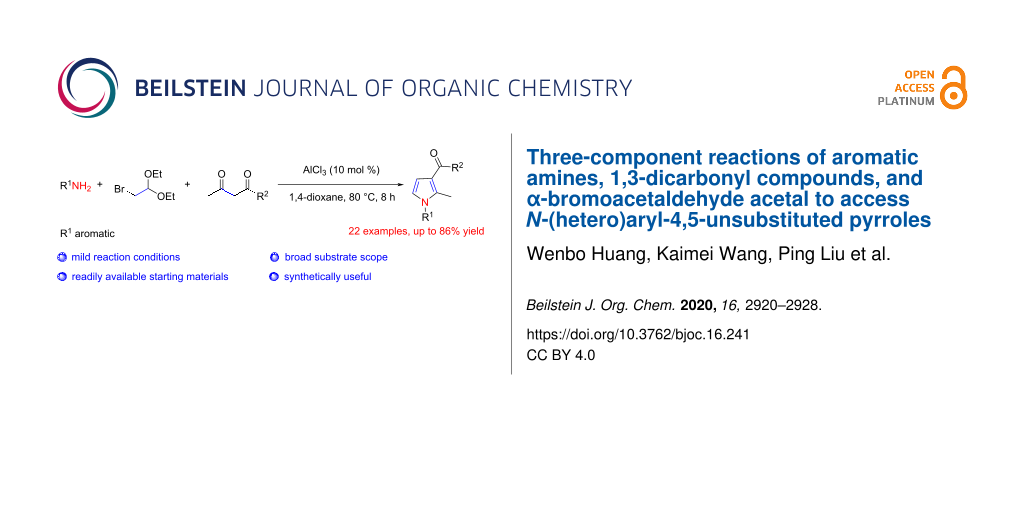
Abstract
N-(Hetero)aryl-4,5-unsubstituted pyrroles were synthesized from (hetero)arylamines, 1,3-dicarbonyl compounds, and α-bromoacetaldehyde acetal by using aluminum(III) chloride as a Lewis acid catalyst through [1 + 2 + 2] annulation. This new versatile methodology provides a wide scope for the synthesis of different functional N-(hetero)aryl-4,5-unsubstituted pyrrole scaffolds, which can be further derived to access multisubstituted pyrrole-3-carboxamides. In the presence of 1.2 equiv of KI, a polysubstituted pyrazolo[3,4-b]pyridine derivative was also successfully synthesized.

Graphical Abstract
Introduction
Among nitrogen-containing heterocycles, pyrroles have garnered significant attention in the literature because of their presence in various natural products [1-4] and pharmaceutically relevant drugs [5,6]. Accordingly, numerous synthetic methods to construct pyrrole skeletons were reported, including the classical Hantzsch [7,8] and the Paal–Knorr pyrrole syntheses [9-11], which have been developed to harvest the pyrrole frameworks. In the past few years, the interest in developing new methods to synthesize this heterocyclic motif has rapidly grown; transition metal-catalyzed cyclization [12-14] and multicomponent reactions [15-18] are some of the commonly used approaches for the construction of pyrrole scaffolds. Additionally, the biocatalytic synthesis of substituted pyrroles was also developed [19]. Though sustained efforts have been achieved to develop efficient synthetic methods for the preparation of this structural motif [20-23], the development of cost-effective methods to access functionalized pyrrole skeletons has remained an ongoing challenge.
N-(Hetero)aryl-4,5-unsubstituted pyrroles are one of the most important types of pyrroles, which are frequently used as a core scaffold in pharmaceuticals (Figure 1) [24,25]. Therefore, many efforts have been paid to the synthesis of these privileged pyrroles. (Hetero)arylamines are readily available chemicals. The direct conversion of (hetero)arylamines into N-(hetero)aryl-4,5-unsubstituted pyrroles has a high intrinsic synthetic potential. At present, the transformations can generally be realized through the following three approaches (Scheme 1): (i) [1 + 1 + 3] annulation, in which (hetero)arylamines are reacted with a C3 donor and a C1 donor to construct pyrrole scaffolds. Kumar et al. [26] developed a proline-catalyzed Mannich reaction–cyclization sequence of succinaldehyde and an in situ-generated arylimine, in which the succinaldehyde contributes three carbon atoms to the pyrrole ring. α,β-Unsaturated aldehydes have also been used as the C3 donor to construct pyrrole scaffolds [27,28]; (ii) [1 + 4] annulation, in which (hetero)arylamines are reacted with a C4 donor to form the pyrrole ring; many functional molecules, such as bioderived furans [29], (Z)-enynols [30], 1-vinylpropargyl alcohols [31], doubly activated cyclopropanes [32], and enynals [33], can be used as C4 counter reagents. The carbon-based 1,4-biselectrophiles, such as the 1,4-dicarbonyl compounds [34,35], γ-carbonyl tert-butyl peroxides [36], and dihydrofurans [37] have also been reported to construct the pyrrole skeletons through this type of annulation; and (iii) [1 + 2 + 2] annulation, in which (hetero)arylamines are reacted with two different molecules, and each of them contributes two carbon atoms to construct a pyrrole ring [38-42]. Among these three approaches, the third is considered the most attractive route for N-(hetero)aryl-4,5-unsubstituted pyrrole synthesis. The reason is twofold: (i) the strategy uses easily available substrates and (ii) permits to synthesize pyrroles with a high potential of molecular diversity and complexity. However, to date, the productivity for creating molecular diversity and complexity has yet to be fully displayed. In addition, some of the reported approaches were established on the basis of using expensive and nonrecyclable homogeneous metal catalysts. To alleviate all these problems, herein, we used easily available α-bromoacetaldehyde acetal (2a) and a simple 1,3-dicarbonyl compound as a reagent couple to react with (hetero)arylamines. The established [1 + 2 + 2] annulation reaction provided a straightforward approach for accessing various N-(hetero)aryl-4,5-unsubstituted pyrroles, and some of the pyrrole products are not accessible with the methods reported hitherto.
