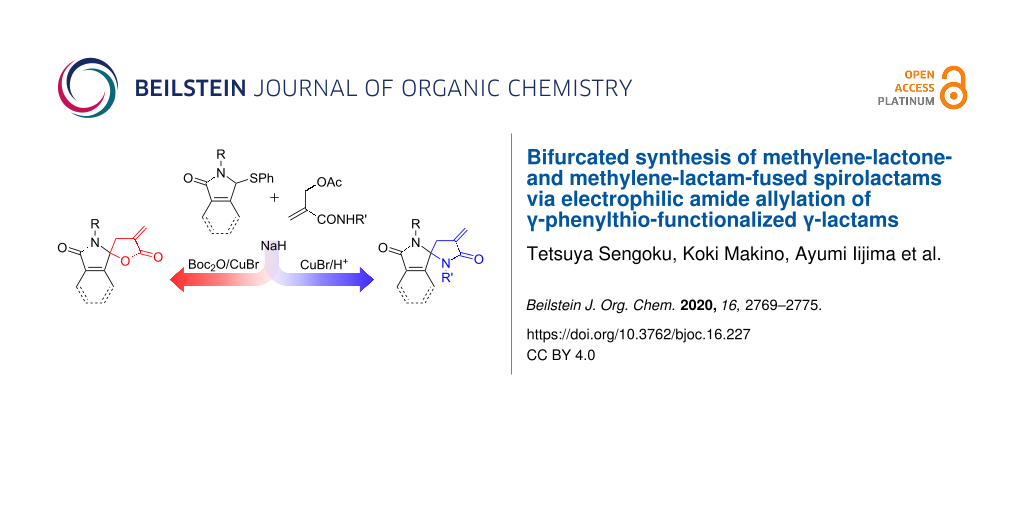
Abstract
New synthetic methods for spirolactams bearing an α-methylene-γ-butyrolactone or its analogous methylene-lactam have been developed. The allylation of γ-phenylthio-functionalized γ-lactams with 2-(acetoxy)methyl acrylamides was accomplished by using 2.5 equivalents of NaH to give the corresponding adducts in excellent yields. The remaining phenylthio group was substituted with a hydroxy group by treatment with CuBr, and the resulting γ-hydroxyamides were cyclized under acidic conditions to afford the corresponding methylene-lactam-fused spirolactams in high yields. On the other hand, methylene-lactone-fused spirolactams could be delivered from the allyl adducts in high yields through a sequential N-Boc protection/desulfinative lactonization.

Graphical Abstract
Introduction
α-Methylene-γ-butyrolactone is a pharmaceutically important motif which is found in a wide variety of bioactive molecules including natural products (Scheme 1a) [1-3]. For example, sesquiterpene lactones represented by parthenolide and helenalin have attracted interest because of their useful biological activities, and many synthetic efforts have been made so far [4-6]. On the other hand, syntheses and biological evaluation of nonnatural methylene-lactones also have been reported [4,6-8]. For instance, Heindel and his co-worker synthesized methylene-lactone derivatives spiro-fused to an oxindole (A), one of which exhibited a potent cytotoxic activity [9]. Our research group succeeded the enantiopure synthesis of these compounds, in which enantioselective allylation of an isatin derivative with an amido-functionalized allylstannane was employed as a key reaction (Scheme 1b) [10,11]. More recently, an amide-functionalized allyl boronate was developed as an alternative reagent for the allylstannane [12-16], which led to the syntheses of not only cytotoxic methylene-lactones spiro-fused to a lactam ring (B) but also their analogous methylene-lactams (C) through zinc-catalyzed addition to N-carbonyl imides [13,14,16].
![[1860-5397-16-227-i1]](/bjoc/content/inline/1860-5397-16-227-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Examples of (a) bioactive compounds bearing an α-methylene-γ-butyrolactone structure, (b) syntheses of spirocyclic compounds through nucleophilic amide allylation, and (c) syntheses of spirocyclic compounds through electrophilic amide allylation.
