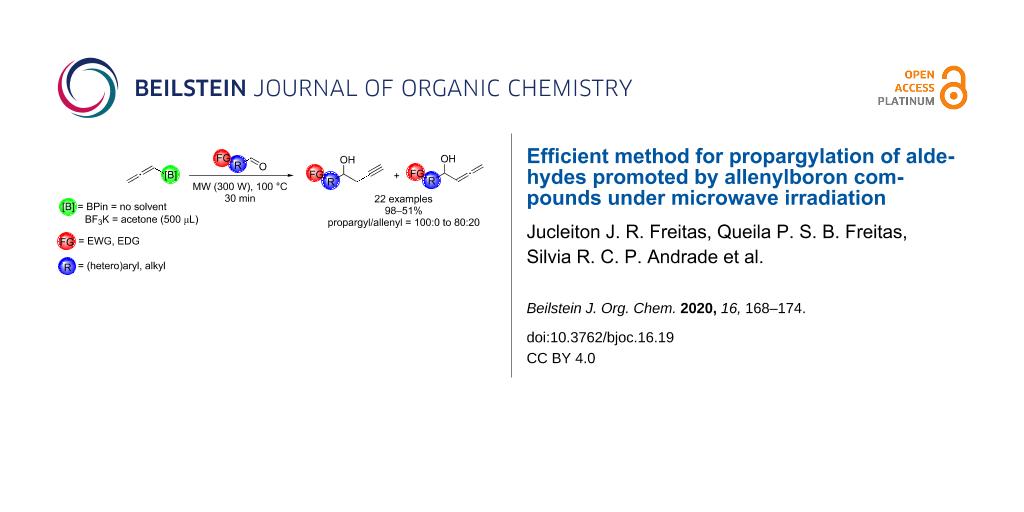
Abstract
The propargylation of aldehydes promoted by microwave irradiation using allenylboron compounds in a chemo- and regioselective way is described. The corresponding products were obtained in short reaction time, high yield and purity without the need of any solvent when allenylboronic acid pinacol ester was used, or using a minimal amount of acetone when potassium allenyltrifluoroborate was used.

Graphical Abstract
Introduction
The propargylation of carbonyl compounds is widely used in the synthesis of biologically active natural products [1]. Some examples can be found in the synthesis of histrionicotoxin [2], rhizopodin [3], bafilomycin [4], bryostatin [5], vancosamine [6] and macrolactin A [7].
Although there are several stereoselective methods described for the reaction of propargyl or allenyl organometallics with carbonyl compounds [8-14], the control of the regioselectivity is still a major concern. This is mainly due to the metallotropic rearrangement of propargyl and allenyl organometallics in solution resulting in mixtures of the two reagents [15]. Thus, upon reaction with an aldehyde, mixtures of propargylic and allenic alcohols can be obtained through a chelate transition state (SE2').
Attempts to improve the regioselectivity of the propargylation reaction by using allenic organometallic species of Cd [16], Ga [17], In [18], Ti [19], Al [20] and Bi [21] were described. However, the majority of these methods involve reagents that are difficult to prepare and to handle due to the sensitivity to air and moisture.
The use of less reactive species based on tin [22-24], silicon [25] or boron [26-28] to perform propargylation reactions typically requires catalysis by Lewis acids or bases and although the utility of allenylstannanes is further indicated by the commercial availability of some of them, the toxicity of these compounds makes them inappropriate for the use in pharmaceutical synthesis [29]. Moreover, the removal of tributyltin residues from reaction mixtures is also a major issue.
The use of microwave irradiation for the formation of new C–C bonds is nowadays widely used and offers several advantages such as the increment in the product yield, reduction of reaction time and the possibility to perform solvent-free reactions [30]. However, the relative “greenness” of microwave-assisted reactions is still a point of discussion. For example, the question about the energy efficiency of microwave vs conventionally heated reactions must, in general, be evaluated with great care on a case-by-case basis. Even so, the search for safer alternatives to current synthetic methodologies avoiding the use of moisture/air-sensitive organometallics and, more important, the development of solvent-free protocols are in accordance with the concept of environmental impact factor (E factor) [31].
Within this context, the development of solvent-free methods is highly desirable since the difficult for solvent recycling in academic laboratories and chemical manufacturing plants is universal. In addition, a reliable method for the propargylation reaction which could involve the use of commercially available and stable allenyl or propargyl compounds without the need of special conditions such as dry solvents or complex catalysts is a subject of great interest.