![[1860-5397-16-241-1]](/bjoc/content/figures/1860-5397-16-241-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative biologically active N-(hetero)aryl-4,5-unsubstituted pyrrole scaffolds.
Figure 1: Representative biologically active N-(hetero)aryl-4,5-unsubstituted pyrrole scaffolds.
![[1860-5397-16-241-i1]](/bjoc/content/inline/1860-5397-16-241-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Typical routes to N-(heteroaryl)-4,5-unsubstituted pyrroles.
Scheme 1: Typical routes to N-(heteroaryl)-4,5-unsubstituted pyrroles.
Results and Discussion
Initially, a mixture of aniline (1a), α-bromoacetaldehyde acetal (2a), and ethyl acetoacetate (3a) was treated under the conditions; the obtained results are listed in Table 1. The mixture was heated in 1,4-dioxane at 80 °C. No reaction occurred in the absence of the catalyst (Table 1, entry 1); however, in the presence of the strong Lewis acid Bi(OTf)3, the expected product 4a was obtained in 36% yield after 6 h of reaction (Table 1, entry 2). To our surprise, the N-aryl-4,5-unsubstituted pyrrole derivative 4a was isolated in 80% yield when 10 mol % of AlCl3 was used as the catalyst (Table 1, entry 3). FeCl3 and NiCl2 were also proven to catalyze this reaction, but the yield of 4a was inferior to those obtained with AlCl3 (Table 1, entries 4 and 5). p-Toluenesulfonic acid (PTSA), a strong Brønsted acid, also exhibited a promising catalytic ability, and the yield of 4a reached 73% (Table 1, entry 6). When HOAc was used, only unreacted starting materials were recovered (Table, entry 7). The effect of the solvent on the model reaction was also examined. Anhydrous ethanol, acetonitrile, toluene, and DMSO did not bring any improvement with respect to the yield of 4a (Table 1, entries 8–11). The decrease of the catalyst loading from 10 to 5 mol % resulted in a slight decrease of the reaction yield (Table 1, entry 12). Further investigations revealed that the reaction was also affected by the temperature and time; a yield of only 51% was obtained at 50 °C (Table 1, entries 13 and 14). Therefore, the optimized reaction conditions were confirmed as the following: 10 mol % of AlCl3 as a catalyst, 1,4-dioxane as a solvent, 6 h, and 80 °C. It is worth noting that the reaction can be effectively scaled up with similar efficiency. In a gram-scale synthesis of 4a, the corresponding pyrrole product was obtained in 72% yield (Table 1, entry 15).
Table 1: Optimization of the conditions for the reaction between 1a, 2a, and 3a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-241-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | catalyst | solvent | yield (%)b |
| 1 | — | 1,4-dioxane | 0 |
| 2 | Bi(OTf)3 | 1,4-dioxane | 36 |
| 3 | AlCl3 | 1,4-dioxane | 80 |
| 4 | FeCl3 | 1,4-dioxane | 44 |
| 5 | NiCl2 | 1,4-dioxane | 21 |
| 6 | PTSA | 1,4-dioxane | 73 |
| 7 | HOAc | 1,4-dioxane | trace |
| 8 | AlCl3 | EtOH | 31 |
| 9 | AlCl3 | MeCN | 50 |
| 10 | AlCl3 | PhMe | 27 |
| 11 | AlCl3 | DMSO | 62 |
| 12c | AlCl3 | 1,4-dioxane | 55 |
| 13d | AlCl3 | 1,4-dioxane | 51 |
| 14e | AlCl3 | 1,4-dioxane | 40 |
| 15f | AlCl3 | 1,4-dioxane | 72 |
a1a: 0.5 mmol, 2a: 0.6 mmol, 3a: 0.6 mmol, catalyst: 0.05 mmol, solvent: 1 mL, 80 °C, 6 h. bIsolated yield, calculated with respect to 1a. cAlCl3: 0.025 mmol. d50 °C. e2 h. f10 mmol-scale reaction.
