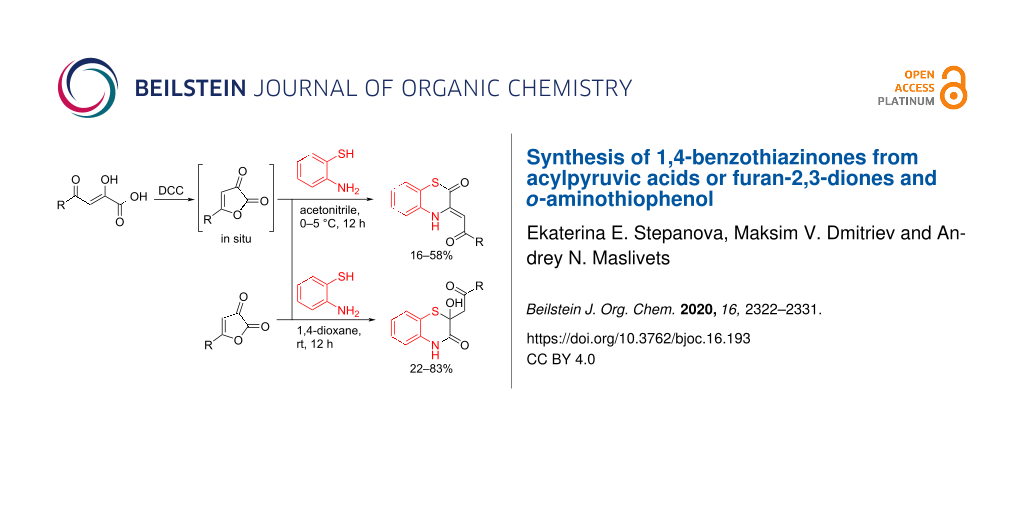
Abstract
Two synthetic approaches to enaminones fused to 1,4-benzothiazin-2-one moiety, which can be interesting in studies on biological activity, chemosensors, and fluorescence, were developed via the reaction of furan-2,3-diones or acylpyruvic acids in the presence of carbodiimides with o-aminothiophenols. The target enaminones were formed together with pharmaceutically interesting 2-hydroxy-2H-1,4-benzothiazin-3(4H)-ones. A selective synthetic approach to 2-hydroxy-2H-1,4-benzothiazin-3(4H)-ones was developed via the solvent-switchable reaction of furan-2,3-diones with o-aminothiophenol. Preliminary biological assays (antimicrobial, acute toxicity) of the new compounds were carried out.

Graphical Abstract
Introduction
Enaminones fused to the 1,4-benzoxazine-2-one I or quinoxaline-2(1H)-one II moiety (Figure 1) represent an intensively investigated class of enamines. Undying interest in these compounds is due to the simplicity of their synthesis and purification, the availability of starting materials and the possibility of their synthesis under mild green conditions [1-6]. Because of their availability in gram-scale quantities, these compounds were thoroughly investigated for the possibility of practical use. Particularly, some enaminones I and II were found to show antioxidant [3], antimycotic [7,8], antimycobacterial [9,10], anti-Alzheimer’s disease (JNK3 inhibitors) [11], platelet aggregation inhibitory [12], antimicrobial [7] and analgesic [13] activities. Enaminones II were reported as potential Cu2+ chemosensors [14], and their BF2 chelates, as multicolor fluorescence complexes, some of which exhibited aggregation-induced emission (AIE) properties [15]. In addition, enaminones I and II proved themselves to be versatile and available building blocks for the synthesis of various heterocyclic systems [10,16-20].
![[1860-5397-16-193-1]](/bjoc/content/figures/1860-5397-16-193-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Enaminones fused to heterocyclic moieties.
Figure 1: Enaminones fused to heterocyclic moieties.
In light of the unquestionable practical value of the above-mentioned 1-oxa- and 1-aza-substituted enaminones I and II, investigations on the properties of their 1-thia analogs, the 1,4-benzothiazin-2-ones (BTAs) III, (Figure 1) seem to be promising. But according to the literature, data on BTAs III have been poorly reported. For example, the only available report [9] on their antimycobacterial activity gives erroneous information, since the structure A of the assayed compound has been established incorrectly (Scheme 1). We reproduced the synthesis from the work [9] and found that the correct structure of the studied compound was structure B (CCDC 2019497) (Scheme 1).
![[1860-5397-16-193-i1]](/bjoc/content/inline/1860-5397-16-193-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reported structure A of assayed compound [9] and its correct structure B.
Scheme 1: Reported structure A of assayed compound [9] and its correct structure B.
In fact, for now, four approaches to BTAs III are known [21-25] (Scheme 2). They include the reaction of dimethyl acetylenedicarboxylate (DMAD) with o-aminothiophenol (1a) on the catalyst SiO2@H3PW12O40 [21], the reaction of tetracarbonyl compounds with o-aminothiophenol (1a) [22,23], the reaction of copper(II) chelate of ethyl pentafluorobenzoylpyruvate with o-aminothiophenol (1a, one example) [24] and the reaction of DMAD with 6-nitro-1,3-benzothiazole (one example) [25]. Our recent research [26] revealed that the approach based on the reaction of tetracarbonyl compounds with o-aminothiophenol (1a) [22,23] is a mistaken one. Considering these facts, it can be concluded that there are no convenient synthetic approaches with a wide substrate scope to BTAs III.
![[1860-5397-16-193-i2]](/bjoc/content/inline/1860-5397-16-193-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Known synthetic approaches to BTAs III.
Scheme 2: Known synthetic approaches to BTAs III.
Herein, we report a comprehensive research on synthetic approaches to BTAs III.
Results and Discussion
To develop an approach to target the BTAs III, we analyzed the general synthetic methods to enaminones I and II, which involve the reaction of the acylpyruvic acids or their esters IV, 5-arylfuran-2,3-diones V, and acetylenedicarboxylates VI with o-aminophenols or o-phenylenediamines (Scheme 3) [1-6]. Since the approach to alkoxy-substituted BTAs III (Figure 1, R = OAlk) has already been reported (Scheme 2) [21], we focused our research on aryl/alkyl-substituted BTAs III (Figure 1, R = Ar, Alk).
