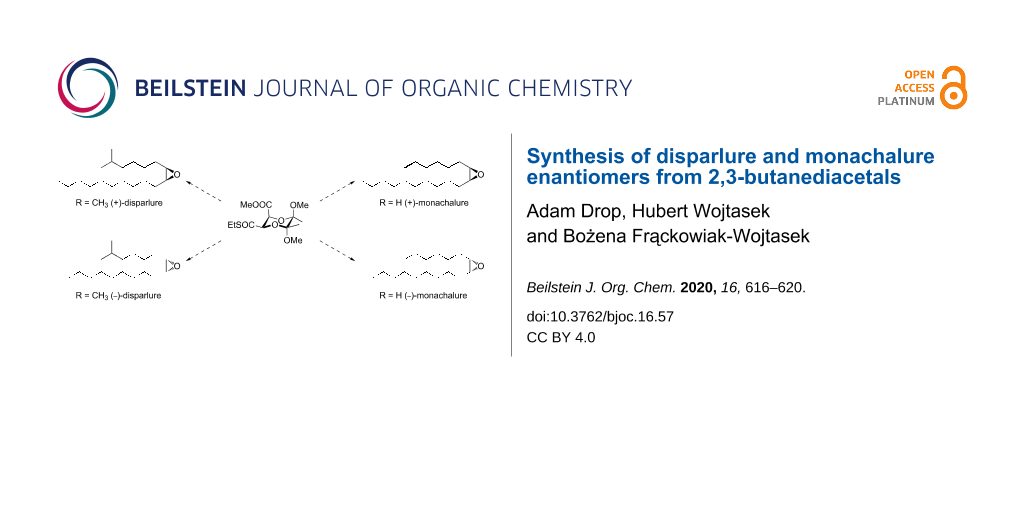
Abstract
2,3-Butanediacetal derivatives were used for the stereoselective synthesis of unsymmetrically substituted cis-epoxides. The procedure was applied for the preparation of both enantiomers of disparlure and monachalure, the components of the sex pheromones of the gypsy moth (Lymantria dispar) and the nun moth (Lymantria monacha) using methyl (2S,3R,5R,6R)-3-ethylsulfanylcarbonyl-5,6-dimethoxy-5,6-dimethyl-1,4-dioxane-2-carboxylate as the starting material.

Graphical Abstract
Introduction
Compounds containing chiral epoxides display a wide range of biological activities and a number of them are semiochemicals. They have been frequently found as pheromones in lepidopteran species of the families Noctuidae, Arctiidae, Lymantridae, Geometridae and Erebidae [1]. (3Z,9Z)-6S,7R-Epoxyheneicosa-3,9-diene, a constituent of the female sex pheromone of the moth Tetanolita mynesalis (Erebidae) [2] is a particularly interesting example, because it is also a component of the luring mixture used by the bolas spider Mastophora hutchinsoni [3,4]. (6R,7S,2E)-6,7-Epoxy-2-nonenal has been identified as a sex-aggregation pheromone of the red-necked longhorn beetle (Aromia bungii) [5] and its synthesis has been recently developed by Mori and co-workers [6,7]. (2E)-trans-4,5-Epoxy-2-decenal is present in mammalian blood and may be used by predators to track their prey [8,9]. A classical example of cis-epoxides with semiochemical activity is the chemical communication system of the gypsy moth (Lymantria dispar) and the nun moth (Lymantria monacha). The sex pheromone of the gypsy moth contains (+)-disparlure (1, (7R,8S)-7,8-epoxy-2-methyloctadecane) [10]. The pheromone blend of the nun moth contains (−)-disparlure (3, (7S,8R)-7,8-epoxy-2-methyloctadecane), (+)-monachalure (2, (7R,8S)-cis-7,8-epoxyoctadecane), (−)-monachalure (4, (7S,8R)-cis-7,8-epoxyoctadecane) and their olefinic precursors in addition to (+)-disparlure (1) [11,12]. For the gypsy moth a high enantiomeric purity of (+)-disparlure (>98% ee) is required to evoke the male’s sexual response and even a small percentage of (−)-disparlure abolishes the biological activity, thus preventing cross-species attraction [11].