The scope of this synthetic protocol for pyrroles was then investigated under the optimized reaction conditions. The substrate scope of the 1,3-dicarbonyl component was first examined (Scheme 2). Acetylacetone reacted smoothly with 1a and 2a to form 4b in 52% yield. 1,3-Dicarbonyl compounds bearing an ester group readily participated in this reaction, affording the desired pyrroles 4c–e in moderate to good yield. Notably, methyl 2-methyl-1-phenyl-1H-pyrrole-3-carboxylate (4c) is a key intermediate in the synthesis of a TRPM8 antagonist [43]. The substrate scope of the aromatic amine component was then examined, and the remarkable efficiency of our pyrrole synthesis was reflected by the tolerance of a broad range of functional groups attached to the aromatic amine. For example, anilines bearing methyl (in 4f), phenyl (in 4g), and halo functionalities (in 4h–m) were readily compatible with the AlCl3 and 1,4-dioxane system. In these cases, the pyrrole products were isolated in moderate to good yield. The presence of an electron-donating group in the phenyl ring facilitated the progress of the reaction to some extent, resulting in 4n. Anilines with electron-withdrawing groups, such as an acetyl or carboxy group, can also be used in the reaction, but the yields obtained for 4o and 4p were slightly inferior. Gratifyingly, a 3,5-dichloro-4-(1,1,2,2-tetrafluoroethoxy)aniline also participated smoothly in this reaction, and the expected product 4q can be obtained in 75% yield. This fluorinated substituent on the aniline ring has been identified as the key precursor to access the insect-growth regulators [44]. It is noteworthy that the sulfonamide group persisted during this transformation, and the desired pyrrole product 4r could be obtained in 69% yield. A high yield was also obtained when the phenyl group was replaced with a naphthyl group in 4s. Subsequently, aliphatic primary amines and ammonia, such as benzylamine and N-butylamine, were also examined as nitrogen donors; however, no desired product was detected.
![[1860-5397-16-241-i2]](/bjoc/content/inline/1860-5397-16-241-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the pyrrole synthesis.
Scheme 2: Substrate scope of the pyrrole synthesis.
Then, we attempted to synthesize pharmaceutically active N-heterocyclic pyrrole derivatives with the aid of this three-component reaction. To our great delight, the N-heterocyclic pyrrole skeletons 4t–v were successfully synthesized in 51–85% yield by using our protocol (Scheme 3). It should be mentioned that the obtained N-pyridylpyrroles 4t and 4u are a class of very important intermediates for synthesizing the soluble guanylyl cyclase (sGC) activator. A reported method for accessing these similar scaffolds suffers from a low product yield and harsh reaction conditions (32%, 130 °C) [45,46].
![[1860-5397-16-241-i3]](/bjoc/content/inline/1860-5397-16-241-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of N-heterocyclic pyrroles.
Scheme 3: Synthesis of N-heterocyclic pyrroles.
One of the obtained N-aryl-4,5-unsubstituted pyrroles, 4l, could undergo hydrolysis to form the (pyrrol-3-yl)carboxylic acid 4w. The latter can react readily with aniline (1a) or amantadine in the presence of HATU or EDCI to form the multisubstituted pyrrole-3-carboxamide derivatives 4x and 4y (Scheme 4). These skeletons have been proven to be promising inhibitors for the production of cytokines [47].
![[1860-5397-16-241-i4]](/bjoc/content/inline/1860-5397-16-241-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Direct synthesis of pyrrole-3-carboxamide derivatives.
Scheme 4: Direct synthesis of pyrrole-3-carboxamide derivatives.
A plausible mechanism for the model reaction was proposed and is depicted in Scheme 5. Initially, a reaction of 1a and 3a occurred, providing an imine intermediate I, which tautomerized to the corresponding enamine form. Meanwhile, the activation of 2a with AlCl3 allowed the formation of a carbocation intermediate II, which was then trapped by the enamine intermediate I to generate another intermediate III. Subsequently, III underwent an intramolecular electrophilic substitution to form the intermediate IV. Finally, IV underwent an elimination of HBr and a spontaneous aromatization to afford the pyrrole product 4a [48,49].
![[1860-5397-16-241-i5]](/bjoc/content/inline/1860-5397-16-241-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism of the three-component reaction.
