Abstract
The thread–link–cut (TLC) approach has previously shown promise as a novel method to synthesize molecular knots. The modular second-generation approach to small trefoil knots described herein involves electrostatic interactions between an electron-rich bis-macrocyclic host compound and electron-deficient guests in the threading step. The bis-macrocyclic host was synthesized in eight steps and 6.6% overall yield. Ammonium and pyridinium guests were synthesized in 4–5 steps. The TLC knot-forming sequence was carried out and produced a product with the expected molecular weight, but, unfortunately, further characterization did not produce conclusive results regarding the topology of the product.

Graphical Abstract
Introduction
Macrocycles have played a central role in the development of molecular recognition, self-assembled molecular devices, and molecular topology [1-6]. For example, early work by Pedersen on crown ethers [1], Lehn on cryptands [2], and Cram on hemicarcerands [3] demonstrated that preorganized macrocycles have the ability to act as hosts for various guest cations and compounds. Their seminal work was recognized with the 1987 Nobel Prize in Chemistry. More recently, the 2016 Nobel Prize in Chemistry was won by Sauvage, Stoddart, and Feringa for their work on molecular machines [4]. Sauvage [5] and Stoddart [6] extensively used macrocycles in their ground-breaking work on catenanes, rotaxanes, knots, and other topologically novel compounds. Exciting advances in the field of molecular topology continue with novel trefoil knots have been prepared with an all-hydrocarbon example by the Itami group [7] and the synthesis of a single enantiomer by Leigh’s group [8]. Complexity has also been achieved with recent work showing that eight-crossing knots [9-11] and a nine-crossing composite knot can be synthesized [12].
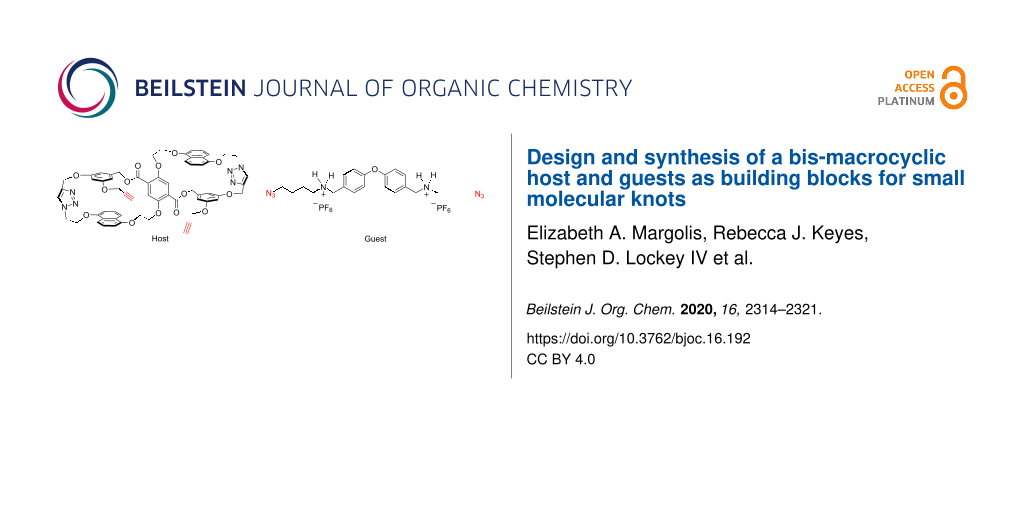
Herein the synthesis of a unique bis-macrocyclic host 1 is described (Figure 1). Host 1 was designed to be a second-generation building block in the thread–link–cut (TLC) approach to molecular knots [13]. The two 25-atom macrocycles are electron-rich and complementary to the electron-deficient guests bis(ammonium) 2 and bis(pyridinium) 3 (Figure 1). The electron-rich macrocycles of host 1 might also render it useful for other molecular recognition applications.
![[1860-5397-16-192-1]](/bjoc/content/figures/1860-5397-16-192-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of electron-rich bis-macrocyclic host 1, and electron-poor guests bis(ammonium) 2, and bis(pyridinium) 3.