Due to the great economic damage caused by the gypsy moth and the nun moth, many synthetic routes for the preparation of (+)-disparlure (1) have been developed to monitor and control their populations. These methods use various approaches to ensure the required configuration of the asymmetric centers determining the biological activity of this compound. They include the use of naturally occurring chiral substrates, such as ʟ-glutamic acid [13], ʟ-tartaric acid [14-16], ᴅ-glucose [17], and sorbitol [18] as starting materials, as well as enantioselective reactions, such as the Sharpless epoxidation [19-24], asymmetric dihydroxylation [25,26], chloroallyloboronation [27], or iodolactonization [28]. Most recently a method using the asymmetric chlorination of dodecanal by LiCl in the presence of a chiral imidazolidinone catalyst has also been described [29]. However, many of these methods have some drawbacks – the most important one being the insufficient enantiomeric purity for biological and commercial applications [29]. (+)-Disparlure used in most commercial lures is prepared by the Sharpless epoxidation reaction, which gives the final product with ees not exceeding 95% [29], which is insufficient to be used as attractant for males of the Gypsy moth. In one of these methods the diols 5 and 7 were used as precursors in the synthesis of the cis-epoxides 1 and 3 (Scheme 1). A few methods for the synthesis of diols 5 and 7 have been described [26,30,31] and we decided to prepare them starting from 2,3-butanediacetal derivatives of ʟ-tartaric acid. This method uses cheap and easily available starting materials, does not require expensive chiral catalysts and offers the possibility of obtaining both enantiomers of disparlure and monachalure with high enantiomeric purity. It can also be adapted for preparation of other chiral cis-epoxides.
![[1860-5397-16-57-i1]](/bjoc/content/inline/1860-5397-16-57-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of (+)-disparlure (1), (−)-disparlure (3), (+)-monachalure (2), and (−)-monachalure (4) from diols 5–8.
Scheme 1: Retrosynthesis of (+)-disparlure (1), (−)-disparlure (3), (+)-monachalure (2), and (−)-monachalure (...
2,3-Butanediacetals obtained from tartaric acid dimethyl diester contain protected hydroxy groups and two functional groups suitable for the attachment of various substituents. trans-2,3-Disubstituted butanediacetal derivatives of dimethyl tartrates 9–11 can be converted to cis-isomers 12–14 (Scheme 2) [32].
![[1860-5397-16-57-i2]](/bjoc/content/inline/1860-5397-16-57-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Isomerization of trans-2,3-butanediacetals 9–11 to cis-2,3-butanediacetals 12–14.
Scheme 2: Isomerization of trans-2,3-butanediacetals 9–11 to cis-2,3-butanediacetals 12–14.
Compound 12 can be obtained from trans-dimethyl ester 9 in a two-step procedure either following a traditional way or using continuous flow chemistry [32-34]. The isomerization of trans-2,3-butanediacetals to the cis-isomers has also been performed via dithiolate derivative 10 to obtain compound 13 (Scheme 2) [35]. However, attempts to isomerize dimethyl ester 9 in the same way gave a cyclic compound as a major product [36]. We have recently developed a simple and efficient method for the isomerization of trans-disubstituted butanediacetal derivative 11 with two different substituents (one ester group and one thioester group) to its cis derivative 14 which proceeded with 94% yield [37]. The cis-2,3-butanediacetals 12–14 have the appropriate configuration of the chiral centers for the synthesis of both enantiomers of disparlure 1 and 3 and monachalure 2 and 4. Compound 14 also offers the possibility of selective modification of one of the substituents as well as sequential introduction of aliphatic chains. After deprotection of the butanediacetal group, diols 5–8 are obtained. We therefore decided to apply this compound for the synthesis of both enantiomers of disparlure and both enantiomers of monachalure.