Scheme 5: Plausible mechanism of the three-component reaction.
Apart from the pyrrole synthesis, we also observed an unexpected reaction in which the pyrazolo[3,4-b]pyridine scaffold was constructed under analogous conditions. As shown in Scheme 6, In the presence of a catalytic amount of AlCl3 and 1.2 equiv of KI, 3-methyl-1-phenyl-1H-pyrazol-5-amine (1b) reacted smoothly with 2a and 3a, affording the polysubstituted pyrazolo[3,4-b]pyridine 5a in 53% yield. The formation of an imine intermediate V may be involved in the reaction mechanism, and the enamine form of V contained two nucleophilic sites to react with a molecule of 3a, which generated the intermediate VI. Finally, VI underwent an aromatization to produce 5a. KI may play a key role in the last step, and we suspected that it can promote the removal of bromide. Although 5a can theoretically be synthesized through a three-component reaction of 1b, 2a, and an appropriate aldehyde, for example, acetaldehyde [50,51] owing to the insufficient reactivity of aliphatic aldehydes, the reaction in Scheme 6 should be a wise choice for the synthesis of products similar in type to 5a. This point deserves further investigation.
![[1860-5397-16-241-i6]](/bjoc/content/inline/1860-5397-16-241-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of polysubstituted pyrazolo[3,4-b]pyridine derivatives.
Scheme 6: Synthesis of polysubstituted pyrazolo[3,4-b]pyridine derivatives.
Conclusion
In summary, an efficient and practical one-pot multicomponent reaction of (hetero)arylamines with α-bromoacetaldehyde acetal (2a) and 1,3-dicarbonyl compounds was developed by using AlCl3 as a catalyst. The developed chemistry is also successful for the synthesis of functionalized pyrazolo[3,4-b]pyridine derivatives. This study offered a complementary method to construct pyrrole scaffolds through [1 + 2 + 2] annulation, and thus enriching the product diversity of the pyrrole derivatives.
Supporting Information
| Supporting Information File 1: Experimental procedures and copies of NMR spectra. | ||
| Format: PDF | Size: 4.3 MB | Download |
Funding
The authors thank the Program for Leading Talents of Hubei Academy of Agricultural Sciences (L2018031) and the Natural Science Foundation of Hubei Province (2020CFB717), and the authors also thank the partial support from the Projects in the Youth Science Foundation of Hubei Academy of Agricultural Sciences (2021NKYJJ17) and Hubei Key Laboratory of Crop Diseases, Insect Pests and Weeds Control (2020ZTSJJ5), the Natural Science Foundation of Hubei Province (2019CFB219), and the National Natural Science Foundation of China (2171101076 and 21872060).
References
-
O’Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d
Return to citation in text: [1] -
Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. Nat. Prod. Rep. 2006, 23, 517–531. doi:10.1039/b605245m
Return to citation in text: [1] -
Trost, B. M.; Dong, G. Org. Lett. 2007, 9, 2357–2359. doi:10.1021/ol070742y
Return to citation in text: [1] -
Lindel, T.; Hochgürtel, M.; Assmann, M.; Köck, M. J. Nat. Prod. 2000, 63, 1566–1569. doi:10.1021/np000160o
Return to citation in text: [1] -
Khajuria, R.; Dham, S.; Kapoor, K. K. RSC Adv. 2016, 6, 37039–37066. doi:10.1039/c6ra03411j
Return to citation in text: [1] -