Figure 1: Structures of electron-rich bis-macrocyclic host 1, and electron-poor guests bis(ammonium) 2, and b...
A principle goal driving our second-generation TLC approach was to test the lower limit on the size of a molecular trefoil knot. In 2008 we surveyed the literature and suggested that a knot of 45–50 backbone atoms was theoretically possible [13]. At that time, the world’s smallest knot was an 80 backbone atom trefoil knot–metal complex by the Sauvage group [14]. Shortly thereafter, Hunter’s group published the synthesis of a knot–metal complex with 77 backbone atoms (Figure 2a) [15]. More recently, Leigh’s group disclosed the synthesis of the current record holder for the smallest knot, which has 76 backbone atoms (Figure 2b) [16]. Since the metal atom template has been removed, the Leigh compound is a true knot in strict topological terms [13,17-20].
![[1860-5397-16-192-2]](/bjoc/content/figures/1860-5397-16-192-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Hunter’s 77 backbone-atom trefoil knot–metal complex [9]. (b) The world’s smallest knot: Leigh’s 76 backbone-atom trefoil [10]. (c) Target 73 backbone-atom trefoil knot of this work using host 1 and guest 2.
Figure 2: (a) Hunter’s 77 backbone-atom trefoil knot–metal complex [9]. (b) The world’s smallest knot: Leigh’s 7...
The first-generation TLC approach for the synthesis of molecular knots involved a single knot precursor compound that had two macrocycles and two long tails [13,21]. Solvophobic effects [22,23] were used to promote the tail threading step, ring-closing olefin metathesis (RCM) was the linking step, and ester saponification was the cutting step. A potential problem with this approach was that the macrocycles were highly flexible and thus they might adopt a closed conformation hindering the necessary threading step. The flexible tails made the compounds soluble, but also appeared to hinder the formation of crystals suitable for X-ray diffraction to prove the topology of the product.
The second-generation TLC approach to trefoil knots is presented here. It involves the binding of an electron-poor guest (2 or 3) in an electron-rich host (1) to promote the threading step, an alkyne–azide click cycloaddition as the linking step, and ester saponification as the cutting step [13,21] (Supporting Information File 1). The target trefoil knot using host 1 and guest 2 is shown in Figure 2c. The binding event during the double-threading step was modeled after previous literature precedents from the Stoddart [6,24,25], Gibson [26], Loeb [27], and Sanders [28] groups. Furthermore, host 1 was designed to be rigid so that the two macrocycles would maintain an open conformation which is required for the threading step. The approach is modular, such that one host can be paired with multiple guests in order to systematically explore the lower size limits of trefoil knots. For example, if TLC were successful with host 1 and guest 2, then a 73 backbone-atom trefoil (and the corresponding unknotted macrocycle) would be formed (Figure 3 and Scheme S1 in Supporting Information File 1), whereas host 1 and guest 3 would lead to a 75 backbone-atom trefoil (and unknotted macrocycle).
![[1860-5397-16-192-3]](/bjoc/content/figures/1860-5397-16-192-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Schematic representation of the second-generation TLC approach to a 73 backbone atom trefoil knot.
Figure 3: Schematic representation of the second-generation TLC approach to a 73 backbone atom trefoil knot.
Results and Discussion
The synthesis of bis-macrocyclic host 1 began by breaking the symmetry of naphthalene-1,5-diol (4) by alkylation of one of the alcohols with 2-azidoethyl mesylate to yield azide 5 in 27% yield (Scheme 1). Alkylation of 5 with 1,2-dibromoethane provided key intermediate azido-bromide 6 in 60% yield. This two-step route to 6 is efficient, but the 16% overall yield was lower than desired. An alternate route began by converting diol 4 to bis(2-hydroxyethoxy)naphthalene 7 in 92% yield by reaction with ethylene carbonate using a modified literature procedure (see Supporting Information File 1). Conversion of 7 to bismesylate 8 proceeded smoothly in 92% yield under standard conditions. The symmetry-breaking step in this route involved treatment of 8 with one equivalent of sodium azide in DMSO to give azide-mesylate 9 in 35–46% yield, which is reasonable based on a maximum statistical yield of 50%. Displacement of the mesylate with bromide provided a 94% yield of 6. This route to 6 is twice as long as the alternative described above, but it is preferred because the 37% overall yield is more than twice as high and this route is more amenable to multigram scale reactions.