Scheme 1: Examples of (a) bioactive compounds bearing an α-methylene-γ-butyrolactone structure, (b) syntheses...
Meanwhile, we also developed an umpolung electrophilic allylation of 3-heterosubstituted oxindole D for the synthesis of lactam analog of A (Scheme 1c) [17]. The oxindole D readily reacted with 2-(acetoxy)methyl acrylamides 1 in the presence of a catalytic amount of Pd(PPh3)4 to give the corresponding adducts which could be delivered into an methylene-lactam-fused oxindole. On the basis of this umpolung strategy, spirolactams 4 and 5, which are N-alkyl or N-phenyl-substituted analogs of B and C and unable to be obtained under the conditions depicted in Scheme 1b, seems to be accessible from the common precursor 3 prepared through the reaction between 1 and 3-heterosubstituted isoindolinones 2 followed by cyclization. Considering that 2 is estimated to be less reactive compared with the oxindole derivative D which show excellent nucleophilicity arising from the oxindole–hydroxyindole tautomerization [18,19], this synthetic approach is undoubtedly attractive because it could complement our previous one using nucleophilic amido-functionalized allyl boronates in terms of the structural diversity as well as understanding of the structure–cytotoxicity relationship [14]. Therefore, we were motivated to develop a new synthetic methodology for N-alkyl and N-phenyl derivatives 4 and 5 starting from 2 with 1.
Results and Discussion
Our studies began with an exploration of a promising substrate for the desired electrophilic allylation. We initially chose N-phenyl-3-(acetoxy)isoindolinone (2a) as a reactant. However, 2a showed no reactivity under the reaction conditions determined for the palladium-catalyzed allylation in our previous work and was recovered in 98% yield [17] (Table 1, entry 1). The use of strong bases such as potassium tert-butoxide or sodium hydride resulted in no formation of the desired product 3a because 2a was decomposed under the harsh reaction conditions (Table 1, entries 2 and 3). Thus we opted to employ another substrate 2b bearing a phenylthio group which polarizes the α-C–H bond to lead the corresponding stabilized anion [20] and is readily transformed into a hydroxy group by treatment with copper bromide according to our previous work [21]. Allylation of 2b with 1a at room temperature proceeded in the presence of a catalytic amount of Pd(PPh3)4 by using potassium tert-butoxide or sodium hydride, affording the corresponding adduct in 50% and 81% yields, respectively (Table 1, entries 6 and 7). Surprisingly, the desired allylation underwent even in the absence of palladium catalyst, probably due to high nucleophilicity of the deprotonated intermediate, to give 3b in 87% yield (Table 1, entry 8). Optimization studies were conducted by screening solvents, reagent amount, and reaction temperature, showing that 3b was produced in the highest yield of 97% when the reaction was carried out with 2.5 equivalents of sodium hydride in THF at −10 °C (Table 1, entries 9–13) [22].
Table 1: Screening of reaction conditions for electrophilic amide allylation.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-227-i3.svg?max-width=637&scale=1.0)
|
|||||
| entry | 2 | reagents (equiv) | solvent | time (h) | 3 (%) |
| 1 | 2a |
Et3N (2.5)
Pd(PPh3)4 (0.1) |
THF | 24 | 3a (0) |
| 2 | 2a |
t-BuOK (2.5)
Pd(PPh3)4 (0.1) |
THF | 24 | 3a (0) |
| 3 | 2a |
NaH (2.5)
Pd(PPh3)4 (0.1) |
THF | 24 | 3a (0) |
| 4 | 2b |
Pyridine (2.5)
Pd(PPh3)4 (0.1) |
THF | 24 | 3b (0) |
| 5 | 2b |
Et3N (2.5)
Pd(PPh3)4 (0.1) |
THF | 24 | 3b (0) |
| 6 | 2b |
t-BuOK (2.5)
Pd(PPh3)4 (0.1) |
THF | 24 | 3b (50) |
| 7 | 2b |
NaH (2.5)
Pd(PPh3)4 (0.1) |
THF | 1 | 3b (81) |
| 8 | 2b | NaH (2.5) | THF | 1 | 3b (87) |
| 9 | 2b | NaH (1.0) | THF | 2 | 3b (62) |
| 10 | 2b | NaH (2.5) | toluene | 1 | 3b (74) |
| 11 | 2b | NaH (2.5) | DMF | 1 | 3b (18) |
| 12a | 2b | NaH (2.5) | THF | 1 | 3b (97) |
| 13b | 2b | NaH (2.5) | THF | 1 | 3b (87) |
aThe reaction was performed at −10 °C. bThe reaction was performed at −20 °C.
