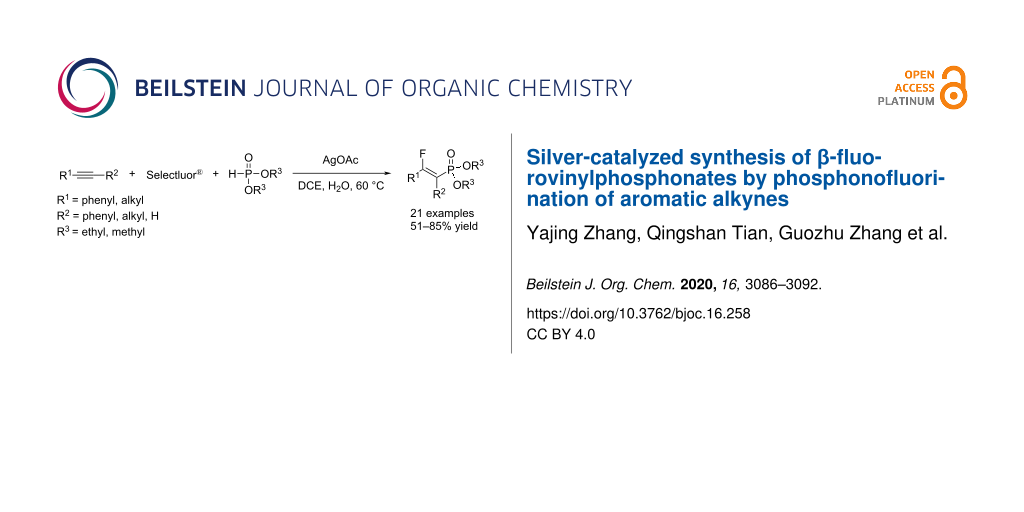
Abstract
A silver-catalyzed three-component reaction involving alkynes, Selectfluor®, and diethyl phosphite was employed for the one-pot formation of C(sp2)–F and C(sp2)–P bonds to provide an efficient access to β-fluorovinylphosphonates in a highly regio- and stereoselective manner under mild reaction conditions. This reaction is operationally simple and offers an excellent functional group tolerance as well as a broad substrate scope that includes both terminal and internal alkynes. The reaction proceeded through the oxidative generation of a P-centered radical and subsequent fluorine atom transfer.

Graphical Abstract
Introduction
As one of the most important topics in organic chemistry, the introduction of fluorine and phosphorus atoms into double bonds is an attractive approach for the synthesis of a variety of valuable organic compounds [1-7]. Although progress has been achieved in the formation of C(sp2)–F bonds from various substrates [8-13], new catalytic reactions to introduce fluorine and phosphorus are seldom reported.
Among the strategies for constructing diverse alkenes containing two-heteroatom bonds, such as disulfonylation [14-16], heterohalogenation [17-20], bis(trifluoromethyl)thiolation [21], and phosphorylation [22], the direct heterodifunctionalization of alkynes using three-component reactions is the most rapid and convenient one (Scheme 1). Although studies on alkyne difunctionalization are ongoing [23], the successful attachment of a fluorine atom to the resulting alkene through transition metal catalysis remains a challenge. In particular, the phosphonofluorination of alkynes for the introduction of two strong electron-withdrawing groups into double bonds has not yet been reported. With our continuous interest in, and inspiration from the well-established transition metal-catalyzed radical difunctionalization of unsaturated carbon–carbon bonds, specifically the work of Li’s group and others concerning the silver-catalyzed phosphonofluorination of alkenes [24-26], we herein present a general silver-catalyzed regio- and stereoselective phosphonofluorination of alkynes using Selectfluor® and phosphonates as reactants (Scheme 1). This new silver-catalyzed approach to fluorinated vinylphosphonates from simple and commercially available alkynes proceeds under mild conditions with high stereoselectivity, and thus enabling the rapid construction of molecular complexity.
![[1860-5397-16-258-i1]](/bjoc/content/inline/1860-5397-16-258-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Metal-catalyzed difunctionalization of unsaturated carbon–carbon bonds.
Scheme 1: Metal-catalyzed difunctionalization of unsaturated carbon–carbon bonds.