![[1860-5397-16-192-i1]](/bjoc/content/inline/1860-5397-16-192-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Azido-bromide 6 can undergo both alkyne–azide click cycloaddition and etherification and the effect of the reaction order on the overall yield was explored next. The click cycloaddition was pursued first and reaction of 6 with an excess of known (see Supporting Information File 1) dialkyne 10 under several of the most common conditions produced triazole 11 in only modest yields (Scheme 2). The best conditions involved using Cu(MeCN)4PF6 as the copper(I) source, tris(2-benzimidazolylmethyl)amine as a ligand, and ascorbic acid to provide a 42% yield of triazole 11. Alkylation of diethyl 2,5-dihydroxyterephthalate (12) with 11 under standard conditions provided low yields (12–18%) of the core diester 13, which contains all the atoms of host 1. The yields for this route were disappointingly low, so it was hoped that changing the order of these steps would be beneficial.
![[1860-5397-16-192-i2]](/bjoc/content/inline/1860-5397-16-192-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Initial route to core diester 13. aLigand = tris(2-benzimidazolylmethyl)amine.
Scheme 2: Initial route to core diester 13. aLigand = tris(2-benzimidazolylmethyl)amine.
Alkylation of terephthalate 12 with azido-bromide 6 using cesium carbonate as the base provided high yields (82%) of core diazide 14 (Scheme 3). Click cycloaddition of 14 with an excess of dialkyne 10 also proceeded in high yields (87%) to give diester 13 which was identical to the material made by the route outlined in Scheme 2. The new route from 6 to 13 proved superior, as the yield for the two-steps improved dramatically from <8% to 71%.
![[1860-5397-16-192-i3]](/bjoc/content/inline/1860-5397-16-192-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Better yielding route to core diester 13. aLigand = tris(2-benzimidazolylmethyl)amine.
Scheme 3: Better yielding route to core diester 13. aLigand = tris(2-benzimidazolylmethyl)amine.
Saponification of diester 13 to diacid 15 was achieved in moderate-to-good yields (67–90%) and the 1H NMR spectrum showed the loss of the ethyl ester peaks (Scheme 4). Bis-macrocyclization of 15 under high-dilution using Shiina’s mixed-anhydride method [29] afforded host 1 in 28% yield. As with the analogous first-generation knot-precursor bis-macrocycle [15], host 1 was formed as a mixture of meta- and ortho-isomers. It is unknown which of the isomers is the major product, but the isomer ratio is approximately 2:1 based on NMR integrations of several aromatic peaks such as naphthlene signals at 6.7 (major)/6.8 ppm (minor) and the phenyl signals at 6.5 (major)/6.6 ppm (minor) and 6.4 (major)/6.3 ppm (minor). This is similar to the first-generation bis-macrocycle which had a 64:36 isomer ratio [15]. The macrocycles in host 1 are rigid by design (vide supra); however, this rigidity appears to also decrease their solubility. The 1H NMR spectrum was obtained in a dilute DMSO-d6 solution and, as expected, several of the aromatic peaks have shifted upfield relative to diacid 15, because of shielding from nearby aromatic rings. The high-resolution mass spectrometric data and IR spectra also support the successful synthesis of 1.
![[1860-5397-16-192-i4]](/bjoc/content/inline/1860-5397-16-192-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Saponification of 13 and bis-macrocyclization to form host 1.
Scheme 4: Saponification of 13 and bis-macrocyclization to form host 1.
The synthesis of bis(ammonium) guest 2 began with selective displacement of the bromide in 1-bromo-4-chlorobutane (16) with sodium azide to give azide 17 in 52% yield (Scheme 5). Treatment of 17 with potassium phthalimide and catalytic potassium iodide was followed by hydrazine unmasking to give the desired aminoazide 18 in 37% over the two steps. Reductive amination of 18 with dial 19 provided diazide 20, albeit in low yield (14%).