![[1860-5397-16-193-i3]](/bjoc/content/inline/1860-5397-16-193-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: General synthetic approaches to enaminones I and II.
Scheme 3: General synthetic approaches to enaminones I and II.
By analogy with the synthesis of enaminones I and II, it can be assumed that the target aryl/alkyl-substituted BTAs III can be accessible via the reaction of acylpyruvic acids or their esters IV and 5-arylfuran-2,3-diones V with o-aminothiophenol (1a). But in fact, this assumption is not correct.
The approach based on the reaction of acylpyruvic acids or their esters IV with o-aminothiophenol (1a) can be supposed as the simplest and expedite, but it was reported to afford other types of products VII–IX (Scheme 4) [27,28] instead of the desired BTAs III.
![[1860-5397-16-193-i4]](/bjoc/content/inline/1860-5397-16-193-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reported reactions of acylpyruvic acids or their esters IV with o-aminothiophenol (1a) [27,28].
Scheme 4: Reported reactions of acylpyruvic acids or their esters IV with o-aminothiophenol (1a) [27,28].
To verify the unsuitability of this approach, we repeated some experiments described previously [27,28] and examined the reaction mixtures by UPLC–UV–MS. We supposed that the peak of the target BTA III would be characterized by appropriate molecular ion signals at ESI+ and ESI− chromatograms and absorbance bands at 400–450 nm (representative for enaminones I and II [28]) at UV chromatograms. The examined reaction mixtures showed peaks characteristic for adducts VII–IX, and no peaks characteristic for target BTAs III were observed, which was in good agreement with previous studies [27,28].
The inability of the reaction of acylpyruvic acids or their esters IV with o-aminothiophenol (1a) to afford the target BTAs III can be explained both by the impossibility of thioesterification/thiotransesterification to proceed under additives-free conditions and by the high nucleophilicity of the o-aminothiophenol’s SH group that attacked the more electrophilic ketone carbonyls of compounds IV firstly. To overcome these issues, we examined the reaction of acylpyruvic acid (2a) with o-aminothiophenol (1a) in the presence of carbodiimides in various solvents (Table 1). Fortunately, in many cases we succeeded to detect the desired BTA 3a by UPLC–UV–MS. The target product 3a was formed along with the concurrent product 4a (Table 1). Both products 3a and 4a were isolated and characterized, and their structures were unequivocally proved by single crystal X-ray analyses (3a, CCDC 2019498; 4a, CCDC 2019501).
Table 1: Reaction of acylpyruvic acid 2a with o-aminothiophenol (1a) in the presence of carbodiimides in various solvents.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-193-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Carbodiimidea | Procedureb | Yieldc, % | |
| 3a | 4a | ||||
| 1 | toluene | DCC | A | 35 | 23 |
| 2 | toluene | DCC | B | 21 | 14 |
| 3 | toluene | DIC | A | 33 | 21 |
| 4 | toluene | DIC | B | 18 | 16 |
| 5 | DMSO | DCC | A | traces | 18 |
| 6 | DMSO | DCC | B | traces | 44 |
| 7 | 1,4-dioxane | DCC | A | 25 | 24 |
| 8 | 1,4-dioxane | DCC | B | 22 | 27 |
| 9 | acetonitrile | DCC | A | 18 | 8 |
| 10 | acetonitrile | DCC | B | 56 | 34 |
| 11 | chloroform | DCC | A | traces | 20 |
| 12 | chloroform | DCC | B | traces | 27 |
| 13 | DMF | DCC | A | 7 | 35 |
| 14 | DMF | DCC | B | 18 | 54 |
| 15 | ethyl acetate | DCC | A | 25 | 21 |
| 16 | ethyl acetate | DCC | B | 30 | 35 |
| 17 | tetrahydrofuran | DCC | A | 31 | 16 |
| 18 | tetrahydrofuran | DCC | B | 25 | 21 |
| 19 | hexane | DCC | A | traces | traces |
| 20 | hexane | DCC | B | traces | traces |
| 21 | acetone | DCC | A | 28 | 18 |
| 22 | acetone | DCC | B | 33 | 20 |
| 23 | N-methyl-2-pyrrolidone | DCC | A | 3 | 35 |
| 24 | N-methyl-2-pyrrolidone | DCC | B | 11 | 54 |
| 25 | acetonitrile | DCC | C | 52 | 30 |
| 26 | acetonitrile | DIC | C | 53 | 33 |
| 27 | 1,4-dioxane | DCC | C | 19 | 46 |
| 28 | DMF | DCC | C | 34 | 24 |
| 29 | acetonitrile | DCC | D | – | traces |
| 30 | acetonitrile | EDC | B | – | – |
aDCC – dicyclohexylcarbodiimide, DIC – diisopropylcarbodiimide, EDC – 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide. bProcedure A: to a stirring suspension of compound 2a (10 mg, 52 µmol) and o-aminothiophenol (1a, 7 µL, 65 µmol) in a solvent (100 µL), a carbodiimide (52 µmol) was added; procedure B: to a stirring suspension of compound 2a (10 mg, 52 µmol) and a carbodiimide (52 µmol) in a solvent (100 µL), o-aminothiophenol (1a, 7 µL, 65 µmol) was added; procedure C: to a stirring suspension of compound 2a (10 mg, 52 µmol) in a solvent (100 µL), a carbodiimide (52 µmol) was added, and right after, N-hydroxybenzotriazole (HOBt) hydrate (8 mg, 52 µmol) was added, and after 5 min, o-aminothiophenol (1a, 7 µL, 65 µmol) was added to the reaction mixture; procedure D: to a stirring suspension of compound 2a (10 mg, 52 µmol) in a solvent (100 µL), a carbodiimide (52 µmol) and 4-dimethylaminopyridine (DMAP, 6.4 mg, 52 µmol) were added, and after 5 min, o-aminothiophenol (1a, 7 µL, 65 µmol) was added to the reaction mixture. cUPLC–UV yields (biphenyl was used as an internal standard; each entry was carried out in triplicate, and the yields are given as mean values).