Results and Discussion
For preliminary optimization of the reaction conditions, 2-naphthaldehyde (1 mmol) and allenylboronic acid pinacol ester (1, 1.5 mmol) in a capped vial were irradiated in a MW synthesizer for 30 minutes under different temperatures. The results are depicted in Table 1.
Table 1: Propargylation of 2-naphthaldehyde with allenylboronic acid pinacol ester (1) at different temperatures.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-19-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Temp. (°C) | 2a/3ab | Yield (%)c |
| 1 | 75 | 97:3 | 94 |
| 2 | 100 | 98:2 | 97 |
| 3 | 125 | 98:2 | 81 |
| 4 | 150 | 98:2 | 84 |
aReaction conditions: Reactions were performed with 2-naphthaldehyde (1 mmol), 1 (1.5 mmol) under MW irradiation (300 W) for 30 min at the temperature indicated. bDetermined by GC analysis. cIsolated yield.
In all cases, the desired propargylic product 2a was obtained together with a small amount of the corresponding allenic product 3a. When the reaction was performed at 75 °C, good conversions of 2-naphthaldehyde into the corresponding products were observed (Table 1, entry 1). The best result was observed when the temperature was increased to 100 °C affording the products in 97% yield in a 98:2 ratio (Table 1, entry 2). Higher temperatures gave lower yields or the decomposition of the boron reagent 1 (Table 1, entries 3 and 4). It is worth to note that when the reaction was carried out with conventional heating (100 °C), the desired product was not observed after 1 h. A similar behavior was previously observed by Schaus [32].
Next, the shortest time necessary for the formation of the products at 100 °C under microwave irradiation was evaluated. The results are shown in Table 2. From Table 2, it can be seen that the increment in the reaction time resulted in higher yields without major changes in the ratio of 2a:3a.
Table 2: Propargylation of 2-naphthaldehyde with allenylboronic acid pinacol ester (1) using different reaction times.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-19-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Time (min) | 2a/3ab | Yield (%)c |
| 1 | 5 | 98:2 | 87 |
| 2 | 10 | 98:2 | 88 |
| 3 | 15 | 97:3 | 88 |
| 4 | 20 | 98:2 | 95 |
| 5 | 30 | 97:3 | 97 |
aReaction conditions: Reactions were performed with 2-naphthaldehyde (1 mmol), 1 (1.5 mmol) under MW irradiation (300 W) at 100 °C for the time indicated. bDetermined by GC analysis. cIsolated yield.
The optimized reaction conditions, namely: 2-naphthaldehyde (1.0 mmol), 1 (1.5 mmol) under microwave irradiation (300 W) were then applied for the propargylation reaction of aldehydes containing a wide range of functional groups and the results are shown in Scheme 1. In all cases the reaction proceeded smoothly leading to the conversion of aldehydes into the corresponding homopropargylic alcohols 2 in moderate to high yields and in a very regioselective way, while the propargylated product was obtained as the major product in all cases.
![[1860-5397-16-19-i1]](/bjoc/content/inline/1860-5397-16-19-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Scope of the propargylation reaction. Reactions were performed with the appropriate aldehyde (1 mmol), 1 (1.5 mmol) under microwave irradiation (300 W) at 100 °C for 30 min. Isolated yields are given. The number in parentheses refers to the mixture of propagyl and allenyl regioisomers determined by GC analysis.
Scheme 1: Scope of the propargylation reaction. Reactions were performed with the appropriate aldehyde (1 mmo...
Aromatic aldehydes such as benzaldehyde, 2-methylbenzaldehyde and 3,5-dimethylbenzaldehyde gave the corresponding products 2b–d in good yields in a regioselective way regardless of the position of the substituent on the aromatic ring.
When aldehydes containing electron-donating groups such as 2-, 3- or 4-methoxybenzaldehyde or 3,4,5-trimethoxybenzaldehyde were used, the corresponding products 2e–h were obtained in good yields and regioselectivities. In the same way, aldehydes containing electron-withdrawing groups such as the nitro group also reacted without influence of the substituent location to give the corresponding homopropargylic alcohols 2m and 2n in good yields. It is interesting to note that the nitro group remained intact under the reaction conditions. Usually, this group is sensitive to reduction when alternative methods involving zinc or tin are used [33].