![[1860-5397-16-192-i5]](/bjoc/content/inline/1860-5397-16-192-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 23 backbone-atom bis(ammonium) guest 2.
Scheme 5: Synthesis of 23 backbone-atom bis(ammonium) guest 2.
Protonation of 20 with HCl followed by anion exchange yielded 72% of the hexafluorophosphate salt of guest 2. The 1H NMR spectrum supports the ammonium ions of 2 as all peaks shifted downfield relative to the chemical shifts in neutral diamine 20. As expected, the methylene groups closest to the nitrogen atom shifted the most (>0.8 ppm for each). The IR spectrum also supports the structure of 2, as strong peaks for both an azide asymmetric stretch (ν 2099 cm−1) and a PF6− stretching vibration (ν 845 cm−1) were observed.
The synthesis of bis(pyridinium) guest 3 started with the conversion of alcohol 21 into the corresponding mesylate followed by substitution with azide to give azidopyridine 22 in 82% yield (Scheme 6). Reaction of 22 with known [18] dibromide 23, followed by anion exchange, yielded 21% of the hexafluorophosphate salt of guest 3. The 1H NMR spectrum supports the pyridinium structure of 3 as the aromatic peaks shifted downfield by 0.6–1.4 ppm relative to the chemical shifts in pyridine 22. The IR spectrum supports the structure of 3 as strong peaks for both an azide asymmetric stretch (ν 2102 cm−1) and a PF6− stretching vibration (ν 835 cm−1) were observed.
![[1860-5397-16-192-i6]](/bjoc/content/inline/1860-5397-16-192-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of 25 backbone-atom bis(pyridinium) guest 3.
Scheme 6: Synthesis of 25 backbone-atom bis(pyridinium) guest 3.
To test the threading step, a 1H NMR experiment was conducted. A mixture of 1 and 2 was dissolved in a 1:1 acetone-d6/DMSO-d6 solution. The spectrum of this solution showed upfield shifts of 0.1 to 0.4 ppm for the methylene protons on the guest and smaller downfield shifts of 0.02 to 0.09 ppm for some aromatic resonances on the host. These shifts suggest at least some host–guest complex was formed, even in this competitive polar solvent mixture. The full TLC sequence was tested by reacting 1 and 2 (1:1 ratio) in a solvent mixture of acetonitrile/dichloromethane/methanol (1 mL/1 mL/0.1 mL) with [(CH3CN)4Cu]PF6 and ascorbic acid (see Scheme S2 in Supporting Information File 1). After removal of the solvent, the cutting step was performed by heating the crude product with KOH in a water/THF/ethanol mixture to saponify the esters. The pH of the mixture was adjusted to neutral by adding HCl and the resulting precipitate was collected. The IR spectrum of the crude reaction product no longer contained an azide peak, consistent with a successful click cycloaddition. The peaks in the 1H NMR spectrum in DMSO-d6 solution at room temperature (≈24 °C) were broad, whereas the spectrum at elevated temperature (e.g., 45, 90, 105 °C) had sharper peaks. This effect of temperature on the spectrum is consistent with restricted dynamics of the backbone such as reputation [30] or slow exchange between different conformers; however, both the trefoil knot and the macrocyclic unknot could exhibit this behavior. Two separate triazole peaks were observed (8.19 and 8.38 ppm), consistent with a new triazole ring being formed. The mass spectrum of the product showed peaks for the expected mass of the TLC product(s) and various cation adducts: (peak mass, assignment, relative intensity): 1563.9 Da, [M + H]+, 88; 1584.9 Da, [M + Na]+, 28; 1601.9 Da, [M + K]+, 52; 1626.8 Da, [M + Cu]+, 42. It is not surprising that the product would sequester metal cations given its electron-rich macrocyclic nature. Of course, the mass spectral data do not address whether the unknot, trefoil knot, or both were formed (see Scheme S1 in Supporting Information File 1). Repeated attempts to grow X-ray crystals of the product failed. To test for topological chirality, a europium chiral shift reagent was added, but the 1H NMR spectrum was too noisy to discern any peak doubling. Additional experiments, such as the TLC sequence with guest 3, were not possible because of a lack of material. Future work will involve making a more soluble host so that NMR and chiral chromatography experiments can be conducted.