During the optimization (Table 1), we found that the yield of BTA 3a depended on the method of the addition of the reagents (Table 1, procedure A or B). In case of some non-polar solvents (toluene, 1,4-dioxane), the addition of o-aminothiophenol (1a) before the carbodiimide resulted in higher yields of BTA 3a, and in case of some polar solvents (acetonitrile, ethyl acetate, DMF, and N-methyl-2-pyrrolidone), addition of the carbodiimide before o-aminothiophenol (1a) afforded higher yields of BTA 3a.
Noteworthy, the solvents strongly influenced the studied reaction. When DMSO was used as a solvent (Table 1, entries 5 and 6), only trace amounts of the target compound 3a were formed, probably, because the combination of DMSO and a carbodiimide induced multiple side-processes similar to the Pfitzner–Moffatt oxidation [29]. In case of hexane as a solvent (Table 1, entries 19 and 20), only traces of compounds 3a and 4a were observed, because of the low solubility of benzoylpyruvic acid (2a) and o-aminothiophenol (1a) in hexane. To our surprise, carrying out the reaction in chloroform (Table 1, entries 11 and 12) resulted in the formation of multiple side-products and only traces of BTA 3a. When acetone was used as a solvent (Table 1, entries 21 and 22), though the target BTA 3a was formed in about 30% yield, side-products were observed, which, possibly, were formed due to the reaction of acetone with o-aminothiophenol (1a). The best yield of BTA 3a was observed in acetonitrile (Table 1, entry 10).
Then we tested the effect of common additives for carbodiimide coupling reactions, DMAP [30] and HOBt [31]. The utilization of HOBt (Table 1, entries 25 and 26) in acetonitrile as an additive did not significantly influence the yield of BTA 3a. But since the solubility of HOBt in acetonitrile is poor [32], we examined the reaction in DMF and 1,4-dioxane (Table 1, entries 27 and 28), which are known to be better solvents for HOBt [32]. Unfortunately, these attempts did not provide any significant results either.
When DMAP (Table 1, entry 29) was employed, the formation of BTA 3a was not observed at all, and the side-product 4a was formed only in trace amounts. This phenomenon can be explained by the formation of intermediates highly sensitive to amines.
We believe that the reaction of acylpyruvic acid 2a with o-aminothiophenol (1a) in the presence of carbodiimides proceeded through the formation of 5-phenylfuran-2,3-dione (5a, CCDC 2019499, Scheme 5).
![[1860-5397-16-193-i5]](/bjoc/content/inline/1860-5397-16-193-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism of the reaction of acylpyruvic acid 2a with o-aminothiophenol (1a) in the presence of carbodiimides.
Scheme 5: Plausible mechanism of the reaction of acylpyruvic acid 2a with o-aminothiophenol (1a) in the prese...
The acid 2a reacted with a carbodiimide to form the reactive isourea derivative X which underwent intramolecular cyclization with the formation of furandione 5a which reacted with o-aminothiophenol (1a). Product 3a was formed when the o-aminothiophenol’s SH group attacked the compound 5a at the lactone carbonyl group, and the product 4a, when o-aminothiophenol’s NH2 group attacked compound 5a at the lactone carbonyl group. The in situ formation of 5-phenylfuran-2,3-dione (5a) was indicated by the appearance of bright yellow color (characteristic for the 5-arylfuran-2,3-diones 5 [6]) after mixing the suspension of acid 2a with a carbodiimide. In addition, after mixing the suspension of acid 2a in acetonitrile with DIC, the formation of the yellow precipitate of 5-phenylfuran-2,3-dione (5a) was observed, and compound 5a could be isolated by simple filtration. Moreover, after mixing the suspension of acid 2a with DCC, formation of the white precipitate of dicyclohexylurea was observed, which gave the evidence of intramolecular cyclization to furandione 5a with dicyclohexylurea elimination occurring. The formation of furandione 5a explained well our observations when DMAP was used as an additive (Table 1, entry 29), since furandiones 5 are well-known to readily react with amines to afford various products [33]. To confirm our assumption, we examined the reaction of furandione 5a with DMAP. For this, equimolar amounts of compound 5a and DMAP were stirred at room temperature in acetonitrile for 1 h, and the reaction mixture was examined by UPLC–UV–MS. As a result, a complex mixture of various products was observed. Similar results were obtained upon the treatment of compound 5a with triethylamine under the same conditions. These meant that 5-arylfuran-2,3-diones 5 were sensitive to tertiary amines and should only be carefully treated with reagents containing tertiary amino groups.
Because of this, we were encouraged to examine EDC as a carbodiimide in the developed procedure (Table 1, entry 30). As expected, we found that the utilization of EDC in this reaction resulted in the formation of a complex mixture containing no target products 3a and 4a. So, EDC cannot be utilized as an activating additive in the studied reaction, while DCC and DIC could be used equally.
Taking these results into account, we examined the direct reaction of furandione 5a with o-aminothiophenol (1a) in various solvents (Table 2). As we expected, the studied process afforded compounds 3a and 4a. The best yields of BTA 3a were observed in acetonitrile (Table 2, entries 4 and 5). Notably, an increase in the reaction temperature (Table 2, entry 6) resulted in a decrease of the BTA 3a yield, which could possibly mean, that BTA 3a was the kinetic product. Unfortunately, conduction of the reaction at −40 °C (Table 2, entry 5) did not increase the BTA 3a yield.
Table 2: Reaction of furandione 5a with o-aminothiophenol (1a) in various solvents.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-193-i10.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Yieldb, % | |
| 3a | 4a | ||
| 1 | toluene | traces | 79 |
| 2 | DMSO | traces | 82 |
| 3 | 1,4-dioxane | 12 | 80 |
| 4 | acetonitrile | 41 | 55 |
| 5 | acetonitrilec | 42 | 50 |
| 6 | acetonitriled | 20 | 55 |
| 7 | chloroform | traces | 79 |
| 8 | DMF | 21 | 72 |
| 9 | ethyl acetate | 25 | 68 |
| 10 | tetrahydrofuran | 15 | 69 |
| 11 | acetic acid | 19 | 69 |
| 12 | acetone | 21 | 50 |
| 13 | N-methyl-2-pyrrolidone | 15 | 81 |
aTo a stirring suspension of furandione 5a (10 mg, 57 µmol) in solvent (100 µL), o-aminothiophenol (1a, 7 µL, 65 µmol) was added. bUPLC–UV yields (biphenyl was used as an internal standard; each entry was carried out in triplicate, and the yields are given as mean values). cThe reaction was carried out at −40 °C. dThe reaction was carried out at 70 °C.