In addition, aldehydes containing halogens also gave the corresponding products 2j–l in moderate yields. These results indicated that the substituent nature, whether electron-donating or electron-withdrawing, has no dramatic influence on the product yields.
When the α,β-unsaturated aldehyde, cinnamaldehyde was used, the corresponding 1,2-addition product 2i was obtained exclusively.
The chemoselectivity of the method was evaluated using aldehydes containing different functionalities. For example, the use of vanillin, an aldehyde containing the acidic phenol group as substituent, gave the corresponding product 2o in 93% yield in an 82:18 ratio of regioisomers. In the same way, when aldehydes containing an ester or nitrile group were used, the corresponding products 2q and 2r were obtained in good yields. The use of a heteroaromatic or an aliphatic aldehyde as substrates under the optimized conditions gave the corresponding homopropargylic alcohols 2s,t in moderate yields.
The development of a solvent-free protocol for propargylation of aldehydes based on microwave irradiation should take into account not only the reaction itself but also an effective method for the extraction of the obtained products. Despite the excellent results described on Scheme 1, a factor that must be taken into consideration is the removal of the desired alcohols 2a–t from pinacol – the byproduct obtained in the reaction. There are some examples in the literature based on the removal of pinacol by distillation in vacuo (50 °C/0.05 mbar) [34]. However, this technique can only be applied for non-volatile samples. More recently, Aggarwal and co-workers [35] described an efficient method for removal of pinacol based on the formation of an azeotrope with water under moderate vacuum. Thus, a test experiment was performed using compound 2h. The crude product obtained from the reaction was dissolved in 50% aqueous methanol and the volatile materials were removed using a rotary evaporator. The procedure was monitored by gas chromatography and was repeated until the crude mixture contained less than 1 mol % of pinacol remaining. The results are described in Table 3.
Table 3: Number of cycles to remove pinacol from the crude product.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-19-i6.svg?max-width=637&scale=1.0)
|
||
| Entry | Cycle | Ratio 2h/pinacol (%)b |
| 1 | 1 | 70:30 |
| 2 | 2 | 77:23 |
| 3 | 3 | 84:16 |
| 4 | 4 | 93:7 |
| 5 | 5 | 99:1 |
aReaction conditions: The reaction was performed using 3,4,5-trimethoxybenzaldehyde (1 mmol) and 1 (1.5 mmol) under microwave irradiation (300 W) at 100 °C for 0.5 h. The crude product was dissolved in 50% aqueous methanol (10 mL) and the solvents were removed on a rotary evaporator (45–50 °C/25–15 mbar). bDetermined by GC analysis.
Although the desired products 2a–t having been obtained along with a small proportion of the corresponding regioisomer in some cases, in the development of new synthetic methods, it is desirable that it gives the corresponding product as a single compound. Our group described the synthesis of homopropargylic alcohols using potassium allenyltrifluoroborate as the propargylating agent in a very regioselective way [36,37]. Thus, allenylboronic acid pinacol ester (1) was converted into the corresponding trifluoroborate using the procedure described by Lloyd-Jones and co-worker [38]. The desired product 4 was obtained in good yield and characterized by 1H, 13C, 11B and 19F NMR [39] (Scheme 2).
![[1860-5397-16-19-i2]](/bjoc/content/inline/1860-5397-16-19-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of potassium allenyltrifluoroborate (4).
Scheme 2: Synthesis of potassium allenyltrifluoroborate (4).
Potassium allenyltrifluoroborate (4) is a crystalline solid and despite several microwave promoted reactions can be conducted without the use of solvents, the propargylation reaction using 2-naphthaldehyde and 4 under the previously optimized conditions gave the desired product in only low conversion (Table 4, entry 1).
Table 4: Propargylation of 2-naphthaldehyde with potassium allenyltrifluoroborate (4) using different solvents.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-19-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Time (min) | 2a/3ab | Yield (%)c |
| 1 | – | – | – | 6 |
| 2 | acetone | 30 | 100:0 | 92 |
| 3 | water | 30 | 100:0 | 3 |
| 4 | acetone/water | 30 | 100:0 | 10 |
| 5 | ethylene glycol | 20 | 100:0 | 45 |
| 6 | ethanol | 20 | 100:0 | 45 |
| 7 | methanol | 20 | 100:0 | 30 |
| 8 | dichloromethane | 20 | 100:0 | 10 |
aReaction conditions: Reactions were performed with 2-naphthaldehyde (1 mmol), 4 (1.5 mmol) under MW irradiation (300 W) at 100 °C for the time indicated using the appropriate solvent (500 μL). bDetermined by GC analysis. cIsolated yield.