Conclusion
The design of a second-generation thread–link–cut (TLC) approach to molecular knots was described. This differed from the first-generation TLC approach [15] in that the threading event was bimolecular, involving rigid electron-rich bis-macrocyclic host 1 and an electron-poor guest molecule (2 or 3), rather than unimolecular threading involving solvophobic forces. The flexible and modular approach involving several guests was designed to test the lower size limits on trefoil molecular knots, as the proposed TLC sequence can produce trefoil knots with 73 or 75 backbone atoms. Several synthetic routes to bis-macrocyclic host 1 were explored and the optimized route required eight steps and proceeded with a 6.6% overall yield. Bis(ammonium) guest 2 was synthesized in five steps with a 1.9% overall yield and bis(pyridinium) guest 3 was prepared in four steps and 17% overall yield. The TLC sequence using host 1 and guest 2 produced a product with the expected molecular weight, but the data were inconclusive on whether this was the unknot, trefoil knot, or a mixture of both.
Supporting Information
| Supporting Information File 1: Conformations of host 1, TLC knot-forming scheme, experimental procedures and copies of 1H NMR spectra. | ||
| Format: PDF | Size: 4.1 MB | Download |
Acknowledgements
We thank Beth Buckwalter for the acquisition of the NMR spectra and Lisa Mertzman for her valuable assistance. We gratefully acknowledge helpful discussions with Sarah Tasker, Scott Van Arman, Dustin Covell, and Scott Brewer. We also thank the peer-review referees for important additions and corrections.
References
-
Pedersen, C. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 1021–1027. doi:10.1002/anie.198810211
Return to citation in text: [1] [2] -
Lehn, J.-M. Angew. Chem., Int. Ed. Engl. 1988, 27, 89–112. doi:10.1002/anie.198800891
Return to citation in text: [1] [2] -
Cram, D. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 1009–1020. doi:10.1002/anie.198810093
Return to citation in text: [1] [2] -
Barnes, J. C.; Mirkin, C. A. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 620–625. doi:10.1073/pnas.1619330114
Return to citation in text: [1] [2] -
Sauvage, J.-P. Angew. Chem., Int. Ed. 2017, 56, 11080–11093. doi:10.1002/anie.201702992
Return to citation in text: [1] [2] -
Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216
Return to citation in text: [1] [2] [3] -
Segawa, Y.; Kuwayama, M.; Hijikata, Y.; Fushimi, M.; Nishihara, T.; Pirillo, J.; Shirasaki, J.; Kubota, N.; Itami, K. Science 2019, 365, 272–276. doi:10.1126/science.aav5021
Return to citation in text: [1] -
Gil-Ramírez, G.; Hoekman, S.; Kitching, M. O.; Leigh, D. A.; Vitorica-Yrezabal, I. J.; Zhang, G. J. Am. Chem. Soc. 2016, 138, 13159–13162. doi:10.1021/jacs.6b08421
Return to citation in text: [1] -
Danon, J. J.; Krüger, A.; Leigh, D. A.; Lemonnier, J.-F.; Stephens, A. J.; Vitorica-Yrezabal, I. J.; Woltering, S. L. Science 2017, 355, 159–162. doi:10.1126/science.aal1619