Thus, we found two approaches to the target BTA 3a. These two approaches could be used as interchangeable in the synthesis of BTAs 3. The furandione-based approach gave less side products, but the yield of the BTA 3 was lower and, sometimes, furandiones 5 were difficult to be synthesized [2]. The acylpyruvic acid-based approach gave higher yields of the target compounds 3, involved available reagents, but produced more waste. In fact, the acylpyruvic acid-based approach could be considered as a furandione-based one with in situ generation of furandiones 5.
Since the acylpyruvic acid-based approach gave higher yields of the target compounds 3, we investigated its substrate scope by involvement of various acylpyruvic acids 2a–m and o-aminothiophenols 1a,b (Scheme 6). It should be noted, that scaled up reactions of acylpyruvic acids 2a–m and o-aminothiophenols 1a,b in the presence of DCC was found to be exothermic, therefore, cooling was applied.
![[1860-5397-16-193-i6]](/bjoc/content/inline/1860-5397-16-193-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: The substrate scope of the optimized approach to BTAs 3a–n. Procedure: to a cooled to 0–5 °C stirring suspension of compound 2 (5 mmol) and DCC (5 mmol) in acetonitrile (10 mL), o-aminothiophenol 1 (5.1 mmol) was added; Isolated yields are shown.
Scheme 6: The substrate scope of the optimized approach to BTAs 3a–n. Procedure: to a cooled to 0–5 °C stirri...
As a result, we found that the developed procedure to BTAs 3a–n worked well with a wide range of substituents on the acylpyruvic acids 2, excepting 4-nitrobenzoylpyruvic acid (2h). In case of acid 2h, target BTA 3h was formed only in trace amounts detected by UPLC–UV–MS. Our attempts to obtain BTA 3h through the alternative approach based on the reaction of furandione 5h with o-aminothiophenol (1a) were unsuccessful. In both cases, a complex mixture of inseparable products was formed. Such a change in reactivity could be explained by the influence of the NO2 substituent in compounds 2h and 5h, which had a strong negative mesomeric effect, affecting the whole conjugated bond system in these molecules.
Notably, the target compounds 3a–n were formed together with the corresponding compounds 4a–n, which can be interesting for pharmaceutics, since compounds bearing a 2-hydroxy-2H-1,4-benzothiazin-3(4H)-one moiety were reported to be potentially useful for treatment of circulatory diseases [34-36]. According to the optimization data, the best yields of compound 4a were observed at the reaction of furandione 5a with o-aminothiophenol (1a) in DMSO or 1,4-dioxane (Table 2, entries 2 and 3). In order to check the substrate scope for compounds 4a–n, we examined the reactions of various furandiones 5a–m and o-aminothiophenols 1a,b (Scheme 7).
![[1860-5397-16-193-i7]](/bjoc/content/inline/1860-5397-16-193-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The substrate scope of the optimized approach to compounds 4a–n. Procedure: to a stirring solution of compound 5 (1 mmol) in 1,4-dioxane (3 mL), o-aminothiophenol 1 (1.05 mmol) was added. Isolated yields are shown.
Scheme 7: The substrate scope of the optimized approach to compounds 4a–n. Procedure: to a stirring solution ...
The examined procedure to compounds 4a–n worked well with a wide range of substituents in furandiones 5. The involvement of 4-nitrophenylfurandione 5h in the reaction afforded only trace amounts of compound 4h possibly because of the above mentioned reasons. A similar situation was observed with tert-butylfurandione 5m. Notably, in case of the reaction of phenylfurandione 5a with chloro-substituted o-aminothiophenol 1b, diketone 6 [26] was isolated as the product instead of the expected compound 4n (Scheme 8). Diketone 6 was formed when the SH group of intermediate XI intramolecularly attacked at the amide carbonyl group (Scheme 8). Such a change in the intramolecular cyclization direction, possibly, was due to the electron-withdrawing effect of the chloro-substituent in the o-aminothiophenol moiety, which decreased the nucleophilicity of the SH group, inducing its attack on the carbonyl group bearing a higher partially positive charge (for partial charges comparison, see Supporting Information File 1).
![[1860-5397-16-193-i8]](/bjoc/content/inline/1860-5397-16-193-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Plausible scheme of the formation of diketone 6.
Scheme 8: Plausible scheme of the formation of diketone 6.
As we were most interested in the biological activity of BTAs 3, preliminary antimicrobial and toxicity assays of these compounds were carried out (detailed data on biological assays is given in Supporting Information File 1). Unfortunately, we found that tested BTAs 3 did not show any significant antimicrobial activity (against Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Candida albicans, Cryptococcus neoformans var. grubii, Mycobacterium tuberculosis) in vitro. Acute toxicity of BTA 3a in mice was determined to be higher than 1000 mg/kg, which means that BTA 3a can be considered as low toxic.
Conclusion
In conclusion, we developed two interchangeable approaches to enaminones fused to 1,4-benzothiazin-2-one moieties, which may be interesting for studies on biological activity, chemosensors, and fluorescence. The first approach is based on the reaction of furan-2,3-diones 5 with o-aminothiophenols 1 in acetonitrile. This approach gave less side products, but the yields of BTAs 3 were lower and, sometimes, starting furandiones 5 are difficult to be synthesized. The second one is based on the reaction of acylpyruvic acids 2 with o-aminothiophenols 1 in the presence of carbodiimides in acetonitrile. This approach gave higher yields of the target compounds 3, involves available precursors, but produced more waste. The acylpyruvic acid-based approach can be considered as furandione-based one with in situ generation of furandiones 5. The BTAs 3 were formed together with the corresponding 2-hydroxy-2H-1,4-benzothiazin-3(4H)-ones 4, which can be interesting for pharmaceutics. A selective synthetic approach to compounds 4 was developed based on the reaction of furan-2,3-diones 5 with o-aminothiophenols 1 in 1,4-dioxane or DMSO. According to antimicrobial assays, BTAs 3 did not show any significant antimicrobial activity in vitro. BTA 3a was found to be low toxic according to the test of acute toxicity in mice.