Results and Discussion
We investigated the reaction of phenylacetylene (1a), Selectfluor®, and diethyl phosphite to afford β-fluorovinylphosphonates under mild conditions. First, we examined the catalytic activity of various transition metal catalysts, including Au, Ag, Cu, Rh, and Pd complexes (Supporting Information File 1). Among these metal salts, silver salts, especially AgOAc, were the most effective catalysts for generating the desired product 2a (Table 1, entries 1–8). The influence of different reaction temperatures on the reaction outcome was also investigated. At room temperature, the product 2a was obtained in 72% yield (Table 1, entry 8). Increasing the reaction temperature to 60 °C further improved the yield to 79% (Table 1, entry 9), although only a low yield was obtained when the reaction was performed at 100 °C (Table 1, entry 10). The reaction outcome was greatly affected by the solvent used. We carried out this reaction in DCE as the only solvent, and no product was obtained (Table 1, entry 15). The use of EtOAc and H2O or of THF and H2O, instead of DCE and H2O, afforded the monofluoroalkene 2a in only a low yield (Table 1, entries 11 and 12), whereas the product 2a was not obtained when using acetonitrile and H2O or when using DMF (Table 1, entries 13 and 14). Similarly, the desired product was not obtained in the absence of a silver catalyst (Table 1, entry 16). The extensive screening of these reaction parameters revealed that the treatment of the alkynes 1 with Selectfluor®, diethyl phosphite, and silver acetate as the catalyst as well as DCE/H2O 1:1 as the solvent at 60 °C afforded the optimal result.
Table 1: Optimization of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-258-i7.svg?max-width=637&scale=1.0)
|
||||
| entrya | catalyst | solvent | T (°C) | yield (%)b |
| 1 | Ag2CO3 | DCE, H2O | rt | 38 |
| 2 | AgF | DCE, H2O | rt | 24 |
| 3 | AgOTf | DCE, H2O | rt | 34 |
| 4 | AgSbF6 | DCE, H2O | rt | 38 |
| 5 | AgNO3 | DCE, H2O | rt | 58 |
| 6 | AgNO3 | DCE, H2O, AcOH | rt | 62 |
| 7 | AgNO3 | DCE, H2O, TFA | rt | 63 |
| 8 | AgOAc | DCE, H2O | rt | 72 |
| 9 | AgOAc | DCE, H2O | 60 | 79 |
| 10 | AgOAc | DCE, H2O | 100 | 20 |
| 11 | AgOAc | EtOAc, H2O | 60 | 33 |
| 12 | AgOAc | THF, H2O | 60 | 40 |
| 13 | AgOAc | CH3CN, H2O | 60 | — |
| 14 | AgOAc | DMF | 60 | — |
| 15 | AgOAc | DCE | 60 | — |
| 16 | — | DCE, H2O | 60 | — |
aReaction conditions: 1a (0.2 mmol), silver salt (0.02 mmol), diethyl phosphite (0.4 mmol), and Selectfluor® (0.4 mmol) were stirred in the reaction solvent (1 mL:1 mL) at 60 °C for 24 h under N2. bIsolated yield.
The substrate scope of the reaction was investigated under the optimized reaction conditions. As shown in Scheme 2, various β-fluorovinylphosphonates could be conveniently and efficiently obtained using the developed method. Aromatic alkynes bearing various functional groups, including both electron-rich and electron-deficient moieties at the para- and meta-positions generally reacted smoothly to afford the desired products 2a–o in moderate to good yield. An aromatic alkyne possessing a Cl substituent at the ortho-position also reacted smoothly to provide the corresponding product 2k in 56% yield. Substrates containing a range of functional groups, including halides (F, Cl, and Br), CF3, and even ester groups, were amenable to this transformation. The reaction was also compatible with a CN group to afford the corresponding product 2n in 69% yield. Heteroaromatic alkynes, such as 2-ethynylpyridine and 2-ethynylthiophene, did not undergo the reaction to form 2r and 2s. Internal aromatic alkynes were tolerated and generated the desired products 2p and 2q in moderate yield. Aliphatic alkynes, such as (prop-2-yn-1-yl)benzene and (but-3-yn-1-yl)benzene, afforded the corresponding products in an extremely low yield. Another H-phosphonate, namely dimethyl phosphite, was a suitable substrate for this transformation and provided the products 3 in good yield (Scheme 3).