Return to citation in text: [1] [2] -
Kim, D. H.; Singh, N.; Oh, J.; Kim, E.-H.; Jung, J.; Kim, H.; Chi, K.-W. Angew. Chem., Int. Ed. 2018, 57, 5669–5673. doi:10.1002/anie.201800638
Return to citation in text: [1] [2] -
Leigh, D. A.; Lemonnier, J.-F.; Woltering, S. L. Angew. Chem., Int. Ed. 2018, 57, 12212–12214. doi:10.1002/anie.201804904
Return to citation in text: [1] -
Zhang, L.; Stephens, A. J.; Nussbaumer, A. L.; Lemonnier, J.-F.; Jurček, P.; Vitorica-Yrezabal, I. J.; Leigh, D. A. Nat. Chem. 2018, 10, 1083–1088. doi:10.1038/s41557-018-0124-6
Return to citation in text: [1] -
Fenlon, E. E. Eur. J. Org. Chem. 2008, 5023–5035. doi:10.1002/ejoc.200800578
Return to citation in text: [1] [2] [3] [4] [5] -
Ditrich-Buchecker, C. O.; Nierengarten, J.-F.; Sauvage, J.-P. Tetrahedron Lett. 1992, 33, 3625–3628. doi:10.1016/s0040-4039(00)92519-x
Return to citation in text: [1] -
Guo, J.; Mayers, P. C.; Breault, G. A.; Hunter, C. A. Nat. Chem. 2010, 2, 218–222. doi:10.1038/nchem.544
Return to citation in text: [1] [2] [3] [4] -
Barran, P. E.; Cole, H. L.; Goldup, S. M.; Leigh, D. A.; McGonigal, P. R.; Symes, M. D.; Wu, J.; Zengerle, M. Angew. Chem., Int. Ed. 2011, 50, 12280–12284. doi:10.1002/anie.201105012
Return to citation in text: [1] -
Walba, D. M. Tetrahedron 1985, 41, 3161–3212. doi:10.1016/s0040-4020(01)96671-2
Return to citation in text: [1] -
Adams, C. C. The Knot Book; W.H. Freeman and Company: New York, NY, USA, 1994.
Return to citation in text: [1] [2] -
Lukin, O.; Vögtle, F. Angew. Chem., Int. Ed. 2005, 44, 1456–1477. doi:10.1002/anie.200460312
Return to citation in text: [1] -
Fielden, S. D. P.; Leigh, D. A.; Woltering, S. L. Angew. Chem., Int. Ed. 2017, 56, 11166–11194. doi:10.1002/anie.201702531
Return to citation in text: [1] -
Fenlon, E. E.; Ito, B. R. Eur. J. Org. Chem. 2008, 3065–3068. doi:10.1002/ejoc.200800387
Return to citation in text: [1] [2] -
Ponnuswamy, N.; Cougnon, F. B. L.; Clough, J. M.; Pantoş, G. D.; Sanders, J. K. M. Science 2012, 338, 783–785. doi:10.1126/science.1227032
Return to citation in text: [1] -
Siegel, J. S. Science 2012, 338, 752–753. doi:10.1126/science.1230319
Return to citation in text: [1] -
Chang, T.; Heiss, A. M.; Cantrill, S. J.; Fyfe, M. C. T.; Pease, A. R.; Rowan, S. J.; Stoddart, J. F.; White, A. J. P.; Williams, D. J. Org. Lett. 2000, 2, 2947–2950. doi:10.1021/ol0061889
Return to citation in text: [1] -
Dichtel, W. R.; Miljanić, O. Š.; Spruell, J. M.; Heath, J. R.; Stoddart, J. F. J. Am. Chem. Soc. 2006, 128, 10388–10390. doi:10.1021/ja063127i
Return to citation in text: [1] -
Bryant, W. S.; Guzei, I. A.; Rheingold, A. L.; Gibson, H. W. Org. Lett. 1999, 1, 47–50. doi:10.1021/ol9905708
Return to citation in text: [1] -
Loeb, S. J.; Tiburcio, J.; Vella, S. J.; Wisner, J. A. Org. Biomol. Chem. 2006, 4, 667–680. doi:10.1039/b514528g
Return to citation in text: [1] -
