Abstract
We present the synthesis and the spin switching efficiencies of Ni(II)-porphyrins substituted with azopyridines as covalently attached photoswitchable ligands. The molecules are designed in such a way that the azopyridines coordinate to the Ni ion if the azo unit is in cis configuration. For steric reasons no intramolecular coordination is possible if the azopyridine unit adopts the trans configuration. Photoisomerization of the azo unit between cis and trans is achieved upon irradiation with 505 nm (trans→cis) and 435 nm (cis→trans). Concurrently with the isomerization and coordination/decoordination, the spin state of the Ni ion switches between singlet (low-spin) and triplet (high-spin). Previous studies have shown that the spin switching efficiency is strongly dependent on the solvent and on the substituent at the 4-position of the pyridine unit. We now introduced thiol, disulfide, thioethers, nitrile and carboxylic acid groups and investigated their spin switching efficiency.
Graphical Abstract
Introduction
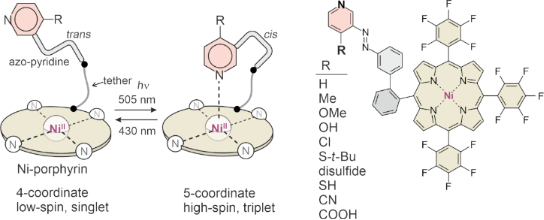
Molecular spin switches operated with visible light in homogeneous solution [1] or on surfaces [2], hold promise for a number of hitherto unprecedented applications such as switchable contrast agents [3-7], switchable NMR relaxation agents [8-10], and building blocks for molecular spintroncis [11]. Our modular design is based on three components: a) a Ni(II)-porphyrin (square planar base complex), b) an azoaryl unit (photoswitchable ligand) and c) a molecular linker or tether (connecting the porphyrin with the switchable ligand). This approach has been dubbed “record player” design for obvious reasons (Figure 1a).
![[1860-5397-16-210-1]](/bjoc/content/figures/1860-5397-16-210-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: “Record player” approach for molecular spin switching. a) General principle b) Variation of the substituent R in 4-position of the pyridine unit. Record player molecules synthesized in this work (1f–j) are highlighted.
Figure 1: “Record player” approach for molecular spin switching. a) General principle b) Variation of the sub...
The Ni-porphyrin (“disk”) is low-spin if the azo-aryl unit (“tone arm”) is in trans configuration. Upon switching to the bent cis configuration, the lone pair of the pyridine (or imidazole) nitrogen (“needle”) is placed onto the Ni ion. The coordination number of the Ni2+ ion changes from 4 (square planar) to 5 (square pyramidal) and concomitantly, the spin state changes from low-spin (singlet, diamagnetic) to high-spin (triplet, paramagnetic). Previous studies have shown that all three components (Ni-porphyrin, azo-aryl unit, and tether) have to be carefully designed and tuned regarding their geometry and electronic properties. The Ni-porphyrin should be electron deficient (electron-withdrawing substituents in meso position), the azo-aryl unit should be electron rich (electron-donating substituents at the pyridine or imidazole part) to increase the Ni–ligand coordination strength. Strong coordination in turn improves the performance of the spin switch, namely the conversion rate to the cis isomer, the conversion rate to the high-spin state, as well as the thermal stability of the high-spin state. 4-Substituted pyridines exhibit a distinguished correlation between basicity and coordination strength as axial ligands with Ni-porphyrins [12]. Hence, Hammet σ values might be used to predict and to systematically optimize the performance of the spin switch. In previous studies we have shown that methoxy substitution (σ = −0.27) improved the spin switching efficiency in organic solvents to >98% in both directions [13]. However, the switching efficiency drops in protic solvents, particularly in water [3,14]. We now synthesized sulfur and carboxylic acid substituted record players aiming at the improvement of the switching efficiency. Thiol (σ = 0.15) and thiolethers (σ ≈ 0.0) are less electron donating than methoxy substituents, however, upon deprotonation, the thiolate (σ = −1.2) should considerably improve coordination and potentially restore switchability in water. Carboxylic acids are electron withdrawing (σ = 0.45), however, they are almost completely dissociated at neutral (physiological) pH. Carboxylate anion substituents are weakly electron donating and should improve both switching efficiency and water solubility.
Results and Discussion
Synthesis
The record player molecules 1f–j (Figure 1) were synthesized by a modular approach described by Heitmann et al. [15]. The boronic ester 22 was prepared according to a mixed aldehyde procedure [16,17] and the substituted azopyridines 14, 18, 20, and 21 were attached using Suzuki conditions.