In order to find an appropriate and suitable solvent that could be used in small amounts to promote the reaction different solvents were screened and the best result was observed when a small amount of acetone was used, where 2a was obtained in 92% yield as a single regioisomer (Table 4, entry 2). Low conversions were observed when water or a 1:1 mixture of acetone and water was used in the reaction (Table 4, entries 3 and 4). The use of alcohols also gave the desired product in lower yields (Table 4, entries 5–7). Finally, when a less polar solvent was used, the observed conversion was only 10% (Table 4, entry 8).
Two factors must be considered when allenylboron species are used in the propargylation reaction. The first is that the atomic efficiency [40] using potassium allenyltrifluoroborate (4, 62%) or allenylboronic acid pinacol ester (1, 58%) is higher when compared to the commercially available allenyltributyltin (22%) for similar reactions.
The second refers to the regioselectivity of the reaction. Despite the reaction using 2-naphthaldehyde led only to the propargyl isomer 2a, when the same reaction conditions were applied to 4-nitrobenzaldehyde, the desired product 2m was once again obtained with a small amount of the corresponding regioisomer 3m in a 97:3 ratio (Scheme 3).
![[1860-5397-16-19-i3]](/bjoc/content/inline/1860-5397-16-19-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Propargylation of aldehydes using potassium allenyltrifluoroborate (4).
Scheme 3: Propargylation of aldehydes using potassium allenyltrifluoroborate (4).
Conclusion
In summary, we have shown an efficient method for the propargylation of aldehydes promoted by microwave irradiation using allenylboron compounds in a chemo- and regioselective way. The corresponding products were obtained after a short reaction time in high yield and purity without the need of a solvent when allenylboronic acid pinacol ester was used, or using a minimal amount of acetone when potassium allenyltrifluoroborate was used. The method is simple, fast and general allowing further applications in the synthesis of more complex compounds.
Supporting Information
| Supporting Information File 1: Experimental procedures and 1H, 13C, 11B and 19F NMR spectra for all synthesized compounds. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Ding, C.-H.; Hou, X.-L. Chem. Rev. 2011, 111, 1914–1937. doi:10.1021/cr100284m
Return to citation in text: [1] -
Adachi, Y.; Kamei, N.; Yokoshima, S.; Fukuyama, T. Org. Lett. 2011, 13, 4446–4449. doi:10.1021/ol2018032
Return to citation in text: [1] -
Pulukuri, K. K.; Chakraborty, T. K. Org. Lett. 2014, 16, 2284–2287. doi:10.1021/ol5008179
Return to citation in text: [1] -
Marshall, J. A.; Adams, N. D. Org. Lett. 2000, 2, 2897–2900. doi:10.1021/ol006344b
Return to citation in text: [1] -
Trost, B. M.; Dong, G. Nature 2008, 456, 485–488. doi:10.1038/nature07543
Return to citation in text: [1] -
Parker, K. A.; Chang, W. Org. Lett. 2003, 5, 3891–3893. doi:10.1021/ol035479p
Return to citation in text: [1] -
Marino, J. P.; McClure, M. S.; Holub, D. P.; Comasseto, J. V.; Tucci, F. C. J. Am. Chem. Soc. 2002, 124, 1664–1668. doi:10.1021/ja017177t