Supporting Information
| Supporting Information File 1: Experimental details, copies of NMR spectra, X-ray crystallographic details, detailed antimicrobial and toxicity assays, results of semi-empirical calculations of partial charges. | ||
| Format: PDF | Size: 3.8 MB | Download |
Acknowledgements
The authors thank Danila Yu. Apushkin and Aleksandr I. Andreev (PSU, Perm, Russia) for toxicity testing in vivo. The authors also thank the Ural Research Institute of Phthisiopulmonology (Yekaterinburg, Russia) and Clinical Phthisiopulmonological Medical Center (Krasnokamsk, Russia) for performing antitubercular assays in vitro.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (FSNF-2020-0008). Antimicrobial screening was performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust (UK) and The University of Queensland (Australia).
References
-
Choudhary, G.; Peddinti, R. K. Green Chem. 2011, 13, 3290–3299. doi:10.1039/c1gc15701a
Return to citation in text: [1] [2] -
Dobiaš, J.; Ondruš, M.; Addová, G.; Boháč, A. Beilstein J. Org. Chem. 2017, 13, 1350–1360. doi:10.3762/bjoc.13.132
Return to citation in text: [1] [2] [3] -
Sharma, V.; Jaiswal, P. K.; Yadav, D. K.; Saran, M.; Prikhodko, J.; Mathur, M.; Swami, A. K.; Mashevskaya, I. V.; Chaudhary, S. Acta Chim. Slov. 2017, 64, 988–1004. doi:10.17344/acsi.2017.3709
Return to citation in text: [1] [2] [3] -
Jaiswal, P. K.; Sharma, V.; Prikhodko, J.; Mashevskaya, I. V.; Chaudhary, S. Tetrahedron Lett. 2017, 58, 2077–2083. doi:10.1016/j.tetlet.2017.03.048
Return to citation in text: [1] [2] -
Petronijević, J.; Bugarčić, Z.; Bogdanović, G. A.; Stefanović, S.; Janković, N. Green Chem. 2017, 19, 707–715. doi:10.1039/c6gc02893d
Return to citation in text: [1] [2] -
Stepanova, E. E.; Lukmanova, D. N.; Kasatkina, S. O.; Dmitriev, M. V.; Maslivets, A. N. ChemistrySelect 2019, 4, 12774–12778. doi:10.1002/slct.201902900
Return to citation in text: [1] [2] [3] -
Sharma, R.; Yadav, L.; Lal, J.; Jaiswal, P. K.; Mathur, M.; Swami, A. K.; Chaudhary, S. Bioorg. Med. Chem. Lett. 2017, 27, 4393–4398. doi:10.1016/j.bmcl.2017.08.017
Return to citation in text: [1] [2] -
Mukovoz, P. P.; Slepukhin, P. A.; Danilova, E. A.; Aysuvakova, O. P.; Glinushkin, A. P. Russ. J. Gen. Chem. 2018, 88, 1363–1368. doi:10.1134/s1070363218070022
Return to citation in text: [1] -
Li, X.; Liu, N.; Zhang, H.; Knudson, S. E.; Slayden, R. A.; Tonge, P. J. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. doi:10.1016/j.bmcl.2010.08.076
Return to citation in text: [1] [2] [3] [4] -
Zampieri, D.; Mamolo, M. G.; Filingeri, J.; Fortuna, S.; De Logu, A.; Sanna, A.; Zanon, D. Bioorg. Med. Chem. Lett. 2019, 29, 2468–2474. doi:10.1016/j.bmcl.2019.07.025
Return to citation in text: [1] [2] -
Dou, X.; Huang, H.; Li, Y.; Jiang, L.; Wang, Y.; Jin, H.; Jiao, N.; Zhang, L.; Zhang, L.; Liu, Z. J. Med. Chem. 2019, 62, 6645–6664. doi:10.1021/acs.jmedchem.9b00537
Return to citation in text: [1] -
Jaiswal, P. K.; Sharma, V.; Kumar, S.; Mathur, M.; Swami, A. K.; Yadav, D. K.; Chaudhary, S. Arch. Pharm. (Weinheim, Ger.) 2018, 351, 1700349. doi:10.1002/ardp.201700349
Return to citation in text: [1] -
Kulakov, I. V.; Karbainova, A. A.; Shulgau, Z. T.; Seilkhanov, T. M.; Gatilov, Yu. V.; Fisyuk, A. S. Chem. Heterocycl. Compd. 2017, 53, 1094–1097. doi:10.1007/s10593-017-2178-6
Return to citation in text: [1] -
Korin, E.; Cohen, B.; Bai, Y.-X.; Zeng, C.-C.; Becker, J. Y. Tetrahedron 2012, 68, 7450–7455. doi:10.1016/j.tet.2012.06.071
Return to citation in text: [1] -
Yao, Q.-C.; Wu, D.-E.; Ma, R.-Z.; Xia, M. J. Organomet. Chem. 2013, 743, 1–9. doi:10.1016/j.jorganchem.2013.06.012
Return to citation in text: [1] -
Bisht, S.; Peddinti, R. K. J. Org. Chem. 2017, 82, 13617–13625. doi:10.1021/acs.joc.7b02207
Return to citation in text: [1] -
Sharma, N.; Peddinti, R. K. J. Org. Chem. 2017, 82, 9360–9366. doi:10.1021/acs.joc.7b01538
Return to citation in text: [1] -
Sharma, S.; Kumar, P.; Sharma, A.; Peddinti, R. K. Eur. J. Org. Chem. 2017, 3059–3071. doi:10.1002/ejoc.201700407
Return to citation in text: [1] -
Quintana, M.; Bilbao, A.; Comas-Barceló, J.; Bujons, J.; Triola, G. Eur. J. Med. Chem. 2019, 178, 648–666. doi:10.1016/j.ejmech.2019.05.086
Return to citation in text: [1] -
Kundu, T.; Bhattacharjee, B.; Hazra, S.; Ghosh, A. K.; Bandyopadhyay, D.; Pramanik, A. J. Med. Chem. 2019, 62, 6315–6329. doi:10.1021/acs.jmedchem.9b00717
Return to citation in text: [1] -
Samzadeh-Kermani, A. J. Sulfur Chem. 2016, 37, 692–701. doi:10.1080/17415993.2016.1187733
Return to citation in text: [1] [2] [3] -
Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Chem. Heterocycl. Compd. 2002, 38, 365–367. doi:10.1023/a:1015660009406
Return to citation in text: [1] [2] [3] -
Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Russ. J. Org. Chem. 2003, 39, 863–868. doi:10.1023/b:rujo.0000003166.58756.fa
Return to citation in text: [1] [2] [3] -
Saloutin, V. I.; Perevalov, S. G.; Chupakhin, O. N. Russ. J. Org. Chem. 2000, 36, 700–705.