Synthesis of azopyridines 10–12, 14, 16–18, 20, and 21
For the synthesis of the azopyridines, Baeyer–Mills reactions [18] with 4-substituted 3-aminopyridines were applied [4]. The nitroso compounds 3 and 6 were synthesized by two different methods. 1-Bromo-3-nitrosobenzene (3) was obtained by oxidation of 3-bromoaniline (2) using Oxone™ (Wegner et al. [19-21], Scheme 1). Isolation of 3 was achieved, however, a one pot approach including a subsequent Baeyer–Mills reaction to yield 10 is preferred. 1-Iodo-3-nitrosobenzene (6) cannot be prepared by oxidation of the corresponding aniline because hypervalent iodine is formed [22-26]. Hence, 3-iodonitrobenzene (4) was reduced to obtain hydroxylamine 5, which was oxidized by iron(III) chloride to yield a mixture of 6 and starting material 4 (Scheme 1), which was directly used as a crude product in the subsequent alkaline Baeyer–Mills reaction.
![[1860-5397-16-210-i1]](/bjoc/content/inline/1860-5397-16-210-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the nitroso compounds 3 and 6 using the two different methods described by Wegner et al. [19-21] and Dommaschk et al. [4,27].
Scheme 1: Synthesis of the nitroso compounds 3 and 6 using the two different methods described by Wegner et a...
It is known that the yields of the Baeyer–Mills reactions, if performed under basic conditions, are higher for electron-deficient amines. 4-Chloropyridines are susceptible to nucleophilic aromatic substitutions [4], which was confirmed by the successful substitution of 3-(3-bromophenylazo)-4-chloropyridine (10) and 3-(3-iodophenylazo)-4-chloropyridine (17) with LiSSiMe3 (8) [28], t-BuSH (13) and HSCH2CH2CO2CH3 (15) [29]. Electron-deficient aromatic, silylated thiols exhibit very labile Si–S bonds [30]. Thus, even bulky silyl protection groups are not suitable as protecting groups for the subsequent Suzuki reaction. Azopyridines 14, 16 and 18 were prepared by nucleophilic substitution with 2-methylpropane-2-thiol (13) and methyl 3-mercaptopropionate (15) as nucleophiles (Scheme 2).
![[1860-5397-16-210-i2]](/bjoc/content/inline/1860-5397-16-210-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of azopyridines 11, 14, 16 and 18 by nucleophilic aromatic substitution.
Scheme 2: Synthesis of azopyridines 11, 14, 16 and 18 by nucleophilic aromatic substitution.
3-(3-Bromophenylazo)-4-cyanopyridine (20) was directly obtained by conversion of 3-amino-4-cyanopyridine (19) and 3. Alkaline hydrolysis as described by Sato et al. [31] of 20 yielded 3-(3-bromophenylazo)-4-pyridinecarboxylic acid (21, Scheme 3).
![[1860-5397-16-210-i3]](/bjoc/content/inline/1860-5397-16-210-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 3-(3-bromophenylazo)-4-cyanopyridine (20), which was hydrolyzed to yield 3-(3-bromophenylazo)-4-pyridinecarboxylic acid (21).
Scheme 3: Synthesis of 3-(3-bromophenylazo)-4-cyanopyridine (20), which was hydrolyzed to yield 3-(3-bromophe...
Synthesis of the Ni(II)-porphyrins
The phenylboronic acid pinacol ester 22 was used as a component for the Suzuki reaction (Scheme 4).
![[1860-5397-16-210-i4]](/bjoc/content/inline/1860-5397-16-210-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Modular approach for the C–C connection of the Ni(II)-porphyrin 22 and the different 4-substituted azopyridines 12, 14, 16, 18, 20, and 21.
Scheme 4: Modular approach for the C–C connection of the Ni(II)-porphyrin 22 and the different 4-substituted ...
No conversion was observed for the Suzuki reaction of the porphyrin 22 and the disulfide 12, obviously because of catalyst poisoning by sulfur [32,33]. To circumvent these problems, protection groups were introduced. Protection with methyl 3-mercaptopropionate (15) [29] was successful, however, inhibition of the Suzuki reaction was observed. We assume an alkaline cleavage of the methyl 3-mercaptopropionate under the Suzuki conditions. Nevertheless, azopyridines 14, 18 and 20 were converted almost quantitatively to the corresponding cross-coupling products, while the yield with 21 to 1j is only 52%. The tert-butyl protection group on 1f was cleaved according to a procedure of Nishimura et al. using mercury acetate in trifluoroacetic acid and anisole as radical scavenger (Scheme 5) [34]. Cleavage of the tert-butyl protection group yielded the disulfide 1g, which was reduced with catecholborane in tetrahydrofuran to give the free thiol 1h, which readily reoxidizes under air to form the disulfide.