Osman, N. A.; Ligresti, A.; Klein, C. D.; Allarà, M.; Rabbito, A.; Di Marzo, V.; Abouzid, K. A.; Abadi, A. H. Eur. J. Med. Chem. 2016, 122, 619–634. doi:10.1016/j.ejmech.2016.07.012
Return to citation in text: [1] -
Hantzsch, A. Ber. Dtsch. Chem. Ges. 1890, 23, 1474–1476. doi:10.1002/cber.189002301243
Return to citation in text: [1] -
Leonardi, M.; Estévez, V.; Villacampa, M.; Menéndez, J. C. Synthesis 2019, 51, 816–828. doi:10.1055/s-0037-1610320
Return to citation in text: [1] -
Knorr, L. Ber. Dtsch. Chem. Ges. 1884, 17, 1635–1642. doi:10.1002/cber.18840170220
Return to citation in text: [1] -
Paal, C. Ber. Dtsch. Chem. Ges. 1885, 18, 367–371. doi:10.1002/cber.18850180175
Return to citation in text: [1] -
Balakrishna, A.; Aguiar, A.; Sobral, P. J. M.; Wani, M. Y.; Almeida e Silva, J.; Sobral, A. J. F. N. Catal. Rev.: Sci. Eng. 2019, 61, 84–110. doi:10.1080/01614940.2018.1529932
Return to citation in text: [1] -
Bunrit, A.; Sawadjoon, S.; Tšupova, S.; Sjöberg, P. J. R.; Samec, J. S. M. J. Org. Chem. 2016, 81, 1450–1460. doi:10.1021/acs.joc.5b02581
Return to citation in text: [1] -
Billedeau, R. J.; Klein, K. R.; Kaplan, D.; Lou, Y. Org. Lett. 2013, 15, 1421–1423. doi:10.1021/ol400062w
Return to citation in text: [1] -
Andreou, D.; Kallitsakis, M. G.; Loukopoulos, E.; Gabriel, C.; Kostakis, G. E.; Lykakis, I. N. J. Org. Chem. 2018, 83, 2104–2113. doi:10.1021/acs.joc.7b03051
Return to citation in text: [1] -
Firoozi, N.; Torres, G. M.; Arndtsen, B. A. J. Org. Chem. 2016, 81, 11145–11152. doi:10.1021/acs.joc.6b02102
Return to citation in text: [1] -
Zheng, Y.; Wang, Y.; Zhou, Z. Chem. Commun. 2015, 51, 16652–16655. doi:10.1039/c5cc05624a
Return to citation in text: [1] -
Singh, K.; Kabadwal, L. M.; Bera, S.; Alanthadka, A.; Banerjee, D. J. Org. Chem. 2018, 83, 15406–15414. doi:10.1021/acs.joc.8b02666
Return to citation in text: [1] -
Herath, A.; Cosford, N. D. P. Org. Lett. 2010, 12, 5182–5185. doi:10.1021/ol102216x
Return to citation in text: [1] -
Xu, J.; Green, A. P.; Turner, N. J. Angew. Chem., Int. Ed. 2018, 57, 16760–16763. doi:10.1002/anie.201810555
Return to citation in text: [1] -
Chelucci, G. Coord. Chem. Rev. 2017, 331, 37–53. doi:10.1016/j.ccr.2016.09.014
Return to citation in text: [1] -
Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2014, 43, 4633–4657. doi:10.1039/c3cs60015g
Return to citation in text: [1] -
Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084–3213. doi:10.1021/cr300333u
Return to citation in text: [1] -
Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2010, 39, 4402–4421. doi:10.1039/b917644f
Return to citation in text: [1] -
Ahmad, S.; Alam, O.; Naim, M. J.; Shaquiquzzaman, M.; Alam, M. M.; Iqbal, M. Eur. J. Med. Chem. 2018, 157, 527–561. doi:10.1016/j.ejmech.2018.08.002
Return to citation in text: [1] -
Winters, M. P.; Sui, Z.; Wall, M.; Wang, Y.; Gunnet, J.; Leonard, J.; Hua, H.; Yan, W.; Suckow, A.; Bell, A.; Clapper, W.; Jenkinson, C.; Haug, P.; Koudriakova, T.; Huebert, N.; Murray, W. V. Bioorg. Med. Chem. Lett. 2018, 28, 841–846. doi:10.1016/j.bmcl.2018.02.013
Return to citation in text: [1] -
Singh, A.; Mir, N. A.; Choudhary, S.; Singh, D.; Sharma, P.; Kant, R.; Kumar, I. RSC Adv. 2018, 8, 15448–15458. doi:10.1039/c8ra01637b
Return to citation in text: [1] -
Thompson, B. B.; Montgomery, J. Org. Lett. 2011, 13, 3289–3291. doi:10.1021/ol201133n
Return to citation in text: [1] -
Biletzki, T.; Imhof, W. Synthesis 2011, 3979–3990. doi:10.1055/s-0031-1289584
