2023 | HOMO→LUMO, 96.6 | |
|
DCM
(40.7c) |
531 | 617 | 86 | 2632 | 0.01 | 36.8 | 506 | 1.1985 | HOMO→LUMO, 96.5 | |
|
THF
(37.4c) |
513 | 604 | 91 | 2961 | 0.01 | 45.9 | 504 | 1.1916 | HOMO→LUMO, 96.4 | |
|
PhMe
(33.9c) |
512 | 584 | 72 | 2414 | <0.01 | 41.4 | 494 | 1.2083 | HOMO→LUMO, 96.6 | |
| 4 | MeOH | 498 | 606 | 108 | 3568 | <0.01 | 55.4 | 491 | 1.1403 | HOMO→LUMO, 89.9 |
| ACN | 495 | 611 | 116 | 3819 | <0.01 | 48.6 | 492 | 1.1469 | HOMO→LUMO, 90.2 | |
| DMSO | 510 | 622 | 112 | 3515 | <0.01 | 49.6 | 495 | 1.1750 | HOMO→LUMO, 91.3 | |
| DCM | 502 | 589 | 87 | 2954 | 0.01 | 30.1 | 490 | 1.1746 | HOMO→LUMO, 90.9 | |
| THF | 486 | 582 | 96 | 3403 | <0.01 | 33.7 | 488 | 1.1674 | HOMO→LUMO, 90.5 | |
| PhMe | 488 | 558 | 70 | 2592 | <0.01 | 41.9 | 478 | 1.2037 | HOMO→LUMO, 91.1 | |
| 5 | MeOH | 476 | 609 | 133 | 4599 | <0.01 | 39.9 | 488 | 1.0314 |
HOMO-1→LUMO, 15.2
HOMO→LUMO, 84.6 |
| ACN | 473 | 611 | 134 | 4770 | <0.01 | 35.9 | 489 | 1.0384 |
HOMO-1→LUMO, 14.8
HOMO→LUMO, 85.0 |
|
| DMSO | 486 | 624 | 138 | 4561 | <0.01 | 42.1 | 492 | 1.0686 |
HOMO-1→LUMO, 13.2
HOMO→LUMO, 86.7 |
|
| DCM | 483 | 592 | 109 | 3828 | 0.01 | 25.3 | 487 | 1.0750 | HOMO-1→LUMO, 13.4 HOMO→LUMO, 86.4 | |
| THF | 457 | 583 | 126 | 4741 | <0.01 | 31.4 | 485 | 1.0687 |
HOMO-1→LUMO, 13.9
HOMO→LUMO, 85.9 |
|
| PhMe | 467 | 557 | 90 | 3476 | <0.01 | 33.9 | 476 | 1.1242 | HOMO-1→LUMO, 12.2 HOMO→LUMO, 87.5 | |
| 6 | MeOH | 446 | 600 | 154 | 5786 | <0.01 | 40.8 | 451 | 0.5104 |
HOMO-1→LUMO, 62.3
HOMO→LUMO, 37.0 |
| ACN | 442 | 603 | 161 | 6052 | <0.01 | 34.6 | 451 | 0.5053 |
HOMO-1→LUMO, 63.2
HOMO→LUMO, 36.1 |
|
| DMSO | 463 | 614 | 151 | 5346 | <0.01 | 42.7 | 453 | 0.4843 | HOMO-1→LUMO, 66.7 HOMO→LUMO, 32.6 | |
| DCM | 448 | 580 | 132 | 5099 | <0.01 | 27.8 | 451 | 0.4988 |
HOMO-1→LUMO, 63.2
HOMO→LUMO, 36.1 |
|
| THF | 439 | 573 | 134 | 5359 | <0.01 | 34.7 | 450 | 0.5077 |
HOMO-1→LUMO, 61.4
HOMO→LUMO, 37.9 |
|
| PhMe | 440 | 546 | 106 | 4399 | <0.01 | 30.6 | 446 | 0.4885 |
HOMO-1→LUMO, 57.8
HOMO→LUMO, 41.4 |
|
| 7 | MeOH | 374 | – | – | – | – | 51.4 | 419 | 1.2899 | HOMO-1→LUMO, 95.0 |
| ACN | 376 | – | – | – | – | 43.4 | 419 | 1.2939 | HOMO-1→LUMO, 95.0 | |
| DMSO | 386 | – | – | – | – | 40.4 | 422 | 1.3123 | HOMO-1→LUMO, 95.2 | |
| DCM | 382 | 606 | 224 | 9705 | n.d. | 42.1 | 421 | 1.3265 | HOMO-1→LUMO, 94.0 | |
| THF | 379 | 434 | 55 | 3357 | n.d | 40.4 | 420 | 1.3246 | HOMO-1→LUMO, 93.7 | |
| PhMe | 382 | 577 | 195 | 8853 | n.d | 53.1 | 419 | 1.3788 | HOMO-1→LUMO, 88.8 | |
aSolvents arranged in order of decreasing ET30 values. bFluorescence quantum yield (±10%) determined relative to fluorescein in pH 9 solution (ΦF = 0.95) as standard. cThe values for relative polarity are taken from [38]. n.d, could not be determined.
![[1860-5397-16-189-3]](/bjoc/content/figures/1860-5397-16-189-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Emission spectra of dyes 3 (a, left) and 8 (b, right). Inset: Color of dyes 3 and 8 in the indicated solvents of different polarities under UV irradiation (λex = 365 nm, c = 1 μM).
Figure 3: Emission spectra of dyes 3 (a, left) and 8 (b, right). Inset: Color of dyes 3 and 8 in the indicate...
Based on theoretical calculations, the main peaks were found to correspond to transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) with the highest contributions from 3–5 and 8–11. For dyes 6, 7, and 12, the highest contributions were from transitions HOMO-1→LUMO. The picture of the frontier molecular orbitals of the dyes are shown in Figure S92 in Supporting Information File 1.
Solvatochromic studies on emissions were also done to gain insights into the photophysical behavior of the new push-pull dyes bearing free amino and azomethine groups. Therefore, the fluorescence spectra of all dyes 3–7 and 8–12 were recorded in the same solvents of various polarities that were used in the UV–vis studies, and the results are summarized in Table 1 and Table S1 (Supporting Information File 1). All dyes showed fluorescence properties and there was no direct correlation of the increase of maximum emission wavelengths with increasing polarity parameters of the solvents. However, the largest bathochromic shifts of the emission maxima for both series of dyes were observed in DMSO. This phenomenon is observed when highly fluorescent polar excited states of push-pull dyes are stabilized by solvents with different polarity [39-45]. The emission spectra of the dyes 3 and 8 and the color changes observed upon UV irradiation of both dyes in various solvents are shown in Figure 3. Both series of the dyes showed fluorosolvatochromic behavior in all solvents used except for MeOH and ACN, and the emission band of dye 3 was red-shifted by 60 nm from PhMe (λemmax = 584 nm) to DMSO (λemmax = 644 nm) (Figure 3). The dyes showed considerably different fluorescence color magnitudes with changing polarity except for compound 12 that was non-fluorescent. The fluorescence intensity of all dyes was high in solvents with low polarities such as PhMe and THF, while it decreased with increasing solvent polarities such as DMSO and MeOH. The fluorescence quantum yields of the dyes were found to be very low as compared to fluorescein (ΦF= 0.95 at pH 9). For example, the quantum yield for 3 in DCM was 0.01 and that of 8 in PhMe was 0.11.