With the optimal reaction conditions for electrophilic allylation of 2b with 1a in hand, we turned to demonstrate the versatility of this method. In our previous work relating to nucleophilic allylation of imide derivatives, we succeeded the syntheses of N-carbonyl-functionalized γ-hydroxy amides bearing an amide side chain [14]. Therefore, we here examined reactions using N-alkyl derivatives in order to gain the structural diversity. Reactions of 3-(phenylthio)isoindolinone derivatives bearing an N-benzyl (2c), N-methyl (2d), and N-(n-pentyl) group (2d) with 1a readily proceeded under the optimized reaction conditions for 3b to afford the corresponding products 3c–e in 90–95% yields (Figure 1). As for the substituents on the methacrylamide side chain, not only aryl groups (p-tolyl and p-anisyl groups) but also an n-pentyl group were tolerated, providing 3f–h in excellent yields. On the other hand, a reaction of a nonaromatic lactam derivative prepared from N-phenyl-2,3-dimethylmaleimide was accompanied by the formation of structure-unidentified side products, resulting in relatively low product yield (3i, 78% yield). This problem was overcome by carrying out the reaction at −20 °C. Under these conditions, 3i–o were predominantly formed in 92–99% yields. Thus, we could obtain a wide variety of 3-phenylthio lactams bearing an amide functionality by electrophilic allylation with 1.
![[1860-5397-16-227-1]](/bjoc/content/figures/1860-5397-16-227-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Syntheses of 3b–o via electrophilic amide allylation of γ-phenylthio lactams. Reactions were carried out with 1 (1.2 equiv), 2 (1.0 equiv), and NaH (2.5 equiv) in THF (0.2 M for 2) at −10 °C for 1 h. aReactions were performed at −20 °C.
Figure 1: Syntheses of 3b–o via electrophilic amide allylation of γ-phenylthio lactams. Reactions were carrie...
Having completed the installation of an acrylamide side chain into 3-phenylthio lactams, we subsequently proceeded to the construction of the spiro skeleton of 4 and 5. For this purpose, we initially attempted to transform 3b to 5a through the reaction sequence consisting of copper-mediated hydroxylation and acid-mediated lactonization based on our previous reports [10,11,14,21]. Substitution of the phenylthio group of 3b with a hydroxy group was readily achieved by treatment with CuBr in aqueous media (THF/H2O) to afford the corresponding hydroxylactam in 90% yield (Scheme 2). However, the subsequent spirolactonization with p-toluenesulfonic acid (p-TsOH) was unsuccessful, leading to predominant production of bislactam derivative 4a (97%) [23-25]. Although acidic spirolactamization was contrary to our initial expectations, a new type of N-phenyl spirobislactam, which could not be obtained by our previous method, became accessible through this reaction. Therefore, we decided to evaluate its applicability to other substrates. Reactions of isoindolinone derivatives 3c–h under the comparable conditions used for 3b were successful, furnishing the corresponding N-alkyl and N-phenyl bislactams 4b–g in 79–91% two-step yields. Similarly, non-aromatic substrates 3i–o were also converted into 4h–n in 84–94% yields, indicating widespread applicability of this sequential transformation.
![[1860-5397-16-227-i2]](/bjoc/content/inline/1860-5397-16-227-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syntheses of N-phenyl and N-alkyl-substituted spirolactams (two-step yields from 3).