Return to citation in text: [1] [2] -
McKillop, A.; Sayer, T. S. B.; Bellinger, G. C. A. J. Org. Chem. 1976, 41, 1328–1331. doi:10.1021/jo00870a007
Return to citation in text: [1] [2] -
Stepanova, E. E.; Krasokha, M. O.; Galeev, A. R.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1735–1738. doi:10.1134/s1070428018110209
Return to citation in text: [1] [2] -
Singh, Y. P.; Shanker, D.; Sharma, R. K.; Sharma, J.; Rai, A. K. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 141–148. doi:10.1080/104265090508055
Return to citation in text: [1] [2] [3] [4] -
Andreichikov, Y. S.; Tendryakova, S. P.; Nalimova, Y. A.; Voronova, L. A. Chem. Heterocycl. Compd. 1977, 13, 611–613. doi:10.1007/bf00480663
Return to citation in text: [1] [2] [3] [4] [5] -
Tidwell, T. T. Synthesis 1990, 857–870. doi:10.1055/s-1990-27036
Return to citation in text: [1] -
Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. doi:10.1002/anie.197805221
Return to citation in text: [1] -
König, W.; Geiger, R. Chem. Ber. 1970, 103, 788–798. doi:10.1002/cber.19701030319
Return to citation in text: [1] -
Chen, J.; Chen, G.; Cheng, C.; Cong, Y.; Du, C.; Zhao, H. J. Chem. Eng. Data 2017, 62, 2191–2197. doi:10.1021/acs.jced.7b00316
Return to citation in text: [1] [2] -
Igidov, N. M.; Koz'minykh, E. N.; Milyutin, A. V.; Berezina, E. S.; Shavkunova, G. A.; Yakovlev, I. B.; Shelenkova, S. A.; Kolla, V. É.; Voronina, É. V.; Koz'minykh, V. O. Pharm. Chem. J. 1996, 30, 685–689. doi:10.1007/bf02223744
Return to citation in text: [1] -
Iwao, J.; Iso, T.; Oya, M. 2-Phenyl-1,4-benzothiazin-3-one derivatives. U.S. Patent US4739050 A, April 19, 1988.
Return to citation in text: [1] -
Fujita, M.; Ito, S.; Ota, A.; Kato, N.; Yamamoto, K.; Kawashima, Y.; Yamauchi, H.; Iwao, J. J. Med. Chem. 1990, 33, 1898–1905. doi:10.1021/jm00169a011
Return to citation in text: [1] -
Matralis, A. N.; Katselou, M. G.; Nikitakis, A.; Kourounakis, A. P. J. Med. Chem. 2011, 54, 5583–5591. doi:10.1021/jm200763k
Return to citation in text: [1]
| 27. | Singh, Y. P.; Shanker, D.; Sharma, R. K.; Sharma, J.; Rai, A. K. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 141–148. doi:10.1080/104265090508055 |
| 28. | Andreichikov, Y. S.; Tendryakova, S. P.; Nalimova, Y. A.; Voronova, L. A. Chem. Heterocycl. Compd. 1977, 13, 611–613. doi:10.1007/bf00480663 |
| 30. | Neises, B.; Steglich, W. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. doi:10.1002/anie.197805221 |
| 1. | Choudhary, G.; Peddinti, R. K. Green Chem. 2011, 13, 3290–3299. doi:10.1039/c1gc15701a |
| 2. | Dobiaš, J.; Ondruš, M.; Addová, G.; Boháč, A. Beilstein J. Org. Chem. 2017, 13, 1350–1360. doi:10.3762/bjoc.13.132 |
| 3. | Sharma, V.; Jaiswal, P. K.; Yadav, D. K.; Saran, M.; Prikhodko, J.; Mathur, M.; Swami, A. K.; Mashevskaya, I. V.; Chaudhary, S. Acta Chim. Slov. 2017, 64, 988–1004. doi:10.17344/acsi.2017.3709 |
| 4. | Jaiswal, P. K.; Sharma, V.; Prikhodko, J.; Mashevskaya, I. V.; Chaudhary, S. Tetrahedron Lett. 2017, 58, 2077–2083. doi:10.1016/j.tetlet.2017.03.048 |
| 5. | Petronijević, J.; Bugarčić, Z.; Bogdanović, G. A.; Stefanović, S.; Janković, N. Green Chem. 2017, 19, 707–715. doi:10.1039/c6gc02893d |
| 6. | Stepanova, E. E.; Lukmanova, D. N.; Kasatkina, S. O.; Dmitriev, M. V.; Maslivets, A. N. ChemistrySelect 2019, 4, 12774–12778. doi:10.1002/slct.201902900 |
| 11. | Dou, X.; Huang, H.; Li, Y.; Jiang, L.; Wang, Y.; Jin, H.; Jiao, N.; Zhang, L.; Zhang, L.; Liu, Z. J. Med. Chem. 2019, 62, 6645–6664. doi:10.1021/acs.jmedchem.9b00537 |
| 21. | Samzadeh-Kermani, A. J. Sulfur Chem. 2016, 37, 692–701. doi:10.1080/17415993.2016.1187733 |
| 22. | Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Chem. Heterocycl. Compd. 2002, 38, 365–367. doi:10.1023/a:1015660009406 |
| 23. | Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Russ. J. Org. Chem. 2003, 39, 863–868. doi:10.1023/b:rujo.0000003166.58756.fa |