Results and Discussion
Compound 14 was prepared and converted to 15 by Fukuyama reduction as described previously [37]. The overall yield for the synthesis of compound 15 from ʟ-dimethyl tartrate and butanone was 32%. We initially attempted to carry out the Wittig reaction of compound 15 with n-pentyltriphenylphosphonium bromide and the conditions of the individual reactions are collected in Table 1. Product 16 was obtained with various yields ranging from 0 to 64% and the results were not reproducible. We could not identify the reason of these problems and could not overcome them. So, although compound 15 seemed an attractive starting material for introducing alkyl chains of pheromones 1–4, we decided to abandon this substrate and use aldehyde 19 instead. This compound was obtained from both aldehyde 15 and its precursor ethyl thioester methyl ester 14, respectively (Scheme 3). Both substrates 14 and 15 were reduced to the corresponding diol 17 with lithium aluminum hydride with 73% or 83% yield, respectively. Next, the selective protection of the axial hydroxymethyl group [32,38,39] was performed affording the product with a diastereomeric ratio of 6:1. Isomer 18 was isolated by column chromatography and subjected to a Swern oxidation to give the desired aldehyde 19 in nearly quantitative yield.
Table 1: Conditions of the Wittig reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-57-i6.svg?max-width=637&scale=1.0)
|
|||
| entry | base (equiv) | ylide (equiv) | yield (%) |
| 1 |
n-BuLi
1.2–2.0 |
1.2–2.0 | 10–53a |
| 2 |
LiHMDS
1.1–2.1 |
1.1–2.2 | 17–64a |
| 3 |
KHMDS
1.2–1.5 |
1.2-1.5 | 0-46a |
aThe yields were in a wide range and were not reproducible.
![[1860-5397-16-57-i3]](/bjoc/content/inline/1860-5397-16-57-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of diol 17 and its subsequent modifications.
Scheme 3: Synthesis of diol 17 and its subsequent modifications.
This aldehyde derivative 19 was then used in the synthesis of (+)-disparlure (1), (−)-disparlure (3), (+)-monachalure (2), and (−)-monachalure (4). First, the (+)-enantiomers 1 and 2 were synthesized (Scheme 4).
![[1860-5397-16-57-i4]](/bjoc/content/inline/1860-5397-16-57-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of (+)-disparlure (1) and (+)-monachalure (2).
Scheme 4: Synthesis of (+)-disparlure (1) and (+)-monachalure (2).
In a Wittig reaction with n-nonyltriphenylphosphonium bromide followed by reduction of the double bond, the longer chain of the pheromone structures was attached giving compound 20 in 74% yield. Next, removal of the protecting group from the secondary alcohol followed by a Swern oxidation gave aldehyde 21 which was subjected to Wittig reactions with either of the two different alkyl derivatives and catalytic hydrogenation, giving compounds 22 and 23. The butanediacetal groups were then removed with p-toluenesulfonic acid and diols 5 and 6 were obtained with 79% and 64% yield, respectively. They were then used in a well-established three-step, one-pot procedure for epoxide ring closure [26,30,40,41]. Pure (+)-disparlure (1) and (+)-monachalure (2) were obtained after column chromatography with 87% and 98% yield, respectively.
For the synthesis of the (−)-enantiomers 3 and 4 the sequence of attachment of alkyl chains to aldehyde 19 was reversed (Scheme 5). The Wittig reactions with 4-methylpentyltriphenylphosphonium bromide or n-pentyltriphenylphosphonium bromide followed by reduction of the double bonds were performed first giving derivatives 24 and 25 containing the shorter carbon chains of the pheromones. Aldehydes 26 and 27 were obtained after removal of the protecting groups from the alcohols in compounds 24 and 25, followed by Swern oxidation. The second Wittig reactions with the longer alkyl derivative and hydrogenation gave compounds 28 and 29. The diols 7 and 8 were then obtained by cleavage of the butanediacetals and were used for the synthesis of pheromones 3 and 4 using the procedure described previously for the synthesis of the (+)-enantiomers 1 and 2 with 81% and 85% yield, respectively.