![[1860-5397-16-258-i2]](/bjoc/content/inline/1860-5397-16-258-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope for the synthesis of the β-fluorovinylphosphonates 2 using diethyl phosphite. Reaction conditions: aromatic alkyne 1 (0.2 mmol), AgOAc (0.02 mmol), diethyl phosphite (0.4 mmol), and Selectfluor® (0.4 mmol) were stirred in DCE/H2O 1 mL:1 mL at 60 °C for 24 h under N2. The yields of the isolated products are shown. aThe E/Z ratio was determined by 1H NMR analysis of the reaction mixture after the reaction reached completion.
Scheme 2: Substrate scope for the synthesis of the β-fluorovinylphosphonates 2 using diethyl phosphite. React...
![[1860-5397-16-258-i3]](/bjoc/content/inline/1860-5397-16-258-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope for the synthesis of the β-fluorovinylphosphonates 3 using dimethyl phosphite. Reaction conditions: aromatic alkyne 1 (0.2 mmol), AgOAc (0.02 mmol), dimethyl phosphite (0.4 mmol), and Selectfluor® (0.4 mmol) were stirred in DCE/H2O 1 mL:1 mL at 60 °C for 24 h under N2. The yields of the isolated products are shown.
Scheme 3: Substrate scope for the synthesis of the β-fluorovinylphosphonates 3 using dimethyl phosphite. Reac...
The well-known radical-trapping reagent 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) was used to gain an insight into the reaction mechanism. As illustrated in Scheme 4, the addition of 1 equiv TEMPO suppressed the reaction. In addition, 4-ethynylaniline (1f) reacted with Selectfluor® and dimethyl phosphite under the optimized conditions to afford the expected product in 13% yield, and no competitive electrophilic compound 4f was observed. These findings imply that the transformation may involve a radical process rather than an ionic process.
![[1860-5397-16-258-i4]](/bjoc/content/inline/1860-5397-16-258-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
On the basis of previous reports [23-26], we propose a radical mechanism involving the silver promoter, as illustrated in Scheme 5. In this mechanism, Ag(I) was oxidized by selectflour to generate Ag(III)F. Subsequently, ethyl phosphite is oxidized by Ag(III)F to generate a P-centered radical (INT-I) and Ag(II)F. The electrophilic phosphonyl radical addition to the triple bond of 1a generates the vinyl-free radical INT-II, which is subsequently trapped by AgF(II) to afford the corresponding product 2a. The vinyl radical INT-II stabilizes the unpaired electron through resonance, and INT-II is more stable than INT-III [27,28]. Finally, we obtained the Z-configured product with respect to the aryl and phosphonyl groups.
![[1860-5397-16-258-i5]](/bjoc/content/inline/1860-5397-16-258-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for the silver-catalyzed phosphonofluorination of alkynes.
Scheme 5: Proposed mechanism for the silver-catalyzed phosphonofluorination of alkynes.
We also performed control experiments under standard conditions to determine whether diethyl fluorophosphonate is a key intermediate in the reaction with aromatic alkynes. Based on a method by Gupta et al. [29], we obtained diethyl fluorophosphonate. Subsequently, the diethyl fluorophosphonate did not react with an aromatic alkyne in the desired phosphonofluorination so that no product was obtained (Scheme 6) [30].
![[1860-5397-16-258-i6]](/bjoc/content/inline/1860-5397-16-258-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Attempted use of a suspected phosphonofluorination intermediate to synthesize a β-fluorovinylphosphonate.
Scheme 6: Attempted use of a suspected phosphonofluorination intermediate to synthesize a β-fluorovinylphosph...
Conclusion
In conclusion, a novel method for the synthesis of β-fluorophosphonates through the direct phosphonofluorination of alkynes with Selectfluor® and H-phosphonates under mild reaction conditions has been successfully developed. Owing to its broad substrate scope and excellent functional group tolerance, this simple protocol may represent a general, one-step approach for the preparation of β-fluorophosphonate frameworks for the use in medicinal chemistry.