Substituent effects
The prepared Schiff bases exhibited greater red shifts than the styryl counterparts (3–7) in both, absorption and emission spectra. For instance, in DCM, dye 8 showed absorption and emission maxima at 546 and 636 nm, whereas the maxima of dye 3 were observed at 531 and 617 nm, respectively. Moreover, both the styryl and the Schiff bases showed greater red shifts compared to the previously reported counterparts [31,32]. As can be seen from Figure 4, the absorption maxima of dyes 3–6 and 8–11 depended on the electron-donating nature of the substituents, and the values of the maximum absorption wavelengths increased in the following order: 4-morpholinyl (6, 11) < 4-piperidinyl (5, 10) < 4-pyrolidinyl (4, 9) < 4-julolidinyl (3, 8). The dyes 3 and 8 containing a julolidinyl group exhibited the most pronounced red shift while the dyes containing a morpholinyl substituent (6 and 11) showed the least one. In addition, dyes 7 and 12 bear a proton-sensitive and electron-accepting pyridin-2-yl group and therefore, the absorption maxima for dyes 7 and 12 were lower than the maximum absorption wavelengths of the other dyes. The maximum absorption wavelengths of these dyes were in the range of 374–386 nm and 377–385 nm. Moreover, almost the same trends which were obtained from the UV–vis absorption spectra were also observed in the emission maxima. The photophysical properties of dyes 3–7 were similar to those of the corresponding Schiff base derivatives 8–12. However, the Schiff bases 8–12 had higher maximum absorption and emission wavelength values than the dyes 3–7 because of the extended conjugation system present in the Schiff bases.
![[1860-5397-16-189-4]](/bjoc/content/figures/1860-5397-16-189-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Red shift phenomena with changing substituents in absorption (a, left) and emission (b, right) spectra of dyes 3–6 (top) and 8–11 (bottom) in DMSO.
Figure 4: Red shift phenomena with changing substituents in absorption (a, left) and emission (b, right) spec...
The observation of an intense red shift might be related to a significant contribution of ICT for electronic state modification in the excited state of the molecules [45]. In this state, all dyes have a notably higher dipole moment and a lower LUMO energy level as compared to their ground state [46-49]. Increasing the electron-donating strength of the various heterocyclic amines led to the increase in the Stokes shift and a significant red shift between the absorption and the emission maxima. This was due to the varying electronic contributions to the host structure induced by the heterocyclic amino constituents [50-52] (Figure 4).
Hydroxide anion sensing properties
The dyes 8–12 could have the ability to detect hydroxide anions due to the presence of an OH substituent at the salicylidene moiety. Therefore, the OH− sensing activity of compounds 8–12 was investigated by the addition of hydroxide anions in the form of the corresponding tetrabutylammonium (TBA) salt. An interaction between the dyes and hydroxide anion was investigated in DMSO as the solvent. The presence of hydroxide led to small changes in the UV–vis absorption spectra of the dyes. As presented in Figure 5, the interaction of dye 12 with the hydroxide anion, led to a small hyperchromic effect in the UV–vis absorption spectrum. Moreover, dyes 8–12 showed an enhancement in fluorescence intensity upon the incremental addition of hydroxide anions to their solutions in DMSO. As shown in Figure 5 and Figure 6, dye 12 showed the highest increase in fluorescence (approx. a 20-fold fluorescence enhancement) upon addition of 15 equiv OH−, thus reflecting its superior sensitivity among the others (Supporting Information File 1). The increased emission intensity and the formation of a new absorption band could likely be due to the formation of a phenoxide group, which is a more pronounced electron donor than the phenol group [52,53]. Furthermore, trifluoroacetic acid (TFA) was added to a solution containing the deprotonated forms of dyes 8–12 in DMSO, so as to investigate the reversible protonation of the dyes. As can be seen in Figure 5, the addition of 5 equiv of TFA to a basic solution of 12 led to the restoration of its emission intensity. Figure 6 shows the deprotonation and reverse protonation of dye 12 by color changes of the dye followed under a UV lamp (λex = 365 nm). Since under the ambient light all solutions seemed to have the same color, a UV lamp was used to observe the color changes. As shown in Figure 6, the color of the free dye 12 dissolved in DMSO was light yellow. The addition of 15 equiv of TBAOH to the solution led to a change of the colorless solution to blue under UV light, whereas no color change was observed in ambient light. The observed color change under UV light indicated that the dye was deprotonated. Indeed, the addition of only 5 equiv of TFA regenerated the light yellow color of the solution indicative for the reversed protonation of 12. All dyes, except 8, showed this reversibility. Furthermore, the limits of detection (LOD) and quantification (LOQ) were determined for dyes 8–12. The LOD and the LOQ were respectively found to be as follow: 73.1 μM and 244 μM for 8, 47.2 μM and 157 μM for 9, 47.3 μM and 158. μM for 10, 50.9 μM and 170 μM for 11, 33.1 μM, and 110 μM for 12 (Figures S87–S91, Supporting Information File 1) [25].
![[1860-5397-16-189-5]](/bjoc/content/figures/1860-5397-16-189-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Absorption (a, left) and emission (b, right) change of dye 12 upon addition of 15 equiv of TBAOH and reverse protonation by adding 5 equiv of TFA in DMSO solution.
Figure 5: Absorption (a, left) and emission (b, right) change of dye 12 upon addition of 15 equiv of TBAOH an...
![[1860-5397-16-189-6]](/bjoc/content/figures/1860-5397-16-189-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Photographs of dye 12 (left, ambient light), without, after the addition of 15 equiv of TBAOH (middle), and reverse protonation by 5 equiv of TFA in DMSO solution (right photograph: the same solutions under UV light (UV lamp, λex = 365 nm)).
Figure 6: Photographs of dye 12 (left, ambient light), without, after the addition of 15 equiv of TBAOH (midd...
pH-Sensitive study and pKa determination of 8 in aqueous solution
The pH value of aqueous systems is important to maintain the physiological stability, both inside and outside a living organism. The hydroxide anion plays a special and critical role in the ecosystem. Therefore, the necessity to develop new pH sensors is vital to control pH conditions in various environments. Due to their low-cost of manufacturing and maintenance, pH sensors were developed and used for both, clinical and environmental analyses [54-56]. Nevertheless, fluorescent pH indicators offer higher selectivity and sensitivity than other classes [28]. Therefore, we investigated dye 8 as a prospect for a fluorescent pH sensor in a basic environment.
After having determined the solubility of dye 8 in aqueous solution, the sensing capability of 8 toward hydroxide anions was tested. Several Britton–Robinson buffer solutions, with different pH values ranging from 5.5–11, were used to mimic basic environments and to preserve pH stability in solution. As depicted in Figure 7, increasing the pH value of the solution led to the formation of a new absorption band at 393 nm (in UV region) and a weak fluorescence increase at 505 nm. This phenomenon was accompanied by darkening purple color shifts of the solutions with increased pH value. However, the slight increase in the fluorescence was hardly noticeable below 365 nm UV light as is shown in Figure 8. The pKa of 8 was calculated by using a spectrophotometric method and was 8.22 ± 0.03 (Figure 9).
![[1860-5397-16-189-7]](/bjoc/content/figures/1860-5397-16-189-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Absorption (a, left) and emission (b, right) change of 8 in Britton–Robinson buffer solutions at different pH values.
Figure 7: Absorption (a, left) and emission (b, right) change of 8 in Britton–Robinson buffer solutions at di...
![[1860-5397-16-189-8]](/bjoc/content/figures/1860-5397-16-189-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Photographs of dye 8 in Britton–Robinson buffer solutions at different pH values.
Figure 8: Photographs of dye 8 in Britton–Robinson buffer solutions at different pH values.
![[1860-5397-16-189-9]](/bjoc/content/figures/1860-5397-16-189-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Sigmoid function obtained from dye 8 UV–vis absorption spectra during pH investigation.
Figure 9: Sigmoid function obtained from dye 8 UV–vis absorption spectra during pH investigation.
NLO properties
The second-order NLO responses of all prepared dyes were measured by the EFISH technique in CHCl3 solution with a nonresonant incident wavelength of 1907 nm. Experimental details on the EFISH measurements are given elsewhere [57]. EFISH measurements provide information about the scalar product, μβ(2ω), of the vector component of the first hyperpolarizability tensor, β, and the dipole moment vector [58-61]. This product is derived according to Equation 1 considering γ0(−2ω,ω,ω,0), the third-order term, as negligible for the push-pull dyes under consideration. This approximation is usually used for push-pull organic and organometallic molecules.