Return to citation in text: [1] -
Tao, L.; Wang, Z.-J.; Yan, T.-H.; Liu, Y.-M.; He, H.-Y.; Cao, Y. ACS Catal. 2017, 7, 959–964. doi:10.1021/acscatal.6b02953
Return to citation in text: [1] -
Lu, Y.; Fu, X.; Chen, H.; Du, X.; Jia, X.; Liu, Y. Adv. Synth. Catal. 2009, 351, 129–134. doi:10.1002/adsc.200800490
Return to citation in text: [1] -
Thies, N.; Gerlach, M.; Haak, E. Eur. J. Org. Chem. 2013, 7354–7365. doi:10.1002/ejoc.201300803
Return to citation in text: [1] -
Zhang, Z.; Zhang, W.; Li, J.; Liu, Q.; Liu, T.; Zhang, G. J. Org. Chem. 2014, 79, 11226–11233. doi:10.1021/jo5018487
Return to citation in text: [1] -
Kong, H.-H.; Pan, H.-L.; Ding, M.-W. J. Org. Chem. 2018, 83, 12921–12930. doi:10.1021/acs.joc.8b01984
Return to citation in text: [1] -
Tracey, M. R.; Hsung, R. P.; Lambeth, R. H. Synthesis 2004, 918–922. doi:10.1055/s-2004-815976
Return to citation in text: [1] -
Delayen, A.; Goossens, L.; Goossens, R.; Henichart, J.-P. Heterocycles 2005, 65, 1673–1678. doi:10.3987/com-05-10386
Return to citation in text: [1] -
Lu, S.; Qi, L.; Li, Z. Asian J. Org. Chem. 2017, 6, 313–321. doi:10.1002/ajoc.201600608
Return to citation in text: [1] -
Wang, M.; Liu, C.; Gu, Y. Tetrahedron 2016, 72, 6854–6865. doi:10.1016/j.tet.2016.09.014
Return to citation in text: [1] -
Zhang, M.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 597–601. doi:10.1002/anie.201206082
Return to citation in text: [1] -
Zhang, M.; Fang, X.; Neumann, H.; Beller, M. J. Am. Chem. Soc. 2013, 135, 11384–11388. doi:10.1021/ja406666r
Return to citation in text: [1] -
Chandrasekhar, S.; Patro, V.; Chavan, L. N.; Chegondi, R.; Grée, R. Tetrahedron Lett. 2014, 55, 5932–5935. doi:10.1016/j.tetlet.2014.08.105
Return to citation in text: [1] -
Dang, T. T.; Seayad, A. M. Chem. – Asian J. 2017, 12, 2383–2387. doi:10.1002/asia.201701045
Return to citation in text: [1] -
Zeng, J.-C.; Xu, H.; Huang, R.-L.; Yu, F.; Zhang, Z. Tetrahedron Lett. 2018, 59, 1576–1580. doi:10.1016/j.tetlet.2018.03.025
Return to citation in text: [1] -
Shishido, Y.; Ohmi, M.; Ando, K. Azaspiro derivatives as trpm8 antagonists. WO Patent WO2015136947A1, Sept 17, 2015.
Return to citation in text: [1] -
Sun, R.; Liu, Y.; Zhang, Y.; Xiong, L.; Wang, Q. J. Agric. Food Chem. 2011, 59, 2471–2477. doi:10.1021/jf104578j
Return to citation in text: [1] -
Hirth-Dietrich, C.; Sandner, P.; Stasch, J.-P.; Hahn, M.; Follmann, M. The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc). Eur. Patent EP2594270A2, May 22, 2013.
Return to citation in text: [1] -
Boyer, T.; Dodic, N.; Evans, B.; Kirk, B. E. 2,6-disubstituted pyridines and 2,4-disubstituted pyrimidines as soluble guanylate cyclase activators. WO Patent WO2009068652A1, June 4, 2009.
Return to citation in text: [1] -
Ushio, H.; Hamada, M.; Watanabe, M.; Numata, A.; Fujie, N.; Takashima, T.; Furukawa, H.; Ando, J. Amide derivative and use thereof. WO Patent WO2012050159A1, April 19, 2012.
Return to citation in text: [1] -
Huang, W.; Liu, C.; Gu, Y. Adv. Synth. Catal. 2017, 359, 1811–1818. doi:10.1002/adsc.201700074
Return to citation in text: [1] -
Huang, W.; Chen, S.; Chen, Z.; Yue, M.; Li, M.; Gu, Y. J. Org. Chem. 2019, 84, 5655–5666. doi:10.1021/acs.joc.9b00596
Return to citation in text: [1] -
Tu, S.; Wang, Q.; Zhang, Y.; Xu, J.; Zhang, J.; Zhu, X.; Shi, F. J. Heterocycl. Chem. 2007, 44, 811–814. doi:10.1002/jhet.5570440409