Scheme 2: Syntheses of N-phenyl and N-alkyl-substituted spirolactams (two-step yields from 3).
Since the cyclization under acidic conditions led to the predominant formation of the methylene-lactam-fused spirolactam, we set another route toward 5 via cyclization of N-Boc-functionalized γ-hydroxymethacrylamide [26]. We initially treated the hydroxylactam prepared through CuBr-mediated hydroxylation of 3b with di-tert-butyl dicarbonate (Boc2O) in the presence of N,N-dimethyl-4-aminopyridine (DMAP), but a mixture of structurally unidentified products was formed. Meanwhile, when the reaction was carried out with 2.0 equivalents of n-butyllithium and 1.1 equivalents of Boc2O in THF at −78 °C, 5a was obtained as a mixture including unreacted starting material (<11% yield), indicating that installation of an N-Boc group on the terminus amide should trigger the desired lactonization. Therefore, we next examined the transposed reaction sequence, N-Boc protection followed by hydroxylation (Scheme 2). N-Boc amides were readily obtained by treatment of 3b–o with Boc2O and DMAP, which were successively subjected to hydroxylation in the presence of CuBr. Expectedly, the desired lactonization occurred spontaneously under the reaction conditions and led to methylene-lactone-fused spirolactams 5a–h in excellent yields without exception (88–98% two-step yields). Thus, we established the bifurcated synthetic routes toward lactams spiro-fused to α-methylene-γ-butyrolactone or α-methylene-γ-butyrolactam by using 3-phenylthiolactams bearing an acrylamide side chain as a common intermediate.
Finally, we subjected methylene-lactone-fused spirolactams to the assay of cytotoxicity on P388 cells (Figure 2). N-Methyl-substituted spirolactam 5c exhibited potent cytotoxicity (IC50 0.32 μg/mL) which was comparable to that of the N-acetyl analog E (0.16 μg/mL) [14]. It should be noted that the IC50 values of our recently reported non-conjugated lactone F [27] was >100 μg/mL. In addition to 5c, we tested the cytotoxicity of methylene-lactam-based compound 4o, which exhibited weak activity (IC50 12.4 μg/mL) against the P388 cell line. These results suggest that cytotoxic activity of this type of spirolactams against P388 cells is simply related to the methylene lactone structure [28].
![[1860-5397-16-227-2]](/bjoc/content/figures/1860-5397-16-227-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cytotoxicity of spirolactams on P388 cells (IC50 values).
Figure 2: Cytotoxicity of spirolactams on P388 cells (IC50 values).
Conclusion
In conclusion, we have achieved a new bifurcated syntheses of N-alkyl and N-phenyl-substituted spirolactams bearing a methylene-lactone or methylene-lactam structure. The key intermediates were synthesized by electrophilic allylation of γ-phenylthio-functionalized γ-lactam derivatives with 2-(acetoxy)methyl acrylamides in the absence of any metal catalyst and delivered into each type of spirolactams through desulfinative hydroxylation/lactamization or N-Boc protection/desulfinative lactonization. The increase of diversity of structure accessible led to further understanding of the structure–cytotoxicity relationship of spirolactams on P388 cells.