![[1860-5397-16-189-i2]](/bjoc/content/inline/1860-5397-16-189-i2.svg?max-width=590&scale=1.18182)
The results of the EFISH measurements are presented in Table 2. The obtained positive µβ values indicated that the excited states are more polarized than the ground states and that both, the ground and the excited states were polarized in the same direction for all dyes. All studied dyes exhibited a higher NLO response than Disperse Red 1, which is generally used as a reference (µβ = 450 × 10−48 esu), except for dyes 7 and 12. For the latter two compounds (7 and 12), relatively low µβ values were observed. However, the dyes 8–12 exhibited a higher NLO response than their analogs 3–7.
Table 2: Experimental and calculated NLO properties and energy gap values for dyes 3–12.
| Compound |
ΔEa
(eV) |
µβb
(× 10−48) (esu) |
µa
(Debye) |
βa
(× 10−30) (esu) |
Compound |
ΔEa
(eV) |
µβb
(× 10−48) (esu) |
µa
(Debye) |
βa
(× 10−30) (esu) |
| 3 | 2.64 | 1430 | 19.42 | 337 | 8 | 2.56 | 1950 | 19.45 | 354 |
| 4 | 2.77 | 1150 | 18.64 | 320 | 9 | 2.68 | 1540 | 18.56 | 342 |
| 5 | 2.76 | 1080 | 18.59 | 339 | 10 | 2.70 | 1340 | 17.18 | 363 |
| 6 | 2.83 | 600 | 16.07 | 314 | 11 | 2.74 | 800 | 17.59 | 328 |
| 7 | 3.00 | 260 | 15.62 | 141 | 12 | 3.12 | 250 | 14.00 | 176 |
aDFT results at the B3LYP/6-31+G(d,p) level of theory in CHCl3. bEFISH: µβ (2ω) at 1907 nm in CHCl3, molecular concentrations used for the measurements were in the range of 10−3 to 10−2 M. µβ ± 10% (for 3–11), µβ ± 50% (for 12); esu: electrostatic unit.
In order to obtain an insight into the NLO properties of the studied dyes, the first-order hyperpolarizability (β) and dipole moment (µ) were calculated at the B3LYP/6-31+G(d,p) level of theory in CHCl3. In general, a high NLO response for a typical organic NLO chromophore is related to the presence of donor (D) and acceptor (A) groups linked through a π-conjugation path and is characterized by a large first-order hyperpolarizability value (β). However, a small energy gap between the HOMO and the LUMO (Egap) is an important indicator for high NLO responses. Based on the results in Table 2, the Egap values obtained from the ground state geometries of the dyes changed as: 3 < 4 ≈ 5 < 6 < 7 for dyes 3–7 and 8 < 9 ≈ 10 < 11 < 12 for dyes 8–12. The smallest Egap was obtained for dyes 3 and 8 due to the presence of the strongest electron-donor group (julolidine), and a high NLO response could be expected. Indeed, dyes 3 and 8 had the highest µ and β values while 7 and 12 had the lowest ones. It should be noted that the hyperpolarizability values were dominated by the component βxxx (for 3–7, 10, and 11) and βxxy (for 8, 9, and 12), which indicated a substantial delocalization of charges in this direction (Tables S2 and S3, Supporting Information File 1).
Thermal properties
The thermal stability is quite important for NLO chromophores when using them in electro-optic materials. Therefore, we conducted a thermogravimetric analysis to understand whether the dyes were suitable as NLO candidate chromophores. NLO candidate chromophores must be thermally stable (>200 °C) and should have a high thermal dissociation value to be applied in electric field poling [62]. The thermal properties of the chromophores were evaluated by thermal gravimetric analysis (TGA) under a nitrogen atmosphere and the results are shown in Figure 10. As depicted in Figure 10, mass wise, all the dyes show noncomplete dissociation under inert environment up to 600 °C. The dyes showed zero weight loss from 0 up to 150 °C which indicated that there was no water or other organic solvent residues on the surface of the dyes.
![[1860-5397-16-189-10]](/bjoc/content/figures/1860-5397-16-189-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: TGA curves of all synthetized dyes.
Figure 10: TGA curves of all synthetized dyes.
Again, the results showed that no dye underwent more than one dissociation step. All dyes dissociated at the very first step but in a nonuniform manner. Regardless, all dyes showed good thermal stability up to around 250 °C, while dye 12, as the least thermally stable dye, started to dissociate at around 310 °C. The Schiff base-based dyes had higher thermal stabilities than the dyes 3–7 having a free amino group. The highest thermal stability was observed for dyes 10–12; their rapid degradation values were above 300 °C.
Conclusion
In summary, we successfully synthesized and characterized a series of new push-pull styryl dyes with free amine functionalities and the corresponding Schiff base analogus. The Schiff bases were found to be able to detect hydroxide anions in DMSO and therefore could act as a pH sensor in a fully aqueous environment. The dyes 8–12 showed increasing fluorescence intensity upon the incremental addition of hydroxide anions in DMSO. Among them, dye 12 showed the highest fluorescence increase (approx. 20-fold fluorescence enhancements upon addition 15 equiv of TBAOH) marking for its superior sensitivity among the other dyes. Furthermore, a reversibility test using TFA showed that all Schiff base-based dyes, except 8, could be brought to the initial state and hence marked their repeatability usage for every single analysis process. The interaction of a Schiff base and hydroxide anion in aqueous environment was conducted successfully using dye 8, which had good solubility in water and a slight rise in fluorescence was observed with increasing pH of the solution.
In addition, the NLO properties of dyes 3–6 and 8–11 were investigated experimentally and theoretically. The dyes 3 and 8 bearing a julolidinyl donor exhibited the highest NLO response (μβ = 1430 × 10−48 esu and 1950 × 10−48 esu, respectively). Thermogravimetric analysis was conducted to understand the thermal stability of the synthesized dyes. In general, all dyes showed good thermal stability up to around 250 °C.
In conclusion, the results indicate that the styryl-based new push-pull dyes are promising candidate materials for optoelectronic device usages and various NLO applications and could be used for applications as water-soluble fluorescent probes for determination of pH.
Experimental
Materials, methods, and instrumentation
Tetrabutylammonium hydroxide (TBAOH) was purchased from Sigma-Aldrich. The solvents were of analytical grade and commercially available chemicals were purchased from Alfa Aeser Chemicals and used without further purification. FTIR (ATR) spectra were recorded on a Thermo Scientific Nicolet iS5 FTIR spectrometer. NMR spectra were recorded on a Bruker Avance 300 (1H: 300 MHz, 13C: 75 MHz) spectrometer at 20 °C (293 K). Chemical shifts (δ) are given in parts per million (ppm) using the residual solvent peaks as reference relative to TMS. Coupling constants (J) are given in hertz (Hz). Signals are abbreviated as follows: broad, br; singlet, s; doublet, d; doublet-doublet, dd; doublet-triplet, dt; triplet, t; multiplet, m. High-resolution mass spectra (HRMS) were recorded at Gazi University Faculty of Pharmacy using electron ionization (EI) mass spectrometry (Waters-LCT-Premier-XE-LTOF (TOF-MS) instruments; in m/z (rel. %). Elemental analysis was performed using a Thermo Scientific Flash 2000 analyzer at the Gazi University Department of Chemistry. The microwave syntheses were carried out in a Milestone Start microwave reaction system. The melting points were measured using an Electrothermal IA9200 apparatus. Absorption spectra were recorded on a Shimadzu 1800 spectrophotometer; fluorescence spectra were recorded on a Hitachi F-7000 fluorescence spectrophotometer. Thermal analyses were performed with a Shimadzu DTG-60H system, up to 700 °C (10 °C min−1) under a dynamic nitrogen atmosphere (15 mL min−1).