![[1860-5397-16-210-i5]](/bjoc/content/inline/1860-5397-16-210-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cleavage of 1f to yield disulfide 1g [34].
Scheme 5: Cleavage of 1f to yield disulfide 1g [34].
Investigation on the spin switching performance of the Ni(II)-porphyrins
The spin switching performance of a record player molecule depends on different parameters: 1. the coordination strength of the axial ligand, 2. the effectivity of the cis–trans isomerization, since only the cis isomer coordinates to the Ni(II) for steric reasons and 3. intermolecular coordination. At higher concentrations dimer formation or coordination oligomers between trans record player molecules occurs [35].
To compare the new molecules (1f–j) with previous systems (1a–d), the switching efficiencies were calculated based on the shift of the pyrrole protons relative to an independently synthesized fivefold coordinated Ni-porphyrin, which was used as a reference for a completely coordinated 5-coordinate Ni-porphyrin [13] (Table 1). Since coordination and decoordination is fast on the NMR time scale, the average pyrrole shift is a very accurate indicator of the proportion of species with and without a coordinating axial ligand (100% coordination (paramagnetic) 48.9 ppm and 0% coordination (diamagnetic) 9.1 ppm in acetone-d6).
Table 1: Switching states and switching efficiencies (Δ) of record player molecules 1a–j in the PSS at 505 nm and 435 nm in acetone as a non-coordinating solvent. %cis-para and %cis-dia are defined as the proportion of the cis isomer in the paramagnetic and diamagnetic state, and %trans is the total proportion of the trans isomer, which is assumed to be completely diamagnetic at low concentrations.
| R | PSS 505 nm | PSS 435 nm | Δ | ||||||
| %cis-para | %cis-dia | %trans | %cis-para | %cis-dia | %trans | % | |||
| 1aa | -H | 54 | 11 | 35 | <5 | >95 | 54 | ||
| 1ba | -Me | 82 | 3 | 15 | <5 | >95 | 82 | ||
| 1ca | -OMe | >95 | <1 | <5 | <5 | >95 | >95 | ||
| 1da | -Cl | 47 | 15 | 38 | <5 | >95 | 47 | ||
| 1eb | -OH | 42 | 2 | 56 | 16 | 1 | 83 | 26 | |
| 1e−b | -O− | 72 | <1 | 28 | 45 | <1 | 55 | 27 | |
| 1f | -S-t-Bu | 70 | 3 | 27 | <5 | >95 | 70 | ||
| 1g | -[S]2- | –c | –c | –c | –c | –c | –c | –c | |
| 1hb | -SH | 41 | <1 | 59 | 13 | <1 | 87 | 28 | |
| 1h−b | -S− | 44 | 2 | 54 | 10 | <1 | 90 | 34 | |
| 1i | -CN | 11 | 16 | 73 | <5 | >95 | 11 | ||
| 1jb | -COOH | 90 | <1 | 10 | 13 | <1 | 87 | 77 | |
| 1j−b | -COO− | 93 | <1 | 7 | 10 | <1 | 90 | 83 | |
aRecalculated from Dommaschk et al. [4], bdetermined by UV–vis spectroscopy, cnot determined because of very strong intramolecular coordination (Supporting Information File 1, Figure S17).
It should be noted that the cis/trans ratio does not necessarily correspond to the coordinated (paramagnetic)/decoordinated (diamagnetic) ratio because the cis isomer does not always coordinate completely and the trans isomer still can coordinate intermolecularly. The cis/trans ratios in the photostationary state (PSS) at 505 nm and 435 nm were investigated by independent NMR experiments. For 1f and 1i the cis/trans ratio was obtained from integration of the signals of H-11, which differ in cis and trans configuration. (Supporting Information File 1, Figure S14). However, NMR spectroscopy was not suitable to determine the cis–trans isomerization yields of 1e, 1g, 1h and 1j because of paramagnetic line broadening and overlapping signals. To circumvent these problems, UV–vis spectroscopy was performed at very low concentrations (Table 1). The porphyrin Soret bands are different for the diamagnetic (λmax ≈ 406 nm) and the paramagnetic species (λmax ≈ 423 nm). The molar extinctions of both species were determined by adding a strong axial ligand such as piperidine (to achieve almost complete coordination) and acid (TFA) (protonation of the pyridine unit to prevent coordination, completely diamagnetic). At concentrations of ≈10 µM, intermolecular coordination is negligible. Thus, the absorption band at 423 nm provides information of the total amount of paramagnetic cis species and the band at 406 nm comprises the total amount of diamagnetic trans and uncoordinated cis isomer. Combined with the information from NMR the cis/trans ratios were determined.