Supporting Information
| Supporting Information File 1: Experimental procedures, full characterization of products, and NMR spectra. | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem. 2010, 122, 7405–7410. doi:10.1002/ange.201003593
Return to citation in text: [1] -
Schuler, M.; Silva, F.; Bobbio, C.; Tessier, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2008, 47, 7927–7930. doi:10.1002/anie.200802162
Return to citation in text: [1] -
Mankad, N. P.; Toste, F. D. Chem. Sci. 2012, 3, 72–76. doi:10.1039/c1sc00515d
Return to citation in text: [1] -
Akana, J. A.; Bhattacharyya, K. X.; Müller, P.; Sadighi, J. P. J. Am. Chem. Soc. 2007, 129, 7736–7737. doi:10.1021/ja0723784
Return to citation in text: [1] -
Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q
Return to citation in text: [1] -
Jiao, X.-Y.; Bentrude, W. G. J. Am. Chem. Soc. 1999, 121, 6088–6089. doi:10.1021/ja984460s
Return to citation in text: [1] -
Palacios, F.; Ochoa de Retana, A. M.; Pascual, S.; Oyarzabal, J. J. Org. Chem. 2004, 69, 8767–8774. doi:10.1021/jo048682m
Return to citation in text: [1] -
Nguyen, T. T.; Koh, M. J.; Shen, X.; Romiti, F.; Schrock, R. R.; Hoveyda, A. H. Science 2016, 352, 569–575. doi:10.1126/science.aaf4622
Return to citation in text: [1] -
Koh, M. J.; Nguyen, T. T.; Zhang, H.; Schrock, R. R.; Hoveyda, A. H. Nature 2016, 531, 459–465. doi:10.1038/nature17396
Return to citation in text: [1] -
Hu, J.; Han, X.; Yuan, Y.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 13342–13346. doi:10.1002/anie.201708224
Return to citation in text: [1] -
Sakaguchi, H.; Uetake, Y.; Ohashi, M.; Niwa, T.; Ogoshi, S.; Hosoya, T. J. Am. Chem. Soc. 2017, 139, 12855–12862. doi:10.1021/jacs.7b08343
Return to citation in text: [1] -
Liu, T.-L.; Wu, J.; Zhao, Y. Chem. Sci. 2017, 8, 3885–3890. doi:10.1039/c7sc00483d
Return to citation in text: [1] -
Liu, Q.; Shen, X.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 619–623. doi:10.1002/anie.201610127
Return to citation in text: [1] -
Liu, Z.; Yang, L.; Zhang, K.; Chen, W.; Yu, T.; Wang, L.; Gao, W.; Tang, B. Org. Lett. 2020, 22, 2081–2086. doi:10.1021/acs.orglett.0c00575
Return to citation in text: [1] -
Dai, C.; Wang, J.; Deng, S.; Zhou, C.; Zhang, W.; Zhu, Q.; Tang, X. RSC Adv. 2017, 7, 36112–36116. doi:10.1039/c7ra07105a
Return to citation in text: [1] -
Fu, H.; Shang, J.-Q.; Yang, T.; Shen, Y.; Gao, C.-Z.; Li, Y.-M. Org. Lett. 2018, 20, 489–492. doi:10.1021/acs.orglett.7b03922
Return to citation in text: [1] -
Wang, J.; Xiong, H.-Y.; Petit, E.; Bailly, L.; Pannecoucke, X.; Poisson, T.; Besset, T. Chem. Commun. 2019, 55, 8784–8787. doi:10.1039/c9cc01851d
Return to citation in text: [1] -
Liang, S.; Jiang, L.; Yi, W.-b.; Wei, J. Org. Lett. 2018, 20, 7024–7028. doi:10.1021/acs.orglett.8b02929
Return to citation in text: [1] -
Gao, M.; Xu, B. Org. Lett. 2016, 18, 4746–4749. doi:10.1021/acs.orglett.6b02464
Return to citation in text: [1] -