Return to citation in text: [1] -
Gunasekaran, P.; Indumathi, S.; Perumal, S. RSC Adv. 2013, 3, 8318–8325. doi:10.1039/c3ra00136a
Return to citation in text: [1]
| 1. | O’Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d |
| 2. | Walsh, C. T.; Garneau-Tsodikova, S.; Howard-Jones, A. R. Nat. Prod. Rep. 2006, 23, 517–531. doi:10.1039/b605245m |
| 3. | Trost, B. M.; Dong, G. Org. Lett. 2007, 9, 2357–2359. doi:10.1021/ol070742y |
| 4. | Lindel, T.; Hochgürtel, M.; Assmann, M.; Köck, M. J. Nat. Prod. 2000, 63, 1566–1569. doi:10.1021/np000160o |
| 12. | Bunrit, A.; Sawadjoon, S.; Tšupova, S.; Sjöberg, P. J. R.; Samec, J. S. M. J. Org. Chem. 2016, 81, 1450–1460. doi:10.1021/acs.joc.5b02581 |
| 13. | Billedeau, R. J.; Klein, K. R.; Kaplan, D.; Lou, Y. Org. Lett. 2013, 15, 1421–1423. doi:10.1021/ol400062w |
| 14. | Andreou, D.; Kallitsakis, M. G.; Loukopoulos, E.; Gabriel, C.; Kostakis, G. E.; Lykakis, I. N. J. Org. Chem. 2018, 83, 2104–2113. doi:10.1021/acs.joc.7b03051 |
| 32. | Zhang, Z.; Zhang, W.; Li, J.; Liu, Q.; Liu, T.; Zhang, G. J. Org. Chem. 2014, 79, 11226–11233. doi:10.1021/jo5018487 |
| 9. | Knorr, L. Ber. Dtsch. Chem. Ges. 1884, 17, 1635–1642. doi:10.1002/cber.18840170220 |
| 10. | Paal, C. Ber. Dtsch. Chem. Ges. 1885, 18, 367–371. doi:10.1002/cber.18850180175 |
| 11. | Balakrishna, A.; Aguiar, A.; Sobral, P. J. M.; Wani, M. Y.; Almeida e Silva, J.; Sobral, A. J. F. N. Catal. Rev.: Sci. Eng. 2019, 61, 84–110. doi:10.1080/01614940.2018.1529932 |
| 33. | Kong, H.-H.; Pan, H.-L.; Ding, M.-W. J. Org. Chem. 2018, 83, 12921–12930. doi:10.1021/acs.joc.8b01984 |
| 7. | Hantzsch, A. Ber. Dtsch. Chem. Ges. 1890, 23, 1474–1476. doi:10.1002/cber.189002301243 |
| 8. | Leonardi, M.; Estévez, V.; Villacampa, M.; Menéndez, J. C. Synthesis 2019, 51, 816–828. doi:10.1055/s-0037-1610320 |
| 30. | Lu, Y.; Fu, X.; Chen, H.; Du, X.; Jia, X.; Liu, Y. Adv. Synth. Catal. 2009, 351, 129–134. doi:10.1002/adsc.200800490 |
| 5. | Khajuria, R.; Dham, S.; Kapoor, K. K. RSC Adv. 2016, 6, 37039–37066. doi:10.1039/c6ra03411j |
| 6. | Osman, N. A.; Ligresti, A.; Klein, C. D.; Allarà, M.; Rabbito, A.; Di Marzo, V.; Abouzid, K. A.; Abadi, A. H. Eur. J. Med. Chem. 2016, 122, 619–634. doi:10.1016/j.ejmech.2016.07.012 |
| 31. | Thies, N.; Gerlach, M.; Haak, E. Eur. J. Org. Chem. 2013, 7354–7365. doi:10.1002/ejoc.201300803 |
| 24. | Ahmad, S.; Alam, O.; Naim, M. J.; Shaquiquzzaman, M.; Alam, M. M.; Iqbal, M. Eur. J. Med. Chem. 2018, 157, 527–561. doi:10.1016/j.ejmech.2018.08.002 |
| 25. | Winters, M. P.; Sui, Z.; Wall, M.; Wang, Y.; Gunnet, J.; Leonard, J.; Hua, H.; Yan, W.; Suckow, A.; Bell, A.; Clapper, W.; Jenkinson, C.; Haug, P.; Koudriakova, T.; Huebert, N.; Murray, W. V. Bioorg. Med. Chem. Lett. 2018, 28, 841–846. doi:10.1016/j.bmcl.2018.02.013 |
| 27. | Thompson, B. B.; Montgomery, J. Org. Lett. 2011, 13, 3289–3291. doi:10.1021/ol201133n |
| 28. | Biletzki, T.; Imhof, W. Synthesis 2011, 3979–3990. doi:10.1055/s-0031-1289584 |
| 20. | Chelucci, G. Coord. Chem. Rev. 2017, 331, 37–53. doi:10.1016/j.ccr.2016.09.014 |
| 21. | Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2014, 43, 4633–4657. doi:10.1039/c3cs60015g |
| 22. | Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084–3213. doi:10.1021/cr300333u |
| 23. | Estévez, V.; Villacampa, M.; Menéndez, J. C. Chem. Soc. Rev. 2010, 39, 4402–4421. doi:10.1039/b917644f |