Disulfide 1g is always in a high paramagnetic state (>80%) and showed only minor switching efficiency after irradiation (4%). This is attributed to intramolecular coordination of the trans state (Supporting Information File 1, Figure S17). Application of density functional theory (DFT) at the B3LYP/def2TZVP//PBE/def2SVP level of theory (for details see Supporting Information File 1) revealed that the thermodynamically most stable structure is a trans configuration, where each of the pyridine units coordinate the nickel of the opposite porphyrin. Reductive cleavage of 1g by catecholeborane yields the photoswitchable thiol 1h.
Table 1 summarizes the photostationary states at 505 and 435 nm, the diamagnetic/paramagnetic ratio (%cis-dia/%cis-para) of the cis isomer for compounds 1a–j and the spin switching efficiency (Δ). The spin switching efficiency is defined as the difference of the percentage of the diamagnetic (or the paramagnetic) species in the two photostationary states. The intermolecular association constants of pyridines as axial ligands to Ni-porphyrins as a function of their para substituents usually follow the Hammett relationship [5,12]. Hence, the association constants should also be reflected in the intramolecular coordination of the covalently bound pyridine units within the cis isomers (1a–j). Whereas the substituents R = H, S-t-Bu, Me, OMe, Cl and CN very roughly comply with this assumption, the protic and deprotonated substituents R = OH, O−, SH, S−, COOH, COO− strongly deviate (Figure 2). The substituents COOH, SH, COO− and O− give rise to almost complete coordination of the cis isomer. In contrast to bimolecular coordination [12] the intramolecular coordination in the substituted cis isomers, obviously, does not follow a clear linear free energy relationship. Steric hindrance or strain might be the reasons for the deviations. We cannot exclude that these groups coordinate directly to the Ni ion.
![[1860-5397-16-210-2]](/bjoc/content/figures/1860-5397-16-210-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Hammett plot of the investigated pyridine substituents [36].
Figure 2: Hammett plot of the investigated pyridine substituents [36].
Whereas the thiol (1h) and thiolate (1h−) exhibit disappointingly low switching efficiencies (Δ = 28% and 34%), the carboxylic acid (1j) and the corresponding carboxylate anion (1j−) perform surprisingly well (Δ = 77% and 83%).
In order to check whether the switching efficiencies would decrease in aqueous environment to a similar extend as in previous cases [3], switching experiments with the protic record players 1e (R = OH), 1h (SH) and 1j (R = COOH) in acetone/water 1:9 were performed. UV experiments revealed that the switching efficiencies of the protonated species indeed dropped dramatically as compared to those in pure acetone. However, the deprotonated 4-hydroxypyridine 1e− and the pyridine-4-carboxylate 1j− still perform with switching efficiencies of 40% and 28% in aqueous environment (Figure 3). If it comes to medical applications, such as switchable contrast agents [3-7], the new carboxylate substituted system 1j has the distinct advantage of being deprotonated at neutral pH, whereas the previously synthesized hydroxylate system 1e exhibits its highest switching efficiency at unphysiological pH 10.5 [14]. Hence, further attempts to optimize our molecular spin switches will be based on pyridine-4-carboxylate-substituted systems.
![[1860-5397-16-210-3]](/bjoc/content/figures/1860-5397-16-210-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of 1e (top), 1h (left) and 1j (right) in acetone water (1:9) (solid line) and after deprotonation using cesium carbonate (dashed line) in the PSS at 505 nm (green) and 435 nm (blue).
Figure 3: UV–vis spectra of 1e (top), 1h (left) and 1j (right) in acetone water (1:9) (solid line) and after ...
Conclusion
Aiming at the optimization of molecular spin switches (record player molecules) towards higher switching efficiencies, particularly in water, we synthesized 5 new record player molecules (1f–j). Water forms H-bonds with the pyridine units, and reduces the coordination power to Ni2+ and thus decreases the efficiency of conversion to the paramagnetic (coordinated) state. Insufficient water solubility is another notorious problem of porphyrin-based spin switches. To kill two birds with one stone, we introduced acidic substituents in 4-position of the pyridines (1h (R = SH), 1j (R = COOH)). Deprotonation to the corresponding anions should increase the coordination power of the pyridine and increase the solubility of the record player molecules. Surprisingly, the thiolate exhibits a low switching efficiency (Δ = 34%), however, the carboxylate performs quite well (Δ = 83%) in acetone and even in aqueous environment.