![[1860-5397-16-57-i5]](/bjoc/content/inline/1860-5397-16-57-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of (−)-disparlure (3) and (−)-monachalure (4).
Scheme 5: Synthesis of (−)-disparlure (3) and (−)-monachalure (4).
Conclusion
The synthesis of pheromones 1–4 was achieved using stable butanediacetal derivatives as the starting materials. All intermediates are stable compounds and could be easily purified. The procedure may also be applied for the preparation of other unsymmetrically substituted cis-epoxides. The overall yields, starting from aldehyde 19, were 21% for (+)-disparlure, 6% for (−)-disparlure, 22% for (+)-monachalure, and 23% for (−)monachalure. The optical rotation of the obtained (+)-disparlure (1) and (−)-disparlure (3) are in agreement with the literature data. For the isomers of monachalure 2 and 4 the optical rotations have been measured for the first time and were low, similar to the values of the disparlure enantiomers 1 and 3.
References
-
Millar, J. G. Annu. Rev. Entomol. 2000, 45, 575–604. doi:10.1146/annurev.ento.45.1.575
Return to citation in text: [1] -
Haynes, K. F.; Yeargan, K. V.; Millar, J. G.; Chastain, B. B. J. Chem. Ecol. 1996, 22, 75–89. doi:10.1007/bf02040201
Return to citation in text: [1] -
Haynes, K. F.; Gemeno, C.; Yeargan, K. V.; Millar, J. G.; Johnson, K. M. Chemoecology 2002, 12, 99–105. doi:10.1007/s00049-002-8332-2
Return to citation in text: [1] -
Olsen, C. A.; Kristensen, A. S.; Strømgaard, K. Angew. Chem., Int. Ed. 2011, 50, 11296–11311. doi:10.1002/anie.201101599
Return to citation in text: [1] -
Xu, T.; Yasui, H.; Teale, S. A.; Fujiwara-Tsujii, N.; Wickham, J. D.; Fukaya, M.; Hansen, L.; Kiriyama, S.; Hao, D.; Nakano, A.; Zhang, L.; Watanabe, T.; Tokoro, M.; Millar, J. G. Sci. Rep. 2017, 7, 7330. doi:10.1038/s41598-017-07520-1
Return to citation in text: [1] -
Yasui, H.; Fujiwara-Tsujii, N.; Yasuda, T.; Fukaya, M.; Kiriyama, S.; Nakano, A.; Watanabe, T.; Mori, K. Appl. Entomol. Zool. 2019, 54, 109–114. doi:10.1007/s13355-018-0600-x
Return to citation in text: [1] -
Mori, K. Tetrahedron 2018, 74, 1444–1448. doi:10.1016/j.tet.2018.01.052
Return to citation in text: [1] -
Nilsson, S.; Sjöberg, J.; Amundin, M.; Hartmann, C.; Buettner, A.; Laska, M. PLoS One 2014, 9, e112694. doi:10.1371/journal.pone.0112694
Return to citation in text: [1] -
Mori, K.; Osada, K.; Amaike, M. Tetrahedron: Asymmetry 2015, 26, 861–867. doi:10.1016/j.tetasy.2015.06.011
Return to citation in text: [1] -
Bierl, B. A.; Beroza, M.; Collier, C. W. Science 1970, 170, 87–89. doi:10.1126/science.170.3953.87
Return to citation in text: [1] -