Return to citation in text: [1] -
Haruta, R.; Ishiguro, M.; Ikeda, N.; Yamamoto, H. J. Am. Chem. Soc. 1982, 104, 7667–7669. doi:10.1021/ja00390a052
Return to citation in text: [1] -
Ikeda, N.; Arai, I.; Yamamoto, H. J. Am. Chem. Soc. 1986, 108, 483–486. doi:10.1021/ja00263a020
Return to citation in text: [1] -
Reddy, L. R. Org. Lett. 2012, 14, 1142–1145. doi:10.1021/ol300075n
Return to citation in text: [1] -
Jain, P.; Wang, H.; Houk, K. N.; Antilla, J. C. Angew. Chem., Int. Ed. 2012, 51, 1391–1394. doi:10.1002/anie.201107407
Return to citation in text: [1] -
Fernández, S.; González, J.; Santamaría, J.; Ballesteros, A. Angew. Chem., Int. Ed. 2019, 58, 10703–10707. doi:10.1002/anie.201905159
Return to citation in text: [1] -
Horino, Y.; Murakami, M.; Ishibashi, M.; Lee, J. H.; Watanabe, A.; Matsumoto, R.; Abe, H. Org. Lett. 2019, 21, 9564–9568. doi:10.1021/acs.orglett.9b03710
Return to citation in text: [1] -
Wisniewska, H. M.; Jarvo, E. R. J. Org. Chem. 2013, 78, 11629–11636. doi:10.1021/jo4019107
Return to citation in text: [1] -
Yamashita, Y.; Cui, Y.; Xie, P.; Kobayashi, S. Org. Lett. 2015, 17, 6042–6045. doi:10.1021/acs.orglett.5b03045
Return to citation in text: [1] -
Zheng, Y.; Bao, W.; Zhang, Y. Synth. Commun. 2000, 30, 3517–3521. doi:10.1080/00397910008087264
Return to citation in text: [1] -
Lee, P. H.; Kim, H.; Lee, K. Adv. Synth. Catal. 2005, 347, 1219–1222. doi:10.1002/adsc.200505046
Return to citation in text: [1] -
Lin, M.-J.; Loh, T.-P. J. Am. Chem. Soc. 2003, 125, 13042–13043. doi:10.1021/ja037410i
Return to citation in text: [1] -
Millán, A.; de Cienfuegos, L. Á.; Martín-Lasanta, A.; Campaña, A. G.; Cuerva, J. M. Adv. Synth. Catal. 2011, 353, 73–78. doi:10.1002/adsc.201000655
Return to citation in text: [1] -
Guo, L.-N.; Gao, H.; Mayer, P.; Knochel, P. Chem. – Eur. J. 2010, 16, 9829–9834. doi:10.1002/chem.201000523
Return to citation in text: [1] -
Kobayashi, S.; Kitanosono, T.; Ueno, M. Synlett 2010, 2033–2036. doi:10.1055/s-0030-1258485
Return to citation in text: [1] -
Keck, G. E.; Krishnamurthy, D.; Chen, X. Tetrahedron Lett. 1994, 35, 8323–8324. doi:10.1016/s0040-4039(00)74397-8
Return to citation in text: [1] -
Yu, C.-M.; Kim, J.-M.; Shin, M.-S.; Cho, D. Tetrahedron Lett. 2003, 44, 5487–5490. doi:10.1016/s0040-4039(03)01033-5
Return to citation in text: [1] -
Yu, C.-M.; Yoon, S.-K.; Baek, K.; Lee, J.-Y. Angew. Chem., Int. Ed. 1998, 37, 2392–2395. doi:10.1002/(sici)1521-3773(19980918)37:17<2392::aid-anie2392>3.0.co;2-d
Return to citation in text: [1] -
Chen, J.; Captain, B.; Takenaka, N. Org. Lett. 2011, 13, 1654–1657. doi:10.1021/ol200102c
Return to citation in text: [1] -
Grayson, M. N.; Goodman, J. M. J. Org. Chem. 2013, 78, 8796–8801. doi:10.1021/jo401611q
Return to citation in text: [1] -
Grayson, M. N.; Goodman, J. M. J. Am. Chem. Soc. 2013, 135, 6142–6148. doi:10.1021/ja3122137
Return to citation in text: [1] -
Wang, M.; Khan, S.; Miliordos, E.; Chen, M. Org. Lett. 2018, 20, 3810–3814. doi:10.1021/acs.orglett.8b01399
Return to citation in text: [1] -
Tri-n-butyltin compounds [MAK Value Documentation, 1990]. The MAK Collection for Occupational Health and Safety (2012) 316.