Experimental
Detailed information on the experimental procedures are given in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental procedures and spectra. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Venkataramani, S.; Jana, U.; Dommaschk, M.; Sönnichsen, F. D.; Tuczek, F.; Herges, R. Science 2011, 331, 445–448. doi:10.1126/science.1201180
Return to citation in text: [1] -
Matino, F.; Schull, G.; Jana, U.; Köhler, F.; Berndt, R.; Herges, R. Chem. Commun. 2010, 46, 6780–6782. doi:10.1039/c0cc00959h
Return to citation in text: [1] -
Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g
Return to citation in text: [1] [2] [3] [4] -
Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d
Return to citation in text: [1] [2] [3] -
Herges, R.; Dommaschk, M.; Heitmann, G.; Groebner, J.; Thoms, V. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. German Patent DE102017119482 A1, Feb 28, 2019.
Return to citation in text: [1] [2] -
Herges, R.; Dommaschk, M.; Heitmann, G.; Groebner, J.; Thoms, V. Neuartiges mrt-kontrastmittel, verwendung zur temperaturmessung und verfahren zur temperaturmessung. WO Patent WO2019038428 A2, Feb 28, 2009.
Return to citation in text: [1] [2] -
Stadler, E.; Tassoti, S.; Lentes, P.; Herges, R.; Glasnov, T.; Zangger, K.; Gescheidt, G. Anal. Chem. (Washington, DC, U. S.) 2019, 91, 11367–11373. doi:10.1021/acs.analchem.9b02613
Return to citation in text: [1] -
Stadler, E.; Dommaschk, M.; Frühwirt, P.; Herges, R.; Gescheidt, G. ChemPhysChem 2018, 19, 571–574. doi:10.1002/cphc.201701304
Return to citation in text: [1] -
Wagner, G. E.; Tassoti, S.; Glanzer, S.; Stadler, E.; Herges, R.; Gescheidt, G.; Zangger, K. Chem. Commun. 2019, 55, 12575–12578. doi:10.1039/c9cc06427c
Return to citation in text: [1] -
Köbke, A.; Gutzeit, F.; Röhricht, F.; Schlimm, A.; Grunwald, J.; Tuczek, F.; Studniarek, M.; Longo, D.; Choueikani, F.; Otero, E.; Ohresser, P.; Rohlf, S.; Johannsen, S.; Diekmann, F.; Rossnagel, K.; Weismann, A.; Jasper-Toennies, T.; Näther, C.; Herges, R.; Berndt, R.; Gruber, M. Nat. Nanotechnol. 2020, 15, 18–21. doi:10.1038/s41565-019-0594-8
Return to citation in text: [1] -
Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F. D.; Näther, C.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2010, 16, 10074–10083. doi:10.1002/chem.201000603
Return to citation in text: [1] [2] [3] -
Gutzeit, F.; Dommaschk, M.; Levin, N.; Buchholz, A.; Schaub, E.; Plass, W.; Näther, C.; Herges, R. Inorg. Chem. 2019, 58, 12542–12546. doi:10.1021/acs.inorgchem.9b00348
Return to citation in text: [1] [2] -
Ludwig, J.; Gröbner, J.; Dommaschk, M.; Huber, L. M.; Peters, M. K.; Hövener, J.-B.; Herges, R. J. Porphyrins Phthalocyanines 2020, 24, 480–488. doi:10.1142/s1088424619501803
Return to citation in text: [1] [2] -
Heitmann, G.; Dommaschk, M.; Löw, R.; Herges, R. Org. Lett. 2016, 18, 5228–5231. doi:10.1021/acs.orglett.6b02507
Return to citation in text: [1] -
Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836. doi:10.1021/jo00381a022
Return to citation in text: [1] -
Lindsey, J. S.; Wagner, R. W. J. Org. Chem. 1989, 54, 828–836. doi:10.1021/jo00265a021
Return to citation in text: [1] -
Baeyer, A. Ber. Dtsch. Chem. Ges. 1874, 7, 1638–1640. doi:10.1002/cber.187400702214
Return to citation in text: [1] -
Reuter, R.; Wegner, H. A. Chem. – Eur. J. 2011, 17, 2987–2995. doi:10.1002/chem.201002671