References
-
Lepoittevin, J.-P.; Berl, V.; Giménez-Arnau, E. Chem. Rec. 2009, 9, 258–270. doi:10.1002/tcr.200900013
Return to citation in text: [1] -
Ramachandran, P. V.; Yip-Schneider, M.; Schmidt, C. M. Future Med. Chem. 2009, 1, 179–200. doi:10.4155/fmc.09.15
Return to citation in text: [1] -
Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Biomed. Pharmacother. 2020, 125, 109955. doi:10.1016/j.biopha.2020.109955
Return to citation in text: [1] -
Hoffmann, H. M. R.; Rabe, J. Angew. Chem., Int. Ed. Engl. 1985, 24, 94–110. doi:10.1002/anie.198500941
Return to citation in text: [1] [2] -
Ando, M. Yuki Gosei Kagaku Kyokaishi 1992, 50, 858–874. doi:10.5059/yukigoseikyokaishi.50.858
Return to citation in text: [1] -
Kitson, R. R. A.; Millemaggi, A.; Taylor, R. J. K. Angew. Chem., Int. Ed. 2009, 48, 9426–9451. doi:10.1002/anie.200903108
Return to citation in text: [1] [2] -
Sarma, J. C.; Sharma, R. P. Heterocycles 1986, 24, 441–457. doi:10.3987/r-1986-02-0441
Return to citation in text: [1] -
NISHITANI, K.; NAKAMURA, Y.; ORII, R.; ARAI, C.; YAMAKAWA, K. Chem. Pharm. Bull. 1993, 41, 822–831. doi:10.1248/cpb.41.822
Return to citation in text: [1] -
Heindel, N. D.; Minatelli, J. A. J. Pharm. Sci. 1981, 70, 84–86. doi:10.1002/jps.2600700117
Return to citation in text: [1] -
Murata, Y.; Takahashi, M.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Org. Lett. 2013, 15, 6182–6185. doi:10.1021/ol403014u
Return to citation in text: [1] [2] -
Takahashi, M.; Murata, Y.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Chem. – Eur. J. 2014, 20, 11091–11100. doi:10.1002/chem.201403357
Return to citation in text: [1] [2] -
Sengoku, T.; Sugiyama, A.; Kamiya, Y.; Maegawa, R.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2017, 1285–1288. doi:10.1002/ejoc.201601612
Return to citation in text: [1] -
Sengoku, T.; Kamiya, Y.; Kawakami, A.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2017, 6096–6098. doi:10.1002/ejoc.201701112
Return to citation in text: [1] [2] -
Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Sengoku, T.; Maegawa, R.; Imamura, H.; Wada, M.; Yoda, H. Adv. Synth. Catal. 2020, 362, 2397–2418. doi:10.1002/adsc.202000195
Return to citation in text: [1] -
Sengoku, T.; Miyoshi, A.; Tsuda, T.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. Tetrahedron 2020, 76, 131252. doi:10.1016/j.tet.2020.131252
Return to citation in text: [1] [2] -
Sengoku, T.; Hayashi, D.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2018, 1813–1820. doi:10.1002/ejoc.201800084
Return to citation in text: [1] [2] -
Sonderegger, O. J.; Bürgi, T.; Limbach, L. K.; Baiker, A. J. Mol. Catal. A: Chem. 2004, 217, 93–101. doi:10.1016/j.molcata.2004.02.018
Return to citation in text: [1] -
Zhang, J.-R.; Jin, H.-S.; Sun, J.; Wang, J.; Zhao, L.-M. Eur. J. Org. Chem. 2020, 4988–4994. doi:10.1002/ejoc.202000830
Return to citation in text: [1] -
Fernández de la Pradilla, R.; Viso, A. Alkylation of α-Sulfur-Containing Carbanions. In Comprehensive Organic Synthesis II; Knochel, P.; Molander, G. A., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 3, pp 157–208. doi:10.1016/b978-0-08-097742-3.00304-9
Return to citation in text: [1] -
Yoda, H.; Inoue, K.-i.; Ujihara, Y.; Mase, N.; Takabe, K. Tetrahedron Lett. 2003, 44, 9057–9060. doi:10.1016/j.tetlet.2003.09.204
Return to citation in text: [1] [2] -
As for the reaction process, it remains unclear whether the reaction proceeds through a SN2 or a SN2’ pathway because the mechanism in this case should be dependent on the reaction conditions (solvent, base, temperature, etc).