Return to citation in text: [1] -
Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. doi:10.1002/anie.200400655
Return to citation in text: [1] -
Sheldon, R. A. Green Chem. 2017, 19, 18–43. doi:10.1039/c6gc02157c
Return to citation in text: [1] -
Barnett, D. S.; Schaus, S. E. Org. Lett. 2011, 13, 4020–4023. doi:10.1021/ol201535b
Return to citation in text: [1] -
Chan, T. H.; Isaac, M. B. Pure Appl. Chem. 1996, 68, 919–924. doi:10.1351/pac199668040919
Return to citation in text: [1] -
Murphy, J. M.; Tzschucke, C. C.; Hartwig, J. F. Org. Lett. 2007, 9, 757–760. doi:10.1021/ol062903o
Return to citation in text: [1] -
Bagutski, V.; Ros, A.; Aggarwal, V. K. Tetrahedron 2009, 65, 9956–9960. doi:10.1016/j.tet.2009.10.002
Return to citation in text: [1] -
Freitas, J. J. R.; Couto, T. R.; Cavalcanti, I. H.; Freitas, J. C. R.; Barbosa, Q. P. S.; Oliveira, R. A. Tetrahedron Lett. 2016, 57, 760–765. doi:10.1016/j.tetlet.2016.01.017
Return to citation in text: [1] -
Couto, T. R.; Freitas, J. J. R.; Freitas, J. C. R.; Cavalcanti, I. H.; Menezes, P. H.; Oliveira, R. A. Synthesis 2015, 47, 71–78. doi:10.1055/s-0034-1379163
Return to citation in text: [1] -
Lennox, A. J. J.; Lloyd-Jones, G. C. Angew. Chem., Int. Ed. 2012, 51, 9385–9388. doi:10.1002/anie.201203930
Return to citation in text: [1] -
Oliveira, R. A.; Silva, R. O.; Molander, G. A.; Menezes, P. H. Magn. Reson. Chem. 2009, 47, 873–878. doi:10.1002/mrc.2467
Return to citation in text: [1] -
Trost, B. M. Science 1991, 254, 1471–1477. doi:10.1126/science.1962206
Return to citation in text: [1]
| 39. | Oliveira, R. A.; Silva, R. O.; Molander, G. A.; Menezes, P. H. Magn. Reson. Chem. 2009, 47, 873–878. doi:10.1002/mrc.2467 |
| 21. | Kobayashi, S.; Kitanosono, T.; Ueno, M. Synlett 2010, 2033–2036. doi:10.1055/s-0030-1258485 |
| 4. | Marshall, J. A.; Adams, N. D. Org. Lett. 2000, 2, 2897–2900. doi:10.1021/ol006344b |
| 22. | Keck, G. E.; Krishnamurthy, D.; Chen, X. Tetrahedron Lett. 1994, 35, 8323–8324. doi:10.1016/s0040-4039(00)74397-8 |
| 23. | Yu, C.-M.; Kim, J.-M.; Shin, M.-S.; Cho, D. Tetrahedron Lett. 2003, 44, 5487–5490. doi:10.1016/s0040-4039(03)01033-5 |
| 24. | Yu, C.-M.; Yoon, S.-K.; Baek, K.; Lee, J.-Y. Angew. Chem., Int. Ed. 1998, 37, 2392–2395. doi:10.1002/(sici)1521-3773(19980918)37:17<2392::aid-anie2392>3.0.co;2-d |
| 3. | Pulukuri, K. K.; Chakraborty, T. K. Org. Lett. 2014, 16, 2284–2287. doi:10.1021/ol5008179 |
| 19. | Millán, A.; de Cienfuegos, L. Á.; Martín-Lasanta, A.; Campaña, A. G.; Cuerva, J. M. Adv. Synth. Catal. 2011, 353, 73–78. doi:10.1002/adsc.201000655 |
| 2. | Adachi, Y.; Kamei, N.; Yokoshima, S.; Fukuyama, T. Org. Lett. 2011, 13, 4446–4449. doi:10.1021/ol2018032 |
| 20. | Guo, L.-N.; Gao, H.; Mayer, P.; Knochel, P. Chem. – Eur. J. 2010, 16, 9829–9834. doi:10.1002/chem.201000523 |
| 15. | Yamashita, Y.; Cui, Y.; Xie, P.; Kobayashi, S. Org. Lett. 2015, 17, 6042–6045. doi:10.1021/acs.orglett.5b03045 |