Gries, G.; Gries, R.; Khaskin, G.; Slessor, K. N.; Grant, G. G.; Liška, J.; Kapitola, P. Naturwissenschaften 1996, 83, 382–385. doi:10.1007/bf01142006
Return to citation in text: [1] [2] -
Gries, R.; Khaskin, G.; Schaefer, P. W.; Hahn, R.; Gotoh, T.; Gries, G. J. Chem. Ecol. 2005, 31, 49–62. doi:10.1007/s10886-005-0973-5
Return to citation in text: [1] -
Iwaki, S.; Marumo, S.; Saito, T.; Yamada, M.; Katagiri, K. J. Am. Chem. Soc. 1974, 96, 7842–7844. doi:10.1021/ja00832a055
Return to citation in text: [1] -
Mori, K.; Takigawa, T.; Matsui, M. Tetrahedron Lett. 1976, 17, 3953–3956. doi:10.1016/s0040-4039(00)92545-0
Return to citation in text: [1] -
Mori, K.; Takigawa, T.; Matsui, M. Tetrahedron 1979, 35, 833–837. doi:10.1016/0040-4020(79)80102-7
Return to citation in text: [1] -
Masaki, Y.; Serizawa, Y.; Nagata, K.; Oda, H.; Nagashima, H.; Kaji, K. Tetrahedron Lett. 1986, 27, 231–234. doi:10.1016/s0040-4039(00)83984-2
Return to citation in text: [1] -
Achmatowicz, O., Jr.; Sadownik, A. J. Carbohydr. Chem. 1985, 4, 435–440. doi:10.1080/07328308508070193
Return to citation in text: [1] -
Paolucci, C.; Mazzini, C.; Fava, A. J. Org. Chem. 1995, 60, 169–175. doi:10.1021/jo00106a030
Return to citation in text: [1] -
Rossiter, B. E.; Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1981, 103, 464–465. doi:10.1021/ja00392a038
Return to citation in text: [1] -
Sato, T.; Itoh, T.; Fujisawa, T. Tetrahedron Lett. 1987, 28, 5677–5680. doi:10.1016/s0040-4039(00)96811-4
Return to citation in text: [1] -
Mori, K.; Ebata, T. Tetrahedron Lett. 1981, 22, 4281–4282. doi:10.1016/s0040-4039(01)82934-8
Return to citation in text: [1] -
Mori, K.; Ebata, T. Tetrahedron 1986, 42, 3471–3478. doi:10.1016/s0040-4020(01)87314-2
Return to citation in text: [1] -
Marczak, S.; Masnyk, M.; Wicha, J. Liebigs Ann. Chem. 1990, 345–347. doi:10.1002/jlac.199019900165
Return to citation in text: [1] -
Li, L. H.; Wang, D.; Chan, T. H. Tetrahedron Lett. 1997, 38, 101–104. doi:10.1016/s0040-4039(96)02226-5
Return to citation in text: [1] -
Keinan, E.; Sinha, S. C.; Sinha-Bagchi, A.; Zhi-Min, W.; Xiu-Lian, Z.; Sharpless, K. B. Tetrahedron Lett. 1992, 33, 6411–6414. doi:10.1016/s0040-4039(00)79002-2
Return to citation in text: [1] -
Sinha-Bagchi, A.; Sinha, S. C.; Keinan, E. Tetrahedron: Asymmetry 1995, 6, 2889–2892. doi:10.1016/0957-4166(95)00405-x
Return to citation in text: [1] [2] [3] -
Hu, S.; Jayaraman, S.; Oehlschlager, A. C. J. Org. Chem. 1999, 64, 3719–3721. doi:10.1021/jo9820871
Return to citation in text: [1] -
Klosowski, D. W.; Martin, S. F. Org. Lett. 2018, 20, 1269–1271. doi:10.1021/acs.orglett.7b03911
Return to citation in text: [1] -
Pinnelli, G. R.; Terrado, M.; Hillier, N. K.; Lance, D. R.; Plettner, E. Eur. J. Org. Chem. 2019, 6807–6821. doi:10.1002/ejoc.201901164