Return to citation in text: [1] [2] -
Yasukawa, N.; Kuwata, M.; Imai, T.; Monguchi, Y.; Sajiki, H.; Sawama, Y. Green Chem. 2018, 20, 4409–4413. doi:10.1039/c8gc01373j
Return to citation in text: [1] [2] -
Purkait, A.; Roy, S. K.; Srivastava, H. K.; Jana, C. K. Org. Lett. 2017, 19, 2540–2543. doi:10.1021/acs.orglett.7b00832
Return to citation in text: [1] [2] -
Zagulyaeva, A. A.; Banek, C. T.; Yusubov, M. S.; Zhdankin, V. V. Org. Lett. 2010, 12, 4644–4647. doi:10.1021/ol101993q
Return to citation in text: [1] -
Yusubov, M. S.; Zagulyaeva, A. A.; Zhdankin, V. V. Chem. – Eur. J. 2009, 15, 11091–11094. doi:10.1002/chem.200901953
Return to citation in text: [1] -
Zagulyaeva, A. A.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2010, 75, 2119–2122. doi:10.1021/jo902733f
Return to citation in text: [1] -
Yoshimura, A.; Neu, H. M.; Nemykin, V. N.; Zhdankin, V. V. Adv. Synth. Catal. 2010, 352, 1455–1460. doi:10.1002/adsc.201000172
Return to citation in text: [1] -
Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V. Tetrahedron 2010, 66, 5745–5752. doi:10.1016/j.tet.2010.04.046
Return to citation in text: [1] -
Fletcher, D. A.; Gowenlock, B. G.; Orrell, K. G. J. Chem. Soc., Perkin Trans. 2 1997, 2201–2206. doi:10.1039/a703760k
Return to citation in text: [1] -
Taher, D.; Wallbank, A. I.; Turner, E. A.; Cuthbert, H. L.; Corrigan, J. F. Eur. J. Inorg. Chem. 2006, 4616–4620. doi:10.1002/ejic.200600583
Return to citation in text: [1] -
Dunn, A. D.; Norrie, R.; L'Hostis, J.; Marjot, S. J. Prakt. Chem. 1992, 334, 119–125. doi:10.1002/prac.19923340204
Return to citation in text: [1] [2] -
Kreis, M.; Bräse, S. Adv. Synth. Catal. 2005, 347, 313–319. doi:10.1002/adsc.200404299
Return to citation in text: [1] -
Sato, C.; Suzuki, S.; Kozaki, M.; Okada, K. Org. Lett. 2016, 18, 1052–1055. doi:10.1021/acs.orglett.6b00171
Return to citation in text: [1] -
Nasri, N. S.; Jones, J. M.; Dupont, V. A.; Williams, A. Energy Fuels 1998, 12, 1130–1134. doi:10.1021/ef980104j
Return to citation in text: [1] -
Rodriguez, J. A.; Hrbek, J. Acc. Chem. Res. 1999, 32, 719–728. doi:10.1021/ar9801191
Return to citation in text: [1] -
Nishimura, O.; Kitada, C.; Fujino, M. Chem. Pharm. Bull. 1978, 26, 1576–1585. doi:10.1248/cpb.26.1576
Return to citation in text: [1] [2] -
Dommaschk, M.; Schütt, C.; Venkataramani, S.; Jana, U.; Näther, C.; Sönnichsen, F. D.; Herges, R. Dalton Trans. 2014, 43, 17395–17405. doi:10.1039/c4dt03048f
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1]
| 4. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 5. | Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d |
| 12. | Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F. D.; Näther, C.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2010, 16, 10074–10083. doi:10.1002/chem.201000603 |
| 12. | Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F. D.; Näther, C.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2010, 16, 10074–10083. doi:10.1002/chem.201000603 |
| 1. | Venkataramani, S.; Jana, U.; Dommaschk, M.; Sönnichsen, F. D.; Tuczek, F.; Herges, R. Science 2011, 331, 445–448. doi:10.1126/science.1201180 |
| 11. | Köbke, A.; Gutzeit, F.; Röhricht, F.; Schlimm, A.; Grunwald, J.; Tuczek, F.; Studniarek, M.; Longo, D.; Choueikani, F.; Otero, E.; Ohresser, P.; Rohlf, S.; Johannsen, S.; Diekmann, F.; Rossnagel, K.; Weismann, A.; Jasper-Toennies, T.; Näther, C.; Herges, R.; Berndt, R.; Gruber, M. Nat. Nanotechnol. 2020, 15, 18–21. doi:10.1038/s41565-019-0594-8 |