Photophysical studies
Dyes 3–7 (10 µM for absorption and 1 µM for emission) were studied in six various solvents with different polarity (toluene, THF, DCM, DMSO, ACN, and MeOH), whereas dyes 8–12 were studied in the same solvents, however, excluding MeOH due to the low solubility of dyes 8–12 in this solvent. All samples were measured in quartz cuvettes (1 cm × 1 cm) with approximately 2 mL in volume. The deprotonation and reverse protonation studies of dyes 8–12 were conducted in DMSO. To each DMSO solution, 5 equiv of TBAOH were introduced and immediately the UV–vis absorption spectra taken at the same concentration mentioned above. This titration was repeated until there was no observable change in the sample spectra. The titrant used in this titration was 10 mM TBAOH in DMSO. The reverse protonation process for the dyes 8–12 was conducted by adding 10 mM TFA to the DMSO solution of the dye and TBAOH. Five equiv of TFA were introduced to fully achieve reprotonation. All of the procedures described above were performed at room temperature (25 °C). Emission changes during the deprotonation and reverse protonation processes for dyes 8–12 were also recorded using a fluorimeter in the same manner with the same concentration as stated earlier for the UV–vis absorption study at room temperature (25 °C). All photographs were taken using a SONY RX100 pocket camera with ISO values of 200 and variable aperture at “Program Auto” mode.
pKa Determination of dye 8
The solubility of dye 8 in deionized water was investigated by studying its calibration graph at 505 nm. The calibration graph was obtained using UV–vis absorption spectrophotometry by the following procedure: A 5 µM solution of 8 was prepared and immediately its absorbance at 505 nm recorded. In the same solution, 10 µL from a 1 mM stock solution of dye 8 was added incrementally, followed by recording its absorbance value after each addition. From the results, the absorbance vs concentration graph was drawn in order to obtain the calibration graph. The linearity was investigated by calculating the R2 value using Microsoft Excel. The spectra of dye 8 were recorded using a Shimadzu 1800 UV–vis spectrophotometer in Britton–Robinson buffer solutions [63] with pH values ranging from 5.5 to 11 at room temperature (25 °C). The pKa values were obtained using the Origin software by recording the curve data.
Computational methods
The geometries of the dyes in their ground states were calculated by Density Functional Theory at the B3LYP/6-31+G(d,p) level of theory in the gas phase [64,65]. This method was also applied to study the nonlinear optic (NLO) properties of the dyes in their ground states. The theoretical absorption spectra in different solvents were calculated using the time dependent DFT (TD-DFT) method at the same level of theory. For calculations in solvents, the Polarizable Continuum Model (PCM) was used [66,67]. All calculations were done using the Gaussian 09 program [68].
General synthetic procedure for dyes 2 and 3–7
The protocol for the synthesis of dye 2 was published previously [31,32]. Dyes 3–7 were synthesized using 2 and the appropriate benzaldehyde. Equimolar (3 mmol) amounts of 2-(1-(4-aminophenyl)ethylidene)malononitrile (2) and the appropriate benzaldehyde derivative in 20 mL of ethanol were refluxed for 2 h. A colored solid formed for all dyes 3–7 which was collected by filtration and recrystallized from ethanol to obtain the pure dyes.
((E)-2-(1-(4-Aminophenyl)-3-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)allylidene)malononitrile) (3)
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-189-i3.svg?max-width=637&scale=1.18182)
Dark purple solid; yield: 64%; mp 236–238 °C; FTIR (cm−1): 3441, 3349, 3224, 2920, 2836, 2202, 1633, 1603, 1568, 1519, 1467; 1H NMR (300 MHz, DMSO-d6) δ 7.15 (d, J = 8.5 Hz, 2H), 7.05 (m, 3H), 6.85 (d, J = 14.9 Hz, 1H), 6.65 (m, 2H), 5.96 (s, 2H), 3.27 (t, 4H), 2.66 (t, 4H), 1.84 (p, J = 6.2 Hz, 4H); 13C NMR (75 MHz, DMSO-d6) δ 171.7 (C10), 153.5 (C1), 153.4 (C16), 149.1 (C3-C5), 132.3 (C12), 131.2 (C11), 124.8 (C13), 120.6 (C4), 120.0 (C14-C18), 116.5 (C15-C17), 115.7 (C7), 114.5 (C8), 113.6 (C2-C6), 72.4 (C9), 66.3 (C21-C22), 47.3 (C19-C24), 44.6 (C20-C23); HRMS (m/z): [M – H]+ calcd for C24H22N4, 367.1923; found, 367.1918; anal. calcd for C24H22N4, C, 78.66; H, 6.05; N, 15.29; found: C, 78.58; H, 6.09; N, 15.18.
General synthetic procedure for dyes 8–12
For the preparation of dyes 8–12, a mixture of 1 mmol of the appropriate styryl dye 3–7 and 8 mmol of salicyaldehyde in 10 mL ethanol was refluxed for half an hour. The colored solid was then collected by filtration and recrystallized from ethanol to obtain the pure dyes.
(2-((E)-1-(4-(((E)-2-Hydroxybenzylidene)amino)phenyl)-3-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)allylidene)malononitrile) (8)
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-189-i4.svg?max-width=637&scale=1.18182)
Dark purple solid; yield 94%; mp 202–203 °C; FTIR (cm−1): 3479 (broad), 3062, 2943, 2835, 2208, 1614, 1560, 1510, 1469; 1H NMR (300 MHz, DMSO-d6) δ 12.80 (s, 1H), 9.05 (s, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.52 (m, 5H), 7.09 (m, 5H), 6.75 (d, J = 14.9 Hz, 1H), 2.65 (t, 4H), 1.83 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 171.0 (C10), 165.2 (C1’), 160.8 (C7’), 153.6 (C1), 150.8 (C16), 150.3 (C3-C5), 136.9 (C5’), 134.2 (C3’), 133.1, 132.3 (C12), 132.1 (C11), 131.0, 123.5 (C13), 123.0 (C2’), 122.3 (C4’), 119.8 (C4), 118.9 (C14-C18), 117.2 (C15-C17), 115.4 (C7), 114.6 (C8), 114.5 (C2-C6), 114.4, 114.3 (C6’), 95.7, 75.8 (C9), 48.1 (C21-C22), 25.4 (C19-24), 24.4 (C20-C23). HRMS (m/z): [M − H]+ calcd for C31H26N4O, 471.2185; found, 471.2162; anal. calcd for C31H26N4O; C, 79.12; H, 5.57; N, 11.91; found: C, 79.01; H, 5.59; N, 11.98.
Supporting Information
| Supporting Information File 1: Additional experimental, photophysical, and calculated data. | ||
| Format: PDF | Size: 5.9 MB | Download |
References
-
Valeur, B.; Berberan-Santos, M. N. Molecular Fluorescence: Principles and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527650002
Return to citation in text: [1] -
Gore, M. Spectrophotometry and Spectrofluorimetry: A Practical Approach; Oxford University Press: Oxford, U.K., 2000.