Hamilton, D. G.; Prodi, L.; Feeder, N.; Sanders, J. K. M. J. Chem. Soc., Perkin Trans. 1 1999, 1057–1066. doi:10.1039/a809816f
Return to citation in text: [1] -
Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. J. Org. Chem. 2004, 69, 1822–1830. doi:10.1021/jo030367x
Return to citation in text: [1] -
de Gennes, P.-G. Macromolecules 1984, 17, 703–704. doi:10.1021/ma00134a030
Return to citation in text: [1]
| 15. | Guo, J.; Mayers, P. C.; Breault, G. A.; Hunter, C. A. Nat. Chem. 2010, 2, 218–222. doi:10.1038/nchem.544 |
| 15. | Guo, J.; Mayers, P. C.; Breault, G. A.; Hunter, C. A. Nat. Chem. 2010, 2, 218–222. doi:10.1038/nchem.544 |
| 18. | Adams, C. C. The Knot Book; W.H. Freeman and Company: New York, NY, USA, 1994. |
| 1. | Pedersen, C. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 1021–1027. doi:10.1002/anie.198810211 |
| 2. | Lehn, J.-M. Angew. Chem., Int. Ed. Engl. 1988, 27, 89–112. doi:10.1002/anie.198800891 |
| 3. | Cram, D. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 1009–1020. doi:10.1002/anie.198810093 |
| 4. | Barnes, J. C.; Mirkin, C. A. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 620–625. doi:10.1073/pnas.1619330114 |
| 5. | Sauvage, J.-P. Angew. Chem., Int. Ed. 2017, 56, 11080–11093. doi:10.1002/anie.201702992 |
| 6. | Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216 |
| 4. | Barnes, J. C.; Mirkin, C. A. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 620–625. doi:10.1073/pnas.1619330114 |
| 15. | Guo, J.; Mayers, P. C.; Breault, G. A.; Hunter, C. A. Nat. Chem. 2010, 2, 218–222. doi:10.1038/nchem.544 |
| 3. | Cram, D. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 1009–1020. doi:10.1002/anie.198810093 |
| 16. | Barran, P. E.; Cole, H. L.; Goldup, S. M.; Leigh, D. A.; McGonigal, P. R.; Symes, M. D.; Wu, J.; Zengerle, M. Angew. Chem., Int. Ed. 2011, 50, 12280–12284. doi:10.1002/anie.201105012 |
| 2. | Lehn, J.-M. Angew. Chem., Int. Ed. Engl. 1988, 27, 89–112. doi:10.1002/anie.198800891 |
| 1. | Pedersen, C. J. Angew. Chem., Int. Ed. Engl. 1988, 27, 1021–1027. doi:10.1002/anie.198810211 |
| 14. | Ditrich-Buchecker, C. O.; Nierengarten, J.-F.; Sauvage, J.-P. Tetrahedron Lett. 1992, 33, 3625–3628. doi:10.1016/s0040-4039(00)92519-x |
| 8. | Gil-Ramírez, G.; Hoekman, S.; Kitching, M. O.; Leigh, D. A.; Vitorica-Yrezabal, I. J.; Zhang, G. J. Am. Chem. Soc. 2016, 138, 13159–13162. doi:10.1021/jacs.6b08421 |
| 12. | Zhang, L.; Stephens, A. J.; Nussbaumer, A. L.; Lemonnier, J.-F.; Jurček, P.; Vitorica-Yrezabal, I. J.; Leigh, D. A. Nat. Chem. 2018, 10, 1083–1088. doi:10.1038/s41557-018-0124-6 |
| 7. | Segawa, Y.; Kuwayama, M.; Hijikata, Y.; Fushimi, M.; Nishihara, T.; Pirillo, J.; Shirasaki, J.; Kubota, N.; Itami, K. Science 2019, 365, 272–276. doi:10.1126/science.aav5021 |
| 6. | Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216 |
| 5. | Sauvage, J.-P. Angew. Chem., Int. Ed. 2017, 56, 11080–11093. doi:10.1002/anie.201702992 |