| 19. | Reuter, R.; Wegner, H. A. Chem. – Eur. J. 2011, 17, 2987–2995. doi:10.1002/chem.201002671 |
| 20. | Yasukawa, N.; Kuwata, M.; Imai, T.; Monguchi, Y.; Sajiki, H.; Sawama, Y. Green Chem. 2018, 20, 4409–4413. doi:10.1039/c8gc01373j |
| 21. | Purkait, A.; Roy, S. K.; Srivastava, H. K.; Jana, C. K. Org. Lett. 2017, 19, 2540–2543. doi:10.1021/acs.orglett.7b00832 |
| 8. | Stadler, E.; Tassoti, S.; Lentes, P.; Herges, R.; Glasnov, T.; Zangger, K.; Gescheidt, G. Anal. Chem. (Washington, DC, U. S.) 2019, 91, 11367–11373. doi:10.1021/acs.analchem.9b02613 |
| 9. | Stadler, E.; Dommaschk, M.; Frühwirt, P.; Herges, R.; Gescheidt, G. ChemPhysChem 2018, 19, 571–574. doi:10.1002/cphc.201701304 |
| 10. | Wagner, G. E.; Tassoti, S.; Glanzer, S.; Stadler, E.; Herges, R.; Gescheidt, G.; Zangger, K. Chem. Commun. 2019, 55, 12575–12578. doi:10.1039/c9cc06427c |
| 4. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 27. | Fletcher, D. A.; Gowenlock, B. G.; Orrell, K. G. J. Chem. Soc., Perkin Trans. 2 1997, 2201–2206. doi:10.1039/a703760k |
| 3. | Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g |
| 4. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 5. | Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d |
| 6. | Herges, R.; Dommaschk, M.; Heitmann, G.; Groebner, J.; Thoms, V. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. German Patent DE102017119482 A1, Feb 28, 2019. |
| 7. | Herges, R.; Dommaschk, M.; Heitmann, G.; Groebner, J.; Thoms, V. Neuartiges mrt-kontrastmittel, verwendung zur temperaturmessung und verfahren zur temperaturmessung. WO Patent WO2019038428 A2, Feb 28, 2009. |
| 19. | Reuter, R.; Wegner, H. A. Chem. – Eur. J. 2011, 17, 2987–2995. doi:10.1002/chem.201002671 |
| 20. | Yasukawa, N.; Kuwata, M.; Imai, T.; Monguchi, Y.; Sajiki, H.; Sawama, Y. Green Chem. 2018, 20, 4409–4413. doi:10.1039/c8gc01373j |
| 21. | Purkait, A.; Roy, S. K.; Srivastava, H. K.; Jana, C. K. Org. Lett. 2017, 19, 2540–2543. doi:10.1021/acs.orglett.7b00832 |
| 2. | Matino, F.; Schull, G.; Jana, U.; Köhler, F.; Berndt, R.; Herges, R. Chem. Commun. 2010, 46, 6780–6782. doi:10.1039/c0cc00959h |
| 22. | Zagulyaeva, A. A.; Banek, C. T.; Yusubov, M. S.; Zhdankin, V. V. Org. Lett. 2010, 12, 4644–4647. doi:10.1021/ol101993q |
| 23. | Yusubov, M. S.; Zagulyaeva, A. A.; Zhdankin, V. V. Chem. – Eur. J. 2009, 15, 11091–11094. doi:10.1002/chem.200901953 |
| 24. | Zagulyaeva, A. A.; Yusubov, M. S.; Zhdankin, V. V. J. Org. Chem. 2010, 75, 2119–2122. doi:10.1021/jo902733f |
| 25. | Yoshimura, A.; Neu, H. M.; Nemykin, V. N.; Zhdankin, V. V. Adv. Synth. Catal. 2010, 352, 1455–1460. doi:10.1002/adsc.201000172 |
| 26. | Yusubov, M. S.; Nemykin, V. N.; Zhdankin, V. V. Tetrahedron 2010, 66, 5745–5752. doi:10.1016/j.tet.2010.04.046 |
| 15. | Heitmann, G.; Dommaschk, M.; Löw, R.; Herges, R. Org. Lett. 2016, 18, 5228–5231. doi:10.1021/acs.orglett.6b02507 |
| 18. | Baeyer, A. Ber. Dtsch. Chem. Ges. 1874, 7, 1638–1640. doi:10.1002/cber.187400702214 |
| 3. | Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g |
| 4. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 5. | Heitmann, G.; Schütt, C.; Gröbner, J.; Huber, L.; Herges, R. Dalton Trans. 2016, 45, 11407–11412. doi:10.1039/c6dt01727d |