Return to citation in text: [1] -
Braslavsky, S. E. Pure Appl. Chem. 2007, 79, 293–465. doi:10.1351/pac200779030293
Return to citation in text: [1] -
Williams, D. J. Angew. Chem., Int. Ed. Engl. 1984, 23, 690–703. doi:10.1002/anie.198406901
Return to citation in text: [1] -
Boyd, R. W. Nonlineaer Optics, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. doi:10.1016/b978-0-12-121682-5.x5000-7
Return to citation in text: [1] -
Marder, S. R.; Beratan, D. N.; Cheng, L.-T. Science 1991, 252, 103–106. doi:10.1126/science.252.5002.103
Return to citation in text: [1] -
Eaton, D. F. Science 1991, 253, 281–287. doi:10.1126/science.253.5017.281
Return to citation in text: [1] -
Deng, G.; Xu, H.; Huang, H.; Jiang, J.; Kun, J.; Zhang, X.; Li, Z.; Liu, J. J. Mol. Struct. 2019, 1196, 439–443. doi:10.1016/j.molstruc.2019.06.106
Return to citation in text: [1] -
Deng, G.; Xu, H.; Kuang, L.; He, C.; Li, B.; Yang, M.; Zhang, X.; Li, Z.; Liu, J. Opt. Mater. 2019, 88, 218–222. doi:10.1016/j.optmat.2018.11.035
Return to citation in text: [1] -
Liu, J.; Ouyang, C.; Huo, F.; He, W.; Cao, A. Dyes Pigm. 2020, 181, 108509. doi:10.1016/j.dyepig.2020.108509
Return to citation in text: [1] -
Liu, J.; Gao, W.; Kityk, I. V.; Liu, X.; Zhen, Z. Dyes Pigm. 2015, 122, 74–84. doi:10.1016/j.dyepig.2015.06.007
Return to citation in text: [1] [2] -
Achelle, S.; Barsella, A.; Baudequin, C.; Caro, B.; Robin-le Guen, F. J. Org. Chem. 2012, 77, 4087–4096. doi:10.1021/jo3004919
Return to citation in text: [1] [2] [3] -
Klikar, M.; le Poul, P.; Růžička, A.; Pytela, O.; Barsella, A.; Dorkenoo, K. D.; Robin-le Guen, F.; Bureš, F.; Achelle, S. J. Org. Chem. 2017, 82, 9435–9451. doi:10.1021/acs.joc.7b01442
Return to citation in text: [1] [2] [3] -
Achelle, S.; Kahlal, S.; Barsella, A.; Saillard, J.-Y.; Che, X.; Vallet, J.; Bureš, F.; Caro, B.; Guen, F. R.-l. Dyes Pigm. 2015, 113, 562–570. doi:10.1016/j.dyepig.2014.09.028
Return to citation in text: [1] [2] [3] -
Kreuer, K.-D. Chem. Mater. 1996, 8, 610–641. doi:10.1021/cm950192a
Return to citation in text: [1] -
Cukierman, S. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 876–885. doi:10.1016/j.bbabio.2005.12.001
Return to citation in text: [1] -
Wraight, C. A. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 886–912. doi:10.1016/j.bbabio.2006.06.017
Return to citation in text: [1] -
Czarnik, A. W. Chem. Biol. 1995, 2, 423–428. doi:10.1016/1074-5521(95)90257-0
Return to citation in text: [1] [2] -
Yan, N. D. Can. J. Fish. Aquat. Sci. 1983, 40, 621–626. doi:10.1139/f83-081
Return to citation in text: [1] -
Gerloff-Elias, A.; Spijkerman, E.; Pröschold, T. Plant, Cell Environ. 2005, 28, 1218–1229. doi:10.1111/j.1365-3040.2005.01357.x
Return to citation in text: [1] -
Lee, K. H.; Lee, H.-Y.; Lee, D. H.; Hong, J.-I. Tetrahedron Lett. 2001, 42, 5447–5449. doi:10.1016/s0040-4039(01)01011-5
Return to citation in text: [1] -
Agmon, N.; Bakker, H. J.; Campen, R. K.; Henchman, R. H.; Pohl, P.; Roke, S.; Thämer, M.; Hassanali, A. Chem. Rev. 2016, 116, 7642–7672. doi:10.1021/acs.chemrev.5b00736
Return to citation in text: [1] -
Fayed, T.; El-Morsi, M.; El-Nahass, M. J. Chem. Sci. 2013, 125, 883–894. doi:10.1007/s12039-013-0428-4
Return to citation in text: [1] -
Achelle, S.; Rodríguez-López, J.; Katan, C.; Robin-le Guen, F. J. Phys. Chem. C 2016, 120, 26986–26995. doi:10.1021/acs.jpcc.6b08401
Return to citation in text: [1] -
Chemchem, M.; Yahaya, I.; Aydıner, B.; Seferoğlu, N.; Doluca, O.; Merabet, N.; Seferoğlu, Z. Tetrahedron 2018, 74, 6897–6906. doi:10.1016/j.tet.2018.10.008
Return to citation in text: [1] [2] [3] -
Wencel, D.; Abel, T.; McDonagh, C. Anal. Chem. (Washington, DC, U. S.) 2014, 86, 15–29. doi:10.1021/ac4035168
Return to citation in text: [1] -
Wu, D.; Sedgwick, A. C.; Gunnlaugsson, T.; Akkaya, E. U.; Yoon, J.; James, T. D. Chem. Soc. Rev. 2017, 46, 7105–7123. doi:10.1039/c7cs00240h
Return to citation in text: [1] -
Fang, M.; Adhikari, R.; Bi, J.; Mazi, W.; Dorh, N.; Wang, J.; Conner, N.; Ainsley, J.; Karabencheva-Christova, T. G.; Luo, F.-T.; Tiwari, A.; Liu, H. J. Mater. Chem. B 2017, 5, 9579–9590. doi:10.1039/c7tb02583a
Return to citation in text: [1] [2] -
Chiannilkulchai, N.; Driouich, Z.; Benoit, J. P.; Parodi, A. L.; Couvreur, P. Sel. Cancer Ther. 1989, 5, 1–11. doi:10.1089/sct.1989.5.1
Return to citation in text: [1] -
Wang, J.; Xia, S.; Bi, J.; Zhang, Y.; Fang, M.; Luck, R. L.; Zeng, Y.; Chen, T.-H.; Lee, H.-M.; Liu, H. J. Mater. Chem. B 2019, 7, 198–209. doi:10.1039/c8tb01524d
Return to citation in text: [1] -
Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Çatal, E.; Keleş, E.; Seferoğlu, N.; Achelle, S.; Barsella, A.; Robin le Guen, F.; Seferoğlu, Z. New J. Chem. 2018, 42, 15052–15060. doi:10.1039/c8nj02794c
Return to citation in text: [1] [2] [3] [4] [5] -
Bardasov, I. N.; Alekseeva, A. U.; Chunikhin, S. S.; Shishlikova, M. A.; Ershov, O. V. Tetrahedron Lett. 2019, 60, 1170–1173. doi:10.1016/j.tetlet.2019.03.054
Return to citation in text: [1] -
Sztanke, K.; Maziarka, A.; Osinka, A.; Sztanke, M. Bioorg. Med. Chem. 2013, 21, 3648–3666. doi:10.1016/j.bmc.2013.04.037
Return to citation in text: [1] -
Di Bella, S.; Fragalà, I.; Ledoux, I.; Diaz-Garcia, M. A.; Marks, T. J. J. Am. Chem. Soc. 1997, 119, 9550–9557. doi:10.1021/ja971349y
Return to citation in text: [1] -
Reimann, M. J.; Salmon, D. R.; Horton, J. T.; Gier, E. C.; Jefferies, L. R. ACS Omega 2019, 4, 2874–2882. doi:10.1021/acsomega.8b02750
Return to citation in text: [1] -
Berhanu, A. L.; Gaurav; Mohiuddin, I.; Malik, A. K.; Aulakh, J. S.; Kumar, V.; Kim, K.-H. TrAC, Trends Anal. Chem. 2019, 116, 74–91. doi:10.1016/j.trac.2019.04.025
Return to citation in text: [1] -
Reichardt, C. Chem. Rev. 1994, 94, 2319–2358. doi:10.1021/cr00032a005
Return to citation in text: [1] [2] -
Tydlitát, J.; Achelle, S.; Rodríguez-López, J.; Pytela, O.; Mikýsek, T.; Cabon, N.; Robin-le Guen, F.; Miklík, D.; Růžičková, Z.; Bureš, F. Dyes Pigm. 2017, 146, 467–478. doi:10.1016/j.dyepig.2017.07.043
Return to citation in text: [1] -
Cvejn, D.; Achelle, S.; Pytela, O.; Malval, J.-P.; Spangenberg, A.; Cabon, N.; Bureš, F.; Robin-le Guen, F. Dyes Pigm. 2016, 124, 101–109. doi:10.1016/j.dyepig.2015.09.012
Return to citation in text: [1] -
Le Droumaguet, C.; Sourdon, A.; Genin, E.; Mongin, O.; Blanchard-Desce, M. Chem. – Asian J. 2013, 8, 2984–3001. doi:10.1002/asia.201300735
Return to citation in text: [1] -
Fonseca, R. D.; Vivas, M. G.; Silva, D. L.; Eucat, G.; Bretonnière, Y.; Andraud, C.; De Boni, L.; Mendonça, C. R. J. Phys. Chem. C 2018, 122, 1770–1778. doi:10.1021/acs.jpcc.7b05829
Return to citation in text: [1] -
Erande, Y.; Kothavale, S.; Sreenath, M. C.; Chitrambalam, S.; Joe, I. H.; Sekar, N. Dyes Pigm. 2018, 148, 474–491. doi:10.1016/j.dyepig.2017.09.045
Return to citation in text: [1] -
Hoffert, K.; Durand, R. J.; Gauthier, S.; Robin-le Guen, F.; Achelle, S. Eur. J. Org. Chem. 2017, 523–529. doi:10.1002/ejoc.201601204
Return to citation in text: [1] -
Denneval, C.; Achelle, S.; Baudequin, C.; Robin-le Guen, F. Dyes Pigm. 2014, 110, 49–55. doi:10.1016/j.dyepig.2014.05.030
Return to citation in text: [1] [2] -
Bergen, A.; Granzhan, A.; Ihmels, H. Photochem. Photobiol. Sci. 2008, 7, 405–407. doi:10.1039/b802018c
Return to citation in text: [1] -
Lakowicz, J. R. Protein Fluorescence. In Principles of Fluorescence Spectroscopy; Lakowicz, J. R., Ed.; Springer US: Boston, MA, USA, 1983; pp 341–381. doi:10.1007/978-1-4615-7658-7_11
Return to citation in text: [1] -
Valeur, B. Molecular Fluorescence. Digital Encyclopedia of Applied Physics; Wiley-VCH: Weinheim, Germany, 2009; pp 477–531. doi:10.1002/3527600434.eap684
Return to citation in text: [1] -
Suppan, P.; Ghoneim, N. Solvatochromism; Royal Society of Chemistry: Cambridge, UK, 1997.