Return to citation in text: [1] -
Yamaguchi, R.; Mochizuki, K.; Kozima, S.; Takaya, H. Chem. Lett. 1994, 23, 1809–1812. doi:10.1246/cl.1994.1809
Return to citation in text: [1] -
Shen, A.; Liu, M.; Jia, Z.-S.; Xu, M.-H.; Lin, G.-Q. Org. Lett. 2010, 12, 5154–5157. doi:10.1021/ol102148b
Return to citation in text: [1] -
Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886
Return to citation in text: [1] -
Valeev, R. F.; Bikzhanov, R. F.; Miftakhov, M. S. Russ. J. Org. Chem. 2014, 50, 1511–1519. doi:10.1134/s1070428014100170
Return to citation in text: [1] -
Sengoku, T.; Nagai, Y.; Inuzuka, T.; Yoda, H. Synlett 2019, 30, 199–202. doi:10.1055/s-0037-1611941
Return to citation in text: [1] -
Cytotoxic evaluation of compound A also supports this suggestion, in which the comparable level of IC50 value (0.27 μg/mL) was obtained.
Return to citation in text: [1]
| 14. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 23. | Yamaguchi, R.; Mochizuki, K.; Kozima, S.; Takaya, H. Chem. Lett. 1994, 23, 1809–1812. doi:10.1246/cl.1994.1809 |
| 24. | Shen, A.; Liu, M.; Jia, Z.-S.; Xu, M.-H.; Lin, G.-Q. Org. Lett. 2010, 12, 5154–5157. doi:10.1021/ol102148b |
| 25. | Donmez, S. E.; Soydaş, E.; Aydın, G.; Şahin, O.; Bozkaya, U.; Türkmen, Y. E. Org. Lett. 2019, 21, 554–558. doi:10.1021/acs.orglett.8b03886 |
| 26. | Valeev, R. F.; Bikzhanov, R. F.; Miftakhov, M. S. Russ. J. Org. Chem. 2014, 50, 1511–1519. doi:10.1134/s1070428014100170 |
| 1. | Lepoittevin, J.-P.; Berl, V.; Giménez-Arnau, E. Chem. Rec. 2009, 9, 258–270. doi:10.1002/tcr.200900013 |
| 2. | Ramachandran, P. V.; Yip-Schneider, M.; Schmidt, C. M. Future Med. Chem. 2009, 1, 179–200. doi:10.4155/fmc.09.15 |
| 3. | Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Biomed. Pharmacother. 2020, 125, 109955. doi:10.1016/j.biopha.2020.109955 |
| 10. | Murata, Y.; Takahashi, M.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Org. Lett. 2013, 15, 6182–6185. doi:10.1021/ol403014u |
| 11. | Takahashi, M.; Murata, Y.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Chem. – Eur. J. 2014, 20, 11091–11100. doi:10.1002/chem.201403357 |
| 14. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 9. | Heindel, N. D.; Minatelli, J. A. J. Pharm. Sci. 1981, 70, 84–86. doi:10.1002/jps.2600700117 |
| 10. | Murata, Y.; Takahashi, M.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Org. Lett. 2013, 15, 6182–6185. doi:10.1021/ol403014u |
| 11. | Takahashi, M.; Murata, Y.; Yagishita, F.; Sakamoto, M.; Sengoku, T.; Yoda, H. Chem. – Eur. J. 2014, 20, 11091–11100. doi:10.1002/chem.201403357 |
| 14. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 21. | Yoda, H.; Inoue, K.-i.; Ujihara, Y.; Mase, N.; Takabe, K. Tetrahedron Lett. 2003, 44, 9057–9060. doi:10.1016/j.tetlet.2003.09.204 |
| 4. | Hoffmann, H. M. R.; Rabe, J. Angew. Chem., Int. Ed. Engl. 1985, 24, 94–110. doi:10.1002/anie.198500941 |
| 6. | Kitson, R. R. A.; Millemaggi, A.; Taylor, R. J. K. Angew. Chem., Int. Ed. 2009, 48, 9426–9451. doi:10.1002/anie.200903108 |
| 7. | Sarma, J. C.; Sharma, R. P. Heterocycles 1986, 24, 441–457. doi:10.3987/r-1986-02-0441 |