| 29. | Tao, L.; Wang, Z.-J.; Yan, T.-H.; Liu, Y.-M.; He, H.-Y.; Cao, Y. ACS Catal. 2017, 7, 959–964. doi:10.1021/acscatal.6b02953 |
| 19. | Xu, J.; Green, A. P.; Turner, N. J. Angew. Chem., Int. Ed. 2018, 57, 16760–16763. doi:10.1002/anie.201810555 |
| 15. | Firoozi, N.; Torres, G. M.; Arndtsen, B. A. J. Org. Chem. 2016, 81, 11145–11152. doi:10.1021/acs.joc.6b02102 |
| 16. | Zheng, Y.; Wang, Y.; Zhou, Z. Chem. Commun. 2015, 51, 16652–16655. doi:10.1039/c5cc05624a |
| 17. | Singh, K.; Kabadwal, L. M.; Bera, S.; Alanthadka, A.; Banerjee, D. J. Org. Chem. 2018, 83, 15406–15414. doi:10.1021/acs.joc.8b02666 |
| 18. | Herath, A.; Cosford, N. D. P. Org. Lett. 2010, 12, 5182–5185. doi:10.1021/ol102216x |
| 26. | Singh, A.; Mir, N. A.; Choudhary, S.; Singh, D.; Sharma, P.; Kant, R.; Kumar, I. RSC Adv. 2018, 8, 15448–15458. doi:10.1039/c8ra01637b |
| 37. | Wang, M.; Liu, C.; Gu, Y. Tetrahedron 2016, 72, 6854–6865. doi:10.1016/j.tet.2016.09.014 |
| 34. | Tracey, M. R.; Hsung, R. P.; Lambeth, R. H. Synthesis 2004, 918–922. doi:10.1055/s-2004-815976 |
| 35. | Delayen, A.; Goossens, L.; Goossens, R.; Henichart, J.-P. Heterocycles 2005, 65, 1673–1678. doi:10.3987/com-05-10386 |
| 36. | Lu, S.; Qi, L.; Li, Z. Asian J. Org. Chem. 2017, 6, 313–321. doi:10.1002/ajoc.201600608 |
| 50. | Tu, S.; Wang, Q.; Zhang, Y.; Xu, J.; Zhang, J.; Zhu, X.; Shi, F. J. Heterocycl. Chem. 2007, 44, 811–814. doi:10.1002/jhet.5570440409 |
| 51. | Gunasekaran, P.; Indumathi, S.; Perumal, S. RSC Adv. 2013, 3, 8318–8325. doi:10.1039/c3ra00136a |
| 47. | Ushio, H.; Hamada, M.; Watanabe, M.; Numata, A.; Fujie, N.; Takashima, T.; Furukawa, H.; Ando, J. Amide derivative and use thereof. WO Patent WO2012050159A1, April 19, 2012. |
| 48. | Huang, W.; Liu, C.; Gu, Y. Adv. Synth. Catal. 2017, 359, 1811–1818. doi:10.1002/adsc.201700074 |
| 49. | Huang, W.; Chen, S.; Chen, Z.; Yue, M.; Li, M.; Gu, Y. J. Org. Chem. 2019, 84, 5655–5666. doi:10.1021/acs.joc.9b00596 |
| 44. | Sun, R.; Liu, Y.; Zhang, Y.; Xiong, L.; Wang, Q. J. Agric. Food Chem. 2011, 59, 2471–2477. doi:10.1021/jf104578j |
| 45. | Hirth-Dietrich, C.; Sandner, P.; Stasch, J.-P.; Hahn, M.; Follmann, M. The use of sGC stimulators, sGC activators, alone and combinations with PDE5 inhibitors for the treatment of systemic sclerosis (SSc). Eur. Patent EP2594270A2, May 22, 2013. |
| 46. | Boyer, T.; Dodic, N.; Evans, B.; Kirk, B. E. 2,6-disubstituted pyridines and 2,4-disubstituted pyrimidines as soluble guanylate cyclase activators. WO Patent WO2009068652A1, June 4, 2009. |
| 38. | Zhang, M.; Neumann, H.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 597–601. doi:10.1002/anie.201206082 |
| 39. | Zhang, M.; Fang, X.; Neumann, H.; Beller, M. J. Am. Chem. Soc. 2013, 135, 11384–11388. doi:10.1021/ja406666r |
| 40. | Chandrasekhar, S.; Patro, V.; Chavan, L. N.; Chegondi, R.; Grée, R. Tetrahedron Lett. 2014, 55, 5932–5935. doi:10.1016/j.tetlet.2014.08.105 |
| 41. | Dang, T. T.; Seayad, A. M. Chem. – Asian J. 2017, 12, 2383–2387. doi:10.1002/asia.201701045 |
| 42. | Zeng, J.-C.; Xu, H.; Huang, R.-L.; Yu, F.; Zhang, Z. Tetrahedron Lett. 2018, 59, 1576–1580. doi:10.1016/j.tetlet.2018.03.025 |
| 43. | Shishido, Y.; Ohmi, M.; Ando, K. Azaspiro derivatives as trpm8 antagonists. WO Patent WO2015136947A1, Sept 17, 2015. |
© 2020 Huang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)