Return to citation in text: [1] -
Coelho, F. L.; de Ávila Braga, C.; Zanotto, G. M.; Gil, E. S.; Campo, L. F.; Gonçalves, P. F. B.; Rodembusch, F. S.; da Silveira Santos, F. Sens. Actuators, B 2018, 259, 514–525. doi:10.1016/j.snb.2017.12.097
Return to citation in text: [1] -
Hu, J.; Xia, B.; Bao, D.; Ferreira, A.; Wan, J.; Jones, G.; Vullev, V. I. J. Phys. Chem. A 2009, 113, 3096–3107. doi:10.1021/jp810909v
Return to citation in text: [1] -
Schäfer, K.; Ihmels, H.; Bohne, C.; Valente, K. P.; Granzhan, A. J. Org. Chem. 2016, 81, 10942–10954. doi:10.1021/acs.joc.6b01991
Return to citation in text: [1] [2] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Lindner, E.; Zwickl, T.; Bakker, E.; Lan, B. T. T.; Tóth, K.; Pretsch, E. Anal. Chem. (Washington, DC, U. S.) 1998, 70, 1176–1181. doi:10.1021/ac970952w
Return to citation in text: [1] -
Hisamoto, H.; Tsubuku, M.; Enomoto, T.; Watanabe, K.; Kawaguchi, H.; Koike, Y.; Suzuki, K. Anal. Chem. (Washington, DC, U. S.) 1996, 68, 3871–3878. doi:10.1021/ac951149+
Return to citation in text: [1] -
Safavi, A.; Bagheri, M. Sens. Actuators, B 2003, 90, 143–150. doi:10.1016/s0925-4005(03)00039-x
Return to citation in text: [1] -
Ulrich, G.; Barsella, A.; Boeglin, A.; Niu, S.; Ziessel, R. ChemPhysChem 2014, 15, 2693–2700. doi:10.1002/cphc.201402123
Return to citation in text: [1] -
Singer, K. D.; Garito, A. F. J. Chem. Phys. 1981, 75, 3572–3580. doi:10.1063/1.442467
Return to citation in text: [1] -
Levine, B. F.; Bethea, C. G. Appl. Phys. Lett. 1974, 24, 445–447. doi:10.1063/1.1655254
Return to citation in text: [1] -
Ledoux, I.; Zyss, J. Chem. Phys. 1982, 73, 203–213. doi:10.1016/0301-0104(82)85161-6
Return to citation in text: [1] -
Thami, T.; Bassoul, P.; Petit, M. A.; Simon, J.; Fort, A.; Barzoukas, M.; Villaeys, A. J. Am. Chem. Soc. 1992, 114, 915–921. doi:10.1021/ja00029a019
Return to citation in text: [1] -
Hu, C.; Liu, F.; Zhang, H.; Huo, F.; Yang, Y.; Wang, H.; Xiao, H.; Chen, Z.; Liu, J.; Qiu, L.; Zhen, Z.; Liu, X.; Bo, S. J. Mater. Chem. C 2015, 3, 11595–11604. doi:10.1039/c5tc02702k
Return to citation in text: [1] -
Britton, H. T. S.; Robinson, R. A. J. Chem. Soc. 1931, 1456–1462. doi:10.1039/jr9310001456
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1992, 96, 2155–2160. doi:10.1063/1.462066
Return to citation in text: [1] -
Cossi, M.; Barone, V. J. Chem. Phys. 2001, 115, 4708–4717. doi:10.1063/1.1394921
Return to citation in text: [1] -
Bauernschmitt, R.; Ahlrichs, R. Chem. Phys. Lett. 1996, 256, 454–464. doi:10.1016/0009-2614(96)00440-x
Return to citation in text: [1] -
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2010.
Return to citation in text: [1]
| 58. | Singer, K. D.; Garito, A. F. J. Chem. Phys. 1981, 75, 3572–3580. doi:10.1063/1.442467 |
| 59. | Levine, B. F.; Bethea, C. G. Appl. Phys. Lett. 1974, 24, 445–447. doi:10.1063/1.1655254 |
| 60. | Ledoux, I.; Zyss, J. Chem. Phys. 1982, 73, 203–213. doi:10.1016/0301-0104(82)85161-6 |
| 61. | Thami, T.; Bassoul, P.; Petit, M. A.; Simon, J.; Fort, A.; Barzoukas, M.; Villaeys, A. J. Am. Chem. Soc. 1992, 114, 915–921. doi:10.1021/ja00029a019 |
| 62. | Hu, C.; Liu, F.; Zhang, H.; Huo, F.; Yang, Y.; Wang, H.; Xiao, H.; Chen, Z.; Liu, J.; Qiu, L.; Zhen, Z.; Liu, X.; Bo, S. J. Mater. Chem. C 2015, 3, 11595–11604. doi:10.1039/c5tc02702k |
| 63. | Britton, H. T. S.; Robinson, R. A. J. Chem. Soc. 1931, 1456–1462. doi:10.1039/jr9310001456 |
| 1. | Valeur, B.; Berberan-Santos, M. N. Molecular Fluorescence: Principles and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527650002 |
| 2. | Gore, M. Spectrophotometry and Spectrofluorimetry: A Practical Approach; Oxford University Press: Oxford, U.K., 2000. |
| 3. | Braslavsky, S. E. Pure Appl. Chem. 2007, 79, 293–465. doi:10.1351/pac200779030293 |
| 18. | Czarnik, A. W. Chem. Biol. 1995, 2, 423–428. doi:10.1016/1074-5521(95)90257-0 |
| 19. | Yan, N. D. Can. J. Fish. Aquat. Sci. 1983, 40, 621–626. doi:10.1139/f83-081 |
| 20. | Gerloff-Elias, A.; Spijkerman, E.; Pröschold, T. Plant, Cell Environ. 2005, 28, 1218–1229. doi:10.1111/j.1365-3040.2005.01357.x |
| 21. | Lee, K. H.; Lee, H.-Y.; Lee, D. H.; Hong, J.-I. Tetrahedron Lett. 2001, 42, 5447–5449. doi:10.1016/s0040-4039(01)01011-5 |
| 22. | Agmon, N.; Bakker, H. J.; Campen, R. K.; Henchman, R. H.; Pohl, P.; Roke, S.; Thämer, M.; Hassanali, A. Chem. Rev. 2016, 116, 7642–7672. doi:10.1021/acs.chemrev.5b00736 |
| 31. | Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133 |
| 15. | Kreuer, K.-D. Chem. Mater. 1996, 8, 610–641. doi:10.1021/cm950192a |
| 16. | Cukierman, S. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 876–885. doi:10.1016/j.bbabio.2005.12.001 |
| 17. | Wraight, C. A. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 886–912. doi:10.1016/j.bbabio.2006.06.017 |
| 11. | Liu, J.; Gao, W.; Kityk, I. V.; Liu, X.; Zhen, Z. Dyes Pigm. 2015, 122, 74–84. doi:10.1016/j.dyepig.2015.06.007 |
| 12. | Achelle, S.; Barsella, A.; Baudequin, C.; Caro, B.; Robin-le Guen, F. J. Org. Chem. 2012, 77, 4087–4096. doi:10.1021/jo3004919 |