| 8. | NISHITANI, K.; NAKAMURA, Y.; ORII, R.; ARAI, C.; YAMAKAWA, K. Chem. Pharm. Bull. 1993, 41, 822–831. doi:10.1248/cpb.41.822 |
| 21. | Yoda, H.; Inoue, K.-i.; Ujihara, Y.; Mase, N.; Takabe, K. Tetrahedron Lett. 2003, 44, 9057–9060. doi:10.1016/j.tetlet.2003.09.204 |
| 4. | Hoffmann, H. M. R.; Rabe, J. Angew. Chem., Int. Ed. Engl. 1985, 24, 94–110. doi:10.1002/anie.198500941 |
| 5. | Ando, M. Yuki Gosei Kagaku Kyokaishi 1992, 50, 858–874. doi:10.5059/yukigoseikyokaishi.50.858 |
| 6. | Kitson, R. R. A.; Millemaggi, A.; Taylor, R. J. K. Angew. Chem., Int. Ed. 2009, 48, 9426–9451. doi:10.1002/anie.200903108 |
| 22. | As for the reaction process, it remains unclear whether the reaction proceeds through a SN2 or a SN2’ pathway because the mechanism in this case should be dependent on the reaction conditions (solvent, base, temperature, etc). |
| 18. | Sonderegger, O. J.; Bürgi, T.; Limbach, L. K.; Baiker, A. J. Mol. Catal. A: Chem. 2004, 217, 93–101. doi:10.1016/j.molcata.2004.02.018 |
| 19. | Zhang, J.-R.; Jin, H.-S.; Sun, J.; Wang, J.; Zhao, L.-M. Eur. J. Org. Chem. 2020, 4988–4994. doi:10.1002/ejoc.202000830 |
| 17. | Sengoku, T.; Hayashi, D.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2018, 1813–1820. doi:10.1002/ejoc.201800084 |
| 17. | Sengoku, T.; Hayashi, D.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2018, 1813–1820. doi:10.1002/ejoc.201800084 |
| 20. | Fernández de la Pradilla, R.; Viso, A. Alkylation of α-Sulfur-Containing Carbanions. In Comprehensive Organic Synthesis II; Knochel, P.; Molander, G. A., Eds.; Elsevier: Amsterdam, Netherlands, 2014; Vol. 3, pp 157–208. doi:10.1016/b978-0-08-097742-3.00304-9 |
| 13. | Sengoku, T.; Kamiya, Y.; Kawakami, A.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2017, 6096–6098. doi:10.1002/ejoc.201701112 |
| 14. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 16. | Sengoku, T.; Miyoshi, A.; Tsuda, T.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. Tetrahedron 2020, 76, 131252. doi:10.1016/j.tet.2020.131252 |
| 27. | Sengoku, T.; Nagai, Y.; Inuzuka, T.; Yoda, H. Synlett 2019, 30, 199–202. doi:10.1055/s-0037-1611941 |
| 12. | Sengoku, T.; Sugiyama, A.; Kamiya, Y.; Maegawa, R.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2017, 1285–1288. doi:10.1002/ejoc.201601612 |
| 13. | Sengoku, T.; Kamiya, Y.; Kawakami, A.; Takahashi, M.; Yoda, H. Eur. J. Org. Chem. 2017, 6096–6098. doi:10.1002/ejoc.201701112 |
| 14. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 15. | Sengoku, T.; Maegawa, R.; Imamura, H.; Wada, M.; Yoda, H. Adv. Synth. Catal. 2020, 362, 2397–2418. doi:10.1002/adsc.202000195 |
| 16. | Sengoku, T.; Miyoshi, A.; Tsuda, T.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. Tetrahedron 2020, 76, 131252. doi:10.1016/j.tet.2020.131252 |
| 14. | Sengoku, T.; Shirai, A.; Takano, A.; Inuzuka, T.; Sakamoto, M.; Takahashi, M.; Yoda, H. J. Org. Chem. 2019, 84, 12532–12541. doi:10.1021/acs.joc.9b02038 |
| 28. | Cytotoxic evaluation of compound A also supports this suggestion, in which the comparable level of IC50 value (0.27 μg/mL) was obtained. |
© 2020 Sengoku et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)