| 17. | Lee, P. H.; Kim, H.; Lee, K. Adv. Synth. Catal. 2005, 347, 1219–1222. doi:10.1002/adsc.200505046 |
| 8. | Haruta, R.; Ishiguro, M.; Ikeda, N.; Yamamoto, H. J. Am. Chem. Soc. 1982, 104, 7667–7669. doi:10.1021/ja00390a052 |
| 9. | Ikeda, N.; Arai, I.; Yamamoto, H. J. Am. Chem. Soc. 1986, 108, 483–486. doi:10.1021/ja00263a020 |
| 10. | Reddy, L. R. Org. Lett. 2012, 14, 1142–1145. doi:10.1021/ol300075n |
| 11. | Jain, P.; Wang, H.; Houk, K. N.; Antilla, J. C. Angew. Chem., Int. Ed. 2012, 51, 1391–1394. doi:10.1002/anie.201107407 |
| 12. | Fernández, S.; González, J.; Santamaría, J.; Ballesteros, A. Angew. Chem., Int. Ed. 2019, 58, 10703–10707. doi:10.1002/anie.201905159 |
| 13. | Horino, Y.; Murakami, M.; Ishibashi, M.; Lee, J. H.; Watanabe, A.; Matsumoto, R.; Abe, H. Org. Lett. 2019, 21, 9564–9568. doi:10.1021/acs.orglett.9b03710 |
| 14. | Wisniewska, H. M.; Jarvo, E. R. J. Org. Chem. 2013, 78, 11629–11636. doi:10.1021/jo4019107 |
| 18. | Lin, M.-J.; Loh, T.-P. J. Am. Chem. Soc. 2003, 125, 13042–13043. doi:10.1021/ja037410i |
| 7. | Marino, J. P.; McClure, M. S.; Holub, D. P.; Comasseto, J. V.; Tucci, F. C. J. Am. Chem. Soc. 2002, 124, 1664–1668. doi:10.1021/ja017177t |
| 16. | Zheng, Y.; Bao, W.; Zhang, Y. Synth. Commun. 2000, 30, 3517–3521. doi:10.1080/00397910008087264 |
| 29. | Tri-n-butyltin compounds [MAK Value Documentation, 1990]. The MAK Collection for Occupational Health and Safety (2012) 316. |
| 25. | Chen, J.; Captain, B.; Takenaka, N. Org. Lett. 2011, 13, 1654–1657. doi:10.1021/ol200102c |
| 26. | Grayson, M. N.; Goodman, J. M. J. Org. Chem. 2013, 78, 8796–8801. doi:10.1021/jo401611q |
| 27. | Grayson, M. N.; Goodman, J. M. J. Am. Chem. Soc. 2013, 135, 6142–6148. doi:10.1021/ja3122137 |
| 28. | Wang, M.; Khan, S.; Miliordos, E.; Chen, M. Org. Lett. 2018, 20, 3810–3814. doi:10.1021/acs.orglett.8b01399 |
| 36. | Freitas, J. J. R.; Couto, T. R.; Cavalcanti, I. H.; Freitas, J. C. R.; Barbosa, Q. P. S.; Oliveira, R. A. Tetrahedron Lett. 2016, 57, 760–765. doi:10.1016/j.tetlet.2016.01.017 |
| 37. | Couto, T. R.; Freitas, J. J. R.; Freitas, J. C. R.; Cavalcanti, I. H.; Menezes, P. H.; Oliveira, R. A. Synthesis 2015, 47, 71–78. doi:10.1055/s-0034-1379163 |
| 38. | Lennox, A. J. J.; Lloyd-Jones, G. C. Angew. Chem., Int. Ed. 2012, 51, 9385–9388. doi:10.1002/anie.201203930 |
| 34. | Murphy, J. M.; Tzschucke, C. C.; Hartwig, J. F. Org. Lett. 2007, 9, 757–760. doi:10.1021/ol062903o |
| 35. | Bagutski, V.; Ros, A.; Aggarwal, V. K. Tetrahedron 2009, 65, 9956–9960. doi:10.1016/j.tet.2009.10.002 |
| 32. | Barnett, D. S.; Schaus, S. E. Org. Lett. 2011, 13, 4020–4023. doi:10.1021/ol201535b |
| 33. | Chan, T. H.; Isaac, M. B. Pure Appl. Chem. 1996, 68, 919–924. doi:10.1351/pac199668040919 |
| 30. | Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. doi:10.1002/anie.200400655 |
© 2020 Freitas et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)