| 13. | Klikar, M.; le Poul, P.; Růžička, A.; Pytela, O.; Barsella, A.; Dorkenoo, K. D.; Robin-le Guen, F.; Bureš, F.; Achelle, S. J. Org. Chem. 2017, 82, 9435–9451. doi:10.1021/acs.joc.7b01442 |
| 14. | Achelle, S.; Kahlal, S.; Barsella, A.; Saillard, J.-Y.; Che, X.; Vallet, J.; Bureš, F.; Caro, B.; Guen, F. R.-l. Dyes Pigm. 2015, 113, 562–570. doi:10.1016/j.dyepig.2014.09.028 |
| 25. | Chemchem, M.; Yahaya, I.; Aydıner, B.; Seferoğlu, N.; Doluca, O.; Merabet, N.; Seferoğlu, Z. Tetrahedron 2018, 74, 6897–6906. doi:10.1016/j.tet.2018.10.008 |
| 34. | Sztanke, K.; Maziarka, A.; Osinka, A.; Sztanke, M. Bioorg. Med. Chem. 2013, 21, 3648–3666. doi:10.1016/j.bmc.2013.04.037 |
| 35. | Di Bella, S.; Fragalà, I.; Ledoux, I.; Diaz-Garcia, M. A.; Marks, T. J. J. Am. Chem. Soc. 1997, 119, 9550–9557. doi:10.1021/ja971349y |
| 36. | Reimann, M. J.; Salmon, D. R.; Horton, J. T.; Gier, E. C.; Jefferies, L. R. ACS Omega 2019, 4, 2874–2882. doi:10.1021/acsomega.8b02750 |
| 37. | Berhanu, A. L.; Gaurav; Mohiuddin, I.; Malik, A. K.; Aulakh, J. S.; Kumar, V.; Kim, K.-H. TrAC, Trends Anal. Chem. 2019, 116, 74–91. doi:10.1016/j.trac.2019.04.025 |
| 4. | Williams, D. J. Angew. Chem., Int. Ed. Engl. 1984, 23, 690–703. doi:10.1002/anie.198406901 |
| 5. | Boyd, R. W. Nonlineaer Optics, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. doi:10.1016/b978-0-12-121682-5.x5000-7 |
| 6. | Marder, S. R.; Beratan, D. N.; Cheng, L.-T. Science 1991, 252, 103–106. doi:10.1126/science.252.5002.103 |
| 7. | Eaton, D. F. Science 1991, 253, 281–287. doi:10.1126/science.253.5017.281 |
| 8. | Deng, G.; Xu, H.; Huang, H.; Jiang, J.; Kun, J.; Zhang, X.; Li, Z.; Liu, J. J. Mol. Struct. 2019, 1196, 439–443. doi:10.1016/j.molstruc.2019.06.106 |
| 9. | Deng, G.; Xu, H.; Kuang, L.; He, C.; Li, B.; Yang, M.; Zhang, X.; Li, Z.; Liu, J. Opt. Mater. 2019, 88, 218–222. doi:10.1016/j.optmat.2018.11.035 |
| 10. | Liu, J.; Ouyang, C.; Huo, F.; He, W.; Cao, A. Dyes Pigm. 2020, 181, 108509. doi:10.1016/j.dyepig.2020.108509 |
| 31. | Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133 |
| 32. | Çatal, E.; Keleş, E.; Seferoğlu, N.; Achelle, S.; Barsella, A.; Robin le Guen, F.; Seferoğlu, Z. New J. Chem. 2018, 42, 15052–15060. doi:10.1039/c8nj02794c |
| 12. | Achelle, S.; Barsella, A.; Baudequin, C.; Caro, B.; Robin-le Guen, F. J. Org. Chem. 2012, 77, 4087–4096. doi:10.1021/jo3004919 |
| 13. | Klikar, M.; le Poul, P.; Růžička, A.; Pytela, O.; Barsella, A.; Dorkenoo, K. D.; Robin-le Guen, F.; Bureš, F.; Achelle, S. J. Org. Chem. 2017, 82, 9435–9451. doi:10.1021/acs.joc.7b01442 |
| 14. | Achelle, S.; Kahlal, S.; Barsella, A.; Saillard, J.-Y.; Che, X.; Vallet, J.; Bureš, F.; Caro, B.; Guen, F. R.-l. Dyes Pigm. 2015, 113, 562–570. doi:10.1016/j.dyepig.2014.09.028 |
| 18. | Czarnik, A. W. Chem. Biol. 1995, 2, 423–428. doi:10.1016/1074-5521(95)90257-0 |
| 31. | Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133 |
| 32. | Çatal, E.; Keleş, E.; Seferoğlu, N.; Achelle, S.; Barsella, A.; Robin le Guen, F.; Seferoğlu, Z. New J. Chem. 2018, 42, 15052–15060. doi:10.1039/c8nj02794c |
| 33. | Bardasov, I. N.; Alekseeva, A. U.; Chunikhin, S. S.; Shishlikova, M. A.; Ershov, O. V. Tetrahedron Lett. 2019, 60, 1170–1173. doi:10.1016/j.tetlet.2019.03.054 |
| 11. | Liu, J.; Gao, W.; Kityk, I. V.; Liu, X.; Zhen, Z. Dyes Pigm. 2015, 122, 74–84. doi:10.1016/j.dyepig.2015.06.007 |
| 12. | Achelle, S.; Barsella, A.; Baudequin, C.; Caro, B.; Robin-le Guen, F. J. Org. Chem. 2012, 77, 4087–4096. doi:10.1021/jo3004919 |
| 13. | Klikar, M.; le Poul, P.; Růžička, A.; Pytela, O.; Barsella, A.; Dorkenoo, K. D.; Robin-le Guen, F.; Bureš, F.; Achelle, S. J. Org. Chem. 2017, 82, 9435–9451. doi:10.1021/acs.joc.7b01442 |
| 14. | Achelle, S.; Kahlal, S.; Barsella, A.; Saillard, J.-Y.; Che, X.; Vallet, J.; Bureš, F.; Caro, B.; Guen, F. R.-l. Dyes Pigm. 2015, 113, 562–570. doi:10.1016/j.dyepig.2014.09.028 |
| 31. | Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133 |
| 32. | Çatal, E.; Keleş, E.; Seferoğlu, N.; Achelle, S.; Barsella, A.; Robin le Guen, F.; Seferoğlu, Z. New J. Chem. 2018, 42, 15052–15060. doi:10.1039/c8nj02794c |
| 31. | Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133 |
| 32. | Çatal, E.; Keleş, E.; Seferoğlu, N.; Achelle, S.; Barsella, A.; Robin le Guen, F.; Seferoğlu, Z. New J. Chem. 2018, 42, 15052–15060. doi:10.1039/c8nj02794c |
| 26. | Wencel, D.; Abel, T.; McDonagh, C. Anal. Chem. (Washington, DC, U. S.) 2014, 86, 15–29. doi:10.1021/ac4035168 |
| 27. | Wu, D.; Sedgwick, A. C.; Gunnlaugsson, T.; Akkaya, E. U.; Yoon, J.; James, T. D. Chem. Soc. Rev. 2017, 46, 7105–7123. doi:10.1039/c7cs00240h |
| 64. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/physrevb.37.785 |
| 65. | Becke, A. D. J. Chem. Phys. 1992, 96, 2155–2160. doi:10.1063/1.462066 |
| 23. | Fayed, T.; El-Morsi, M.; El-Nahass, M. J. Chem. Sci. 2013, 125, 883–894. doi:10.1007/s12039-013-0428-4 |