| 6. | Herges, R.; Dommaschk, M.; Heitmann, G.; Groebner, J.; Thoms, V. Neuartiges MRT-Kontrastmittel, Verwendung zur Temperaturmessung und Verfahren zur Temperaturmessung. German Patent DE102017119482 A1, Feb 28, 2019. |
| 7. | Herges, R.; Dommaschk, M.; Heitmann, G.; Groebner, J.; Thoms, V. Neuartiges mrt-kontrastmittel, verwendung zur temperaturmessung und verfahren zur temperaturmessung. WO Patent WO2019038428 A2, Feb 28, 2009. |
| 3. | Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g |
| 14. | Ludwig, J.; Gröbner, J.; Dommaschk, M.; Huber, L. M.; Peters, M. K.; Hövener, J.-B.; Herges, R. J. Porphyrins Phthalocyanines 2020, 24, 480–488. doi:10.1142/s1088424619501803 |
| 4. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 14. | Ludwig, J.; Gröbner, J.; Dommaschk, M.; Huber, L. M.; Peters, M. K.; Hövener, J.-B.; Herges, R. J. Porphyrins Phthalocyanines 2020, 24, 480–488. doi:10.1142/s1088424619501803 |
| 13. | Gutzeit, F.; Dommaschk, M.; Levin, N.; Buchholz, A.; Schaub, E.; Plass, W.; Näther, C.; Herges, R. Inorg. Chem. 2019, 58, 12542–12546. doi:10.1021/acs.inorgchem.9b00348 |
| 36. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 12. | Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F. D.; Näther, C.; Tuczek, F.; Herges, R. Chem. – Eur. J. 2010, 16, 10074–10083. doi:10.1002/chem.201000603 |
| 16. | Lindsey, J. S.; Schreiman, I. C.; Hsu, H. C.; Kearney, P. C.; Marguerettaz, A. M. J. Org. Chem. 1987, 52, 827–836. doi:10.1021/jo00381a022 |
| 17. | Lindsey, J. S.; Wagner, R. W. J. Org. Chem. 1989, 54, 828–836. doi:10.1021/jo00265a021 |
| 3. | Dommaschk, M.; Gröbner, J.; Wellm, V.; Hövener, J.-B.; Riedel, C.; Herges, R. Phys. Chem. Chem. Phys. 2019, 21, 24296–24299. doi:10.1039/c9cp04156g |
| 29. | Dunn, A. D.; Norrie, R.; L'Hostis, J.; Marjot, S. J. Prakt. Chem. 1992, 334, 119–125. doi:10.1002/prac.19923340204 |
| 4. | Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F. D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. J. Am. Chem. Soc. 2015, 137, 7552–7555. doi:10.1021/jacs.5b00929 |
| 28. | Taher, D.; Wallbank, A. I.; Turner, E. A.; Cuthbert, H. L.; Corrigan, J. F. Eur. J. Inorg. Chem. 2006, 4616–4620. doi:10.1002/ejic.200600583 |
| 35. | Dommaschk, M.; Schütt, C.; Venkataramani, S.; Jana, U.; Näther, C.; Sönnichsen, F. D.; Herges, R. Dalton Trans. 2014, 43, 17395–17405. doi:10.1039/c4dt03048f |
| 13. | Gutzeit, F.; Dommaschk, M.; Levin, N.; Buchholz, A.; Schaub, E.; Plass, W.; Näther, C.; Herges, R. Inorg. Chem. 2019, 58, 12542–12546. doi:10.1021/acs.inorgchem.9b00348 |
| 34. | Nishimura, O.; Kitada, C.; Fujino, M. Chem. Pharm. Bull. 1978, 26, 1576–1585. doi:10.1248/cpb.26.1576 |
| 34. | Nishimura, O.; Kitada, C.; Fujino, M. Chem. Pharm. Bull. 1978, 26, 1576–1585. doi:10.1248/cpb.26.1576 |
| 32. | Nasri, N. S.; Jones, J. M.; Dupont, V. A.; Williams, A. Energy Fuels 1998, 12, 1130–1134. doi:10.1021/ef980104j |
| 33. | Rodriguez, J. A.; Hrbek, J. Acc. Chem. Res. 1999, 32, 719–728. doi:10.1021/ar9801191 |
| 29. | Dunn, A. D.; Norrie, R.; L'Hostis, J.; Marjot, S. J. Prakt. Chem. 1992, 334, 119–125. doi:10.1002/prac.19923340204 |
| 30. | Kreis, M.; Bräse, S. Adv. Synth. Catal. 2005, 347, 313–319. doi:10.1002/adsc.200404299 |
| 31. | Sato, C.; Suzuki, S.; Kozaki, M.; Okada, K. Org. Lett. 2016, 18, 1052–1055. doi:10.1021/acs.orglett.6b00171 |
© 2020 Ludwig et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)