| 24. | Saloutin, V. I.; Perevalov, S. G.; Chupakhin, O. N. Russ. J. Org. Chem. 2000, 36, 700–705. |
| 25. | McKillop, A.; Sayer, T. S. B.; Bellinger, G. C. A. J. Org. Chem. 1976, 41, 1328–1331. doi:10.1021/jo00870a007 |
| 34. | Iwao, J.; Iso, T.; Oya, M. 2-Phenyl-1,4-benzothiazin-3-one derivatives. U.S. Patent US4739050 A, April 19, 1988. |
| 35. | Fujita, M.; Ito, S.; Ota, A.; Kato, N.; Yamamoto, K.; Kawashima, Y.; Yamauchi, H.; Iwao, J. J. Med. Chem. 1990, 33, 1898–1905. doi:10.1021/jm00169a011 |
| 36. | Matralis, A. N.; Katselou, M. G.; Nikitakis, A.; Kourounakis, A. P. J. Med. Chem. 2011, 54, 5583–5591. doi:10.1021/jm200763k |
| 9. | Li, X.; Liu, N.; Zhang, H.; Knudson, S. E.; Slayden, R. A.; Tonge, P. J. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. doi:10.1016/j.bmcl.2010.08.076 |
| 10. | Zampieri, D.; Mamolo, M. G.; Filingeri, J.; Fortuna, S.; De Logu, A.; Sanna, A.; Zanon, D. Bioorg. Med. Chem. Lett. 2019, 29, 2468–2474. doi:10.1016/j.bmcl.2019.07.025 |
| 21. | Samzadeh-Kermani, A. J. Sulfur Chem. 2016, 37, 692–701. doi:10.1080/17415993.2016.1187733 |
| 26. | Stepanova, E. E.; Krasokha, M. O.; Galeev, A. R.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1735–1738. doi:10.1134/s1070428018110209 |
| 7. | Sharma, R.; Yadav, L.; Lal, J.; Jaiswal, P. K.; Mathur, M.; Swami, A. K.; Chaudhary, S. Bioorg. Med. Chem. Lett. 2017, 27, 4393–4398. doi:10.1016/j.bmcl.2017.08.017 |
| 8. | Mukovoz, P. P.; Slepukhin, P. A.; Danilova, E. A.; Aysuvakova, O. P.; Glinushkin, A. P. Russ. J. Gen. Chem. 2018, 88, 1363–1368. doi:10.1134/s1070363218070022 |
| 9. | Li, X.; Liu, N.; Zhang, H.; Knudson, S. E.; Slayden, R. A.; Tonge, P. J. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. doi:10.1016/j.bmcl.2010.08.076 |
| 33. | Igidov, N. M.; Koz'minykh, E. N.; Milyutin, A. V.; Berezina, E. S.; Shavkunova, G. A.; Yakovlev, I. B.; Shelenkova, S. A.; Kolla, V. É.; Voronina, É. V.; Koz'minykh, V. O. Pharm. Chem. J. 1996, 30, 685–689. doi:10.1007/bf02223744 |
| 3. | Sharma, V.; Jaiswal, P. K.; Yadav, D. K.; Saran, M.; Prikhodko, J.; Mathur, M.; Swami, A. K.; Mashevskaya, I. V.; Chaudhary, S. Acta Chim. Slov. 2017, 64, 988–1004. doi:10.17344/acsi.2017.3709 |
| 9. | Li, X.; Liu, N.; Zhang, H.; Knudson, S. E.; Slayden, R. A.; Tonge, P. J. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. doi:10.1016/j.bmcl.2010.08.076 |
| 2. | Dobiaš, J.; Ondruš, M.; Addová, G.; Boháč, A. Beilstein J. Org. Chem. 2017, 13, 1350–1360. doi:10.3762/bjoc.13.132 |
| 14. | Korin, E.; Cohen, B.; Bai, Y.-X.; Zeng, C.-C.; Becker, J. Y. Tetrahedron 2012, 68, 7450–7455. doi:10.1016/j.tet.2012.06.071 |
| 10. | Zampieri, D.; Mamolo, M. G.; Filingeri, J.; Fortuna, S.; De Logu, A.; Sanna, A.; Zanon, D. Bioorg. Med. Chem. Lett. 2019, 29, 2468–2474. doi:10.1016/j.bmcl.2019.07.025 |
| 16. | Bisht, S.; Peddinti, R. K. J. Org. Chem. 2017, 82, 13617–13625. doi:10.1021/acs.joc.7b02207 |
| 17. | Sharma, N.; Peddinti, R. K. J. Org. Chem. 2017, 82, 9360–9366. doi:10.1021/acs.joc.7b01538 |
| 18. | Sharma, S.; Kumar, P.; Sharma, A.; Peddinti, R. K. Eur. J. Org. Chem. 2017, 3059–3071. doi:10.1002/ejoc.201700407 |
| 19. | Quintana, M.; Bilbao, A.; Comas-Barceló, J.; Bujons, J.; Triola, G. Eur. J. Med. Chem. 2019, 178, 648–666. doi:10.1016/j.ejmech.2019.05.086 |
| 20. | Kundu, T.; Bhattacharjee, B.; Hazra, S.; Ghosh, A. K.; Bandyopadhyay, D.; Pramanik, A. J. Med. Chem. 2019, 62, 6315–6329. doi:10.1021/acs.jmedchem.9b00717 |
| 32. | Chen, J.; Chen, G.; Cheng, C.; Cong, Y.; Du, C.; Zhao, H. J. Chem. Eng. Data 2017, 62, 2191–2197. doi:10.1021/acs.jced.7b00316 |
| 13. | Kulakov, I. V.; Karbainova, A. A.; Shulgau, Z. T.; Seilkhanov, T. M.; Gatilov, Yu. V.; Fisyuk, A. S. Chem. Heterocycl. Compd. 2017, 53, 1094–1097. doi:10.1007/s10593-017-2178-6 |
| 9. | Li, X.; Liu, N.; Zhang, H.; Knudson, S. E.; Slayden, R. A.; Tonge, P. J. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. doi:10.1016/j.bmcl.2010.08.076 |