| 24. | Achelle, S.; Rodríguez-López, J.; Katan, C.; Robin-le Guen, F. J. Phys. Chem. C 2016, 120, 26986–26995. doi:10.1021/acs.jpcc.6b08401 |
| 25. | Chemchem, M.; Yahaya, I.; Aydıner, B.; Seferoğlu, N.; Doluca, O.; Merabet, N.; Seferoğlu, Z. Tetrahedron 2018, 74, 6897–6906. doi:10.1016/j.tet.2018.10.008 |
| 28. | Fang, M.; Adhikari, R.; Bi, J.; Mazi, W.; Dorh, N.; Wang, J.; Conner, N.; Ainsley, J.; Karabencheva-Christova, T. G.; Luo, F.-T.; Tiwari, A.; Liu, H. J. Mater. Chem. B 2017, 5, 9579–9590. doi:10.1039/c7tb02583a |
| 29. | Chiannilkulchai, N.; Driouich, Z.; Benoit, J. P.; Parodi, A. L.; Couvreur, P. Sel. Cancer Ther. 1989, 5, 1–11. doi:10.1089/sct.1989.5.1 |
| 30. | Wang, J.; Xia, S.; Bi, J.; Zhang, Y.; Fang, M.; Luck, R. L.; Zeng, Y.; Chen, T.-H.; Lee, H.-M.; Liu, H. J. Mater. Chem. B 2019, 7, 198–209. doi:10.1039/c8tb01524d |
| 66. | Cossi, M.; Barone, V. J. Chem. Phys. 2001, 115, 4708–4717. doi:10.1063/1.1394921 |
| 67. | Bauernschmitt, R.; Ahlrichs, R. Chem. Phys. Lett. 1996, 256, 454–464. doi:10.1016/0009-2614(96)00440-x |
| 31. | Yalçın, E.; Achelle, S.; Bayrak, Y.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Tetrahedron Lett. 2015, 56, 2586–2589. doi:10.1016/j.tetlet.2015.03.133 |
| 32. | Çatal, E.; Keleş, E.; Seferoğlu, N.; Achelle, S.; Barsella, A.; Robin le Guen, F.; Seferoğlu, Z. New J. Chem. 2018, 42, 15052–15060. doi:10.1039/c8nj02794c |
| 39. | Tydlitát, J.; Achelle, S.; Rodríguez-López, J.; Pytela, O.; Mikýsek, T.; Cabon, N.; Robin-le Guen, F.; Miklík, D.; Růžičková, Z.; Bureš, F. Dyes Pigm. 2017, 146, 467–478. doi:10.1016/j.dyepig.2017.07.043 |
| 40. | Cvejn, D.; Achelle, S.; Pytela, O.; Malval, J.-P.; Spangenberg, A.; Cabon, N.; Bureš, F.; Robin-le Guen, F. Dyes Pigm. 2016, 124, 101–109. doi:10.1016/j.dyepig.2015.09.012 |
| 41. | Le Droumaguet, C.; Sourdon, A.; Genin, E.; Mongin, O.; Blanchard-Desce, M. Chem. – Asian J. 2013, 8, 2984–3001. doi:10.1002/asia.201300735 |
| 42. | Fonseca, R. D.; Vivas, M. G.; Silva, D. L.; Eucat, G.; Bretonnière, Y.; Andraud, C.; De Boni, L.; Mendonça, C. R. J. Phys. Chem. C 2018, 122, 1770–1778. doi:10.1021/acs.jpcc.7b05829 |
| 43. | Erande, Y.; Kothavale, S.; Sreenath, M. C.; Chitrambalam, S.; Joe, I. H.; Sekar, N. Dyes Pigm. 2018, 148, 474–491. doi:10.1016/j.dyepig.2017.09.045 |
| 44. | Hoffert, K.; Durand, R. J.; Gauthier, S.; Robin-le Guen, F.; Achelle, S. Eur. J. Org. Chem. 2017, 523–529. doi:10.1002/ejoc.201601204 |
| 45. | Denneval, C.; Achelle, S.; Baudequin, C.; Robin-le Guen, F. Dyes Pigm. 2014, 110, 49–55. doi:10.1016/j.dyepig.2014.05.030 |
| 28. | Fang, M.; Adhikari, R.; Bi, J.; Mazi, W.; Dorh, N.; Wang, J.; Conner, N.; Ainsley, J.; Karabencheva-Christova, T. G.; Luo, F.-T.; Tiwari, A.; Liu, H. J. Mater. Chem. B 2017, 5, 9579–9590. doi:10.1039/c7tb02583a |
| 57. | Ulrich, G.; Barsella, A.; Boeglin, A.; Niu, S.; Ziessel, R. ChemPhysChem 2014, 15, 2693–2700. doi:10.1002/cphc.201402123 |
| 25. | Chemchem, M.; Yahaya, I.; Aydıner, B.; Seferoğlu, N.; Doluca, O.; Merabet, N.; Seferoğlu, Z. Tetrahedron 2018, 74, 6897–6906. doi:10.1016/j.tet.2018.10.008 |
| 54. | Lindner, E.; Zwickl, T.; Bakker, E.; Lan, B. T. T.; Tóth, K.; Pretsch, E. Anal. Chem. (Washington, DC, U. S.) 1998, 70, 1176–1181. doi:10.1021/ac970952w |
| 55. | Hisamoto, H.; Tsubuku, M.; Enomoto, T.; Watanabe, K.; Kawaguchi, H.; Koike, Y.; Suzuki, K. Anal. Chem. (Washington, DC, U. S.) 1996, 68, 3871–3878. doi:10.1021/ac951149+ |
| 56. | Safavi, A.; Bagheri, M. Sens. Actuators, B 2003, 90, 143–150. doi:10.1016/s0925-4005(03)00039-x |
| 50. | Coelho, F. L.; de Ávila Braga, C.; Zanotto, G. M.; Gil, E. S.; Campo, L. F.; Gonçalves, P. F. B.; Rodembusch, F. S.; da Silveira Santos, F. Sens. Actuators, B 2018, 259, 514–525. doi:10.1016/j.snb.2017.12.097 |
| 51. | Hu, J.; Xia, B.; Bao, D.; Ferreira, A.; Wan, J.; Jones, G.; Vullev, V. I. J. Phys. Chem. A 2009, 113, 3096–3107. doi:10.1021/jp810909v |
| 52. | Schäfer, K.; Ihmels, H.; Bohne, C.; Valente, K. P.; Granzhan, A. J. Org. Chem. 2016, 81, 10942–10954. doi:10.1021/acs.joc.6b01991 |
| 52. | Schäfer, K.; Ihmels, H.; Bohne, C.; Valente, K. P.; Granzhan, A. J. Org. Chem. 2016, 81, 10942–10954. doi:10.1021/acs.joc.6b01991 |
| 53. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 45. | Denneval, C.; Achelle, S.; Baudequin, C.; Robin-le Guen, F. Dyes Pigm. 2014, 110, 49–55. doi:10.1016/j.dyepig.2014.05.030 |
| 46. | Bergen, A.; Granzhan, A.; Ihmels, H. Photochem. Photobiol. Sci. 2008, 7, 405–407. doi:10.1039/b802018c |
| 47. | Lakowicz, J. R. Protein Fluorescence. In Principles of Fluorescence Spectroscopy; Lakowicz, J. R., Ed.; Springer US: Boston, MA, USA, 1983; pp 341–381. doi:10.1007/978-1-4615-7658-7_11 |
| 48. | Valeur, B. Molecular Fluorescence. Digital Encyclopedia of Applied Physics; Wiley-VCH: Weinheim, Germany, 2009; pp 477–531. doi:10.1002/3527600434.eap684 |
| 49. | Suppan, P.; Ghoneim, N. Solvatochromism; Royal Society of Chemistry: Cambridge, UK, 1997. |
© 2020 Putra et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