Zhao, M.; Shan, C.-C.; Wang, Z.-L.; Yang, C.; Fu, Y.; Xu, Y.-H. Org. Lett. 2019, 21, 6016–6020. doi:10.1021/acs.orglett.9b02160
Return to citation in text: [1] -
Pan, S.; Li, H.; Huang, Y.; Xu, X.-H.; Qing, F.-L. Org. Lett. 2017, 19, 3247–3250. doi:10.1021/acs.orglett.7b01366
Return to citation in text: [1] -
Zhu, L.; Yu, H.; Guo, Q.; Chen, Q.; Xu, Z.; Wang, R. Org. Lett. 2015, 17, 1978–1981. doi:10.1021/acs.orglett.5b00728
Return to citation in text: [1] -
Tian, Q.; Chen, B.; Zhang, G. Green Chem. 2016, 18, 6236–6240. doi:10.1039/c6gc02656g
Return to citation in text: [1] [2] -
Zhang, C.; Li, Z.; Zhu, L.; Yu, L.; Wang, Z.; Li, C. J. Am. Chem. Soc. 2013, 135, 14082–14085. doi:10.1021/ja408031s
Return to citation in text: [1] [2] -
Sun, B.; Yin, S.; Zhuang, X.; Jin, C.; Su, W. Org. Biomol. Chem. 2018, 16, 6017–6024. doi:10.1039/c8ob01348a
Return to citation in text: [1] [2] -
Lv, Y.; Sun, K.; Pu, W.; Mao, S.; Li, G.; Niu, J.; Chen, Q.; Wang, T. RSC Adv. 2016, 6, 93486–93490. doi:10.1039/c6ra22653a
Return to citation in text: [1] [2] -
Lai, J.; Tian, L.; Liang, Y.; Zhang, Y.; Xie, X.; Fang, B.; Tang, S. Chem. – Eur. J. 2015, 21, 14328–14331. doi:10.1002/chem.201502581
Return to citation in text: [1] -
Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d
Return to citation in text: [1] -
Gupta, A. K.; Acharya, J.; Dubey, D. K.; Kaushik, M. P. J. Fluorine Chem. 2008, 129, 226–229. doi:10.1016/j.jfluchem.2007.11.008
Return to citation in text: [1] -
Chen, Q.; Zeng, J.; Yan, X.; Huang, Y.; Wen, C.; Liu, X.; Zhang, K. J. Org. Chem. 2016, 81, 10043–10048. doi:10.1021/acs.joc.6b01932
Return to citation in text: [1]
| 1. | Wang, W.; Jasinski, J.; Hammond, G. B.; Xu, B. Angew. Chem. 2010, 122, 7405–7410. doi:10.1002/ange.201003593 |
| 2. | Schuler, M.; Silva, F.; Bobbio, C.; Tessier, A.; Gouverneur, V. Angew. Chem., Int. Ed. 2008, 47, 7927–7930. doi:10.1002/anie.200802162 |
| 3. | Mankad, N. P.; Toste, F. D. Chem. Sci. 2012, 3, 72–76. doi:10.1039/c1sc00515d |
| 4. | Akana, J. A.; Bhattacharyya, K. X.; Müller, P.; Sadighi, J. P. J. Am. Chem. Soc. 2007, 129, 7736–7737. doi:10.1021/ja0723784 |
| 5. | Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q |
| 6. | Jiao, X.-Y.; Bentrude, W. G. J. Am. Chem. Soc. 1999, 121, 6088–6089. doi:10.1021/ja984460s |
| 7. | Palacios, F.; Ochoa de Retana, A. M.; Pascual, S.; Oyarzabal, J. J. Org. Chem. 2004, 69, 8767–8774. doi:10.1021/jo048682m |
| 21. | Pan, S.; Li, H.; Huang, Y.; Xu, X.-H.; Qing, F.-L. Org. Lett. 2017, 19, 3247–3250. doi:10.1021/acs.orglett.7b01366 |
| 17. | Wang, J.; Xiong, H.-Y.; Petit, E.; Bailly, L.; Pannecoucke, X.; Poisson, T.; Besset, T. Chem. Commun. 2019, 55, 8784–8787. doi:10.1039/c9cc01851d |
| 18. | Liang, S.; Jiang, L.; Yi, W.-b.; Wei, J. Org. Lett. 2018, 20, 7024–7028. doi:10.1021/acs.orglett.8b02929 |
| 19. | Gao, M.; Xu, B. Org. Lett. 2016, 18, 4746–4749. doi:10.1021/acs.orglett.6b02464 |