| 9. | Danon, J. J.; Krüger, A.; Leigh, D. A.; Lemonnier, J.-F.; Stephens, A. J.; Vitorica-Yrezabal, I. J.; Woltering, S. L. Science 2017, 355, 159–162. doi:10.1126/science.aal1619 |
| 10. | Kim, D. H.; Singh, N.; Oh, J.; Kim, E.-H.; Jung, J.; Kim, H.; Chi, K.-W. Angew. Chem., Int. Ed. 2018, 57, 5669–5673. doi:10.1002/anie.201800638 |
| 11. | Leigh, D. A.; Lemonnier, J.-F.; Woltering, S. L. Angew. Chem., Int. Ed. 2018, 57, 12212–12214. doi:10.1002/anie.201804904 |
| 15. | Guo, J.; Mayers, P. C.; Breault, G. A.; Hunter, C. A. Nat. Chem. 2010, 2, 218–222. doi:10.1038/nchem.544 |
| 10. | Kim, D. H.; Singh, N.; Oh, J.; Kim, E.-H.; Jung, J.; Kim, H.; Chi, K.-W. Angew. Chem., Int. Ed. 2018, 57, 5669–5673. doi:10.1002/anie.201800638 |
| 13. | Fenlon, E. E. Eur. J. Org. Chem. 2008, 5023–5035. doi:10.1002/ejoc.200800578 |
| 17. | Walba, D. M. Tetrahedron 1985, 41, 3161–3212. doi:10.1016/s0040-4020(01)96671-2 |
| 18. | Adams, C. C. The Knot Book; W.H. Freeman and Company: New York, NY, USA, 1994. |
| 19. | Lukin, O.; Vögtle, F. Angew. Chem., Int. Ed. 2005, 44, 1456–1477. doi:10.1002/anie.200460312 |
| 20. | Fielden, S. D. P.; Leigh, D. A.; Woltering, S. L. Angew. Chem., Int. Ed. 2017, 56, 11166–11194. doi:10.1002/anie.201702531 |
| 9. | Danon, J. J.; Krüger, A.; Leigh, D. A.; Lemonnier, J.-F.; Stephens, A. J.; Vitorica-Yrezabal, I. J.; Woltering, S. L. Science 2017, 355, 159–162. doi:10.1126/science.aal1619 |
| 28. | Hamilton, D. G.; Prodi, L.; Feeder, N.; Sanders, J. K. M. J. Chem. Soc., Perkin Trans. 1 1999, 1057–1066. doi:10.1039/a809816f |
| 29. | Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. J. Org. Chem. 2004, 69, 1822–1830. doi:10.1021/jo030367x |
| 26. | Bryant, W. S.; Guzei, I. A.; Rheingold, A. L.; Gibson, H. W. Org. Lett. 1999, 1, 47–50. doi:10.1021/ol9905708 |
| 27. | Loeb, S. J.; Tiburcio, J.; Vella, S. J.; Wisner, J. A. Org. Biomol. Chem. 2006, 4, 667–680. doi:10.1039/b514528g |
| 13. | Fenlon, E. E. Eur. J. Org. Chem. 2008, 5023–5035. doi:10.1002/ejoc.200800578 |
| 21. | Fenlon, E. E.; Ito, B. R. Eur. J. Org. Chem. 2008, 3065–3068. doi:10.1002/ejoc.200800387 |
| 6. | Stoddart, J. F. Angew. Chem., Int. Ed. 2017, 56, 11094–11125. doi:10.1002/anie.201703216 |
| 24. | Chang, T.; Heiss, A. M.; Cantrill, S. J.; Fyfe, M. C. T.; Pease, A. R.; Rowan, S. J.; Stoddart, J. F.; White, A. J. P.; Williams, D. J. Org. Lett. 2000, 2, 2947–2950. doi:10.1021/ol0061889 |
| 25. | Dichtel, W. R.; Miljanić, O. Š.; Spruell, J. M.; Heath, J. R.; Stoddart, J. F. J. Am. Chem. Soc. 2006, 128, 10388–10390. doi:10.1021/ja063127i |
| 13. | Fenlon, E. E. Eur. J. Org. Chem. 2008, 5023–5035. doi:10.1002/ejoc.200800578 |
| 21. | Fenlon, E. E.; Ito, B. R. Eur. J. Org. Chem. 2008, 3065–3068. doi:10.1002/ejoc.200800387 |
| 22. | Ponnuswamy, N.; Cougnon, F. B. L.; Clough, J. M.; Pantoş, G. D.; Sanders, J. K. M. Science 2012, 338, 783–785. doi:10.1126/science.1227032 |
| 23. | Siegel, J. S. Science 2012, 338, 752–753. doi:10.1126/science.1230319 |
© 2020 Margolis et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)