| 6. | Stepanova, E. E.; Lukmanova, D. N.; Kasatkina, S. O.; Dmitriev, M. V.; Maslivets, A. N. ChemistrySelect 2019, 4, 12774–12778. doi:10.1002/slct.201902900 |
| 7. | Sharma, R.; Yadav, L.; Lal, J.; Jaiswal, P. K.; Mathur, M.; Swami, A. K.; Chaudhary, S. Bioorg. Med. Chem. Lett. 2017, 27, 4393–4398. doi:10.1016/j.bmcl.2017.08.017 |
| 31. | König, W.; Geiger, R. Chem. Ber. 1970, 103, 788–798. doi:10.1002/cber.19701030319 |
| 12. | Jaiswal, P. K.; Sharma, V.; Kumar, S.; Mathur, M.; Swami, A. K.; Yadav, D. K.; Chaudhary, S. Arch. Pharm. (Weinheim, Ger.) 2018, 351, 1700349. doi:10.1002/ardp.201700349 |
| 15. | Yao, Q.-C.; Wu, D.-E.; Ma, R.-Z.; Xia, M. J. Organomet. Chem. 2013, 743, 1–9. doi:10.1016/j.jorganchem.2013.06.012 |
| 32. | Chen, J.; Chen, G.; Cheng, C.; Cong, Y.; Du, C.; Zhao, H. J. Chem. Eng. Data 2017, 62, 2191–2197. doi:10.1021/acs.jced.7b00316 |
| 25. | McKillop, A.; Sayer, T. S. B.; Bellinger, G. C. A. J. Org. Chem. 1976, 41, 1328–1331. doi:10.1021/jo00870a007 |
| 22. | Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Chem. Heterocycl. Compd. 2002, 38, 365–367. doi:10.1023/a:1015660009406 |
| 23. | Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Russ. J. Org. Chem. 2003, 39, 863–868. doi:10.1023/b:rujo.0000003166.58756.fa |
| 24. | Saloutin, V. I.; Perevalov, S. G.; Chupakhin, O. N. Russ. J. Org. Chem. 2000, 36, 700–705. |
| 27. | Singh, Y. P.; Shanker, D.; Sharma, R. K.; Sharma, J.; Rai, A. K. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 141–148. doi:10.1080/104265090508055 |
| 28. | Andreichikov, Y. S.; Tendryakova, S. P.; Nalimova, Y. A.; Voronova, L. A. Chem. Heterocycl. Compd. 1977, 13, 611–613. doi:10.1007/bf00480663 |
| 28. | Andreichikov, Y. S.; Tendryakova, S. P.; Nalimova, Y. A.; Voronova, L. A. Chem. Heterocycl. Compd. 1977, 13, 611–613. doi:10.1007/bf00480663 |
| 27. | Singh, Y. P.; Shanker, D.; Sharma, R. K.; Sharma, J.; Rai, A. K. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 141–148. doi:10.1080/104265090508055 |
| 28. | Andreichikov, Y. S.; Tendryakova, S. P.; Nalimova, Y. A.; Voronova, L. A. Chem. Heterocycl. Compd. 1977, 13, 611–613. doi:10.1007/bf00480663 |
| 27. | Singh, Y. P.; Shanker, D.; Sharma, R. K.; Sharma, J.; Rai, A. K. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 141–148. doi:10.1080/104265090508055 |
| 28. | Andreichikov, Y. S.; Tendryakova, S. P.; Nalimova, Y. A.; Voronova, L. A. Chem. Heterocycl. Compd. 1977, 13, 611–613. doi:10.1007/bf00480663 |
| 1. | Choudhary, G.; Peddinti, R. K. Green Chem. 2011, 13, 3290–3299. doi:10.1039/c1gc15701a |
| 2. | Dobiaš, J.; Ondruš, M.; Addová, G.; Boháč, A. Beilstein J. Org. Chem. 2017, 13, 1350–1360. doi:10.3762/bjoc.13.132 |
| 3. | Sharma, V.; Jaiswal, P. K.; Yadav, D. K.; Saran, M.; Prikhodko, J.; Mathur, M.; Swami, A. K.; Mashevskaya, I. V.; Chaudhary, S. Acta Chim. Slov. 2017, 64, 988–1004. doi:10.17344/acsi.2017.3709 |
| 4. | Jaiswal, P. K.; Sharma, V.; Prikhodko, J.; Mashevskaya, I. V.; Chaudhary, S. Tetrahedron Lett. 2017, 58, 2077–2083. doi:10.1016/j.tetlet.2017.03.048 |
| 5. | Petronijević, J.; Bugarčić, Z.; Bogdanović, G. A.; Stefanović, S.; Janković, N. Green Chem. 2017, 19, 707–715. doi:10.1039/c6gc02893d |
| 6. | Stepanova, E. E.; Lukmanova, D. N.; Kasatkina, S. O.; Dmitriev, M. V.; Maslivets, A. N. ChemistrySelect 2019, 4, 12774–12778. doi:10.1002/slct.201902900 |
| 21. | Samzadeh-Kermani, A. J. Sulfur Chem. 2016, 37, 692–701. doi:10.1080/17415993.2016.1187733 |
| 26. | Stepanova, E. E.; Krasokha, M. O.; Galeev, A. R.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1735–1738. doi:10.1134/s1070428018110209 |
| 22. | Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Chem. Heterocycl. Compd. 2002, 38, 365–367. doi:10.1023/a:1015660009406 |
| 23. | Kozminykh, V. O.; Igidov, N. M.; Kozminykh, E. N. Russ. J. Org. Chem. 2003, 39, 863–868. doi:10.1023/b:rujo.0000003166.58756.fa |
© 2020 Stepanova et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)