| 20. | Zhao, M.; Shan, C.-C.; Wang, Z.-L.; Yang, C.; Fu, Y.; Xu, Y.-H. Org. Lett. 2019, 21, 6016–6020. doi:10.1021/acs.orglett.9b02160 |
| 14. | Liu, Z.; Yang, L.; Zhang, K.; Chen, W.; Yu, T.; Wang, L.; Gao, W.; Tang, B. Org. Lett. 2020, 22, 2081–2086. doi:10.1021/acs.orglett.0c00575 |
| 15. | Dai, C.; Wang, J.; Deng, S.; Zhou, C.; Zhang, W.; Zhu, Q.; Tang, X. RSC Adv. 2017, 7, 36112–36116. doi:10.1039/c7ra07105a |
| 16. | Fu, H.; Shang, J.-Q.; Yang, T.; Shen, Y.; Gao, C.-Z.; Li, Y.-M. Org. Lett. 2018, 20, 489–492. doi:10.1021/acs.orglett.7b03922 |
| 8. | Nguyen, T. T.; Koh, M. J.; Shen, X.; Romiti, F.; Schrock, R. R.; Hoveyda, A. H. Science 2016, 352, 569–575. doi:10.1126/science.aaf4622 |
| 9. | Koh, M. J.; Nguyen, T. T.; Zhang, H.; Schrock, R. R.; Hoveyda, A. H. Nature 2016, 531, 459–465. doi:10.1038/nature17396 |
| 10. | Hu, J.; Han, X.; Yuan, Y.; Shi, Z. Angew. Chem., Int. Ed. 2017, 56, 13342–13346. doi:10.1002/anie.201708224 |
| 11. | Sakaguchi, H.; Uetake, Y.; Ohashi, M.; Niwa, T.; Ogoshi, S.; Hosoya, T. J. Am. Chem. Soc. 2017, 139, 12855–12862. doi:10.1021/jacs.7b08343 |
| 12. | Liu, T.-L.; Wu, J.; Zhao, Y. Chem. Sci. 2017, 8, 3885–3890. doi:10.1039/c7sc00483d |
| 13. | Liu, Q.; Shen, X.; Ni, C.; Hu, J. Angew. Chem., Int. Ed. 2017, 56, 619–623. doi:10.1002/anie.201610127 |
| 23. | Tian, Q.; Chen, B.; Zhang, G. Green Chem. 2016, 18, 6236–6240. doi:10.1039/c6gc02656g |
| 24. | Zhang, C.; Li, Z.; Zhu, L.; Yu, L.; Wang, Z.; Li, C. J. Am. Chem. Soc. 2013, 135, 14082–14085. doi:10.1021/ja408031s |
| 25. | Sun, B.; Yin, S.; Zhuang, X.; Jin, C.; Su, W. Org. Biomol. Chem. 2018, 16, 6017–6024. doi:10.1039/c8ob01348a |
| 26. | Lv, Y.; Sun, K.; Pu, W.; Mao, S.; Li, G.; Niu, J.; Chen, Q.; Wang, T. RSC Adv. 2016, 6, 93486–93490. doi:10.1039/c6ra22653a |
| 29. | Gupta, A. K.; Acharya, J.; Dubey, D. K.; Kaushik, M. P. J. Fluorine Chem. 2008, 129, 226–229. doi:10.1016/j.jfluchem.2007.11.008 |
| 24. | Zhang, C.; Li, Z.; Zhu, L.; Yu, L.; Wang, Z.; Li, C. J. Am. Chem. Soc. 2013, 135, 14082–14085. doi:10.1021/ja408031s |
| 25. | Sun, B.; Yin, S.; Zhuang, X.; Jin, C.; Su, W. Org. Biomol. Chem. 2018, 16, 6017–6024. doi:10.1039/c8ob01348a |
| 26. | Lv, Y.; Sun, K.; Pu, W.; Mao, S.; Li, G.; Niu, J.; Chen, Q.; Wang, T. RSC Adv. 2016, 6, 93486–93490. doi:10.1039/c6ra22653a |
| 30. | Chen, Q.; Zeng, J.; Yan, X.; Huang, Y.; Wen, C.; Liu, X.; Zhang, K. J. Org. Chem. 2016, 81, 10043–10048. doi:10.1021/acs.joc.6b01932 |
| 23. | Tian, Q.; Chen, B.; Zhang, G. Green Chem. 2016, 18, 6236–6240. doi:10.1039/c6gc02656g |
| 22. | Zhu, L.; Yu, H.; Guo, Q.; Chen, Q.; Xu, Z.; Wang, R. Org. Lett. 2015, 17, 1978–1981. doi:10.1021/acs.orglett.5b00728 |
| 27. | Lai, J.; Tian, L.; Liang, Y.; Zhang, Y.; Xie, X.; Fang, B.; Tang, S. Chem. – Eur. J. 2015, 21, 14328–14331. doi:10.1002/chem.201502581 |
| 28. | Wille, U. Chem. Rev. 2013, 113, 813–853. doi:10.1021/cr100359d |
© 2020 Zhang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)