Return to citation in text: [1] [2] [3] -
Koumbis, A. E.; Chronopoulos, D. D. Tetrahedron Lett. 2005, 46, 4353–4355. doi:10.1016/j.tetlet.2005.04.081
Return to citation in text: [1] [2] -
Kim, S.-G. Synthesis 2009, 2418–2422. doi:10.1055/s-0029-1216855
Return to citation in text: [1] -
Dixon, D. J.; Foster, A. C.; Ley, S. V. Can. J. Chem. 2001, 79, 1668–1680. doi:10.1139/v01-144
Return to citation in text: [1] [2] [3] -
Carter, C. F.; Baxendale, I. R.; O'Brien, M.; Pavey, J. B. J.; Ley, S. V. Org. Biomol. Chem. 2009, 7, 4594–4597. doi:10.1039/b917289k
Return to citation in text: [1] -
Carter, C. F.; Baxendale, I. R.; Pavey, J. B. J.; Ley, S. V. Org. Biomol. Chem. 2010, 8, 1588–1595. doi:10.1039/b924309g
Return to citation in text: [1] -
Maycock, C. D.; Ventura, M. R. Tetrahedron: Asymmetry 2012, 23, 1262–1271. doi:10.1016/j.tetasy.2012.08.008
Return to citation in text: [1] -
Barros, M. T.; Burke, A. J.; Maycock, C. D. Tetrahedron Lett. 1999, 40, 1583–1586. doi:10.1016/s0040-4039(98)02650-1
Return to citation in text: [1] -
Drop, A.; Wojtasek, H.; Frąckowiak-Wojtasek, B. Tetrahedron Lett. 2017, 58, 1453–1455. doi:10.1016/j.tetlet.2017.02.072
Return to citation in text: [1] [2] -
Barlow, J. S.; Dixon, D. J.; Foster, A. C.; Ley, S. V.; Reynolds, D. J. J. Chem. Soc., Perkin Trans. 1 1999, 1627–1630. doi:10.1039/a902734c
Return to citation in text: [1] -
Dixon, D. J.; Foster, A. C.; Ley, S. V.; Reynolds, D. J. J. Chem. Soc., Perkin Trans. 1 1999, 1635–1638. doi:10.1039/a902732g
Return to citation in text: [1] -
Kolb, H. C.; Sharpless, K. B. Tetrahedron 1992, 48, 10515–10530. doi:10.1016/s0040-4020(01)88349-6
Return to citation in text: [1] -
Sinha, S. C.; Sinha-Bagchi, A.; Keinan, E. J. Org. Chem. 1993, 58, 7789–7796. doi:10.1021/jo00079a024
Return to citation in text: [1]
| 32. | Dixon, D. J.; Foster, A. C.; Ley, S. V. Can. J. Chem. 2001, 79, 1668–1680. doi:10.1139/v01-144 |
| 38. | Barlow, J. S.; Dixon, D. J.; Foster, A. C.; Ley, S. V.; Reynolds, D. J. J. Chem. Soc., Perkin Trans. 1 1999, 1627–1630. doi:10.1039/a902734c |
| 39. | Dixon, D. J.; Foster, A. C.; Ley, S. V.; Reynolds, D. J. J. Chem. Soc., Perkin Trans. 1 1999, 1635–1638. doi:10.1039/a902732g |
| 26. | Sinha-Bagchi, A.; Sinha, S. C.; Keinan, E. Tetrahedron: Asymmetry 1995, 6, 2889–2892. doi:10.1016/0957-4166(95)00405-x |
| 30. | Koumbis, A. E.; Chronopoulos, D. D. Tetrahedron Lett. 2005, 46, 4353–4355. doi:10.1016/j.tetlet.2005.04.081 |
| 40. | Kolb, H. C.; Sharpless, K. B. Tetrahedron 1992, 48, 10515–10530. doi:10.1016/s0040-4020(01)88349-6 |
| 41. | Sinha, S. C.; Sinha-Bagchi, A.; Keinan, E. J. Org. Chem. 1993, 58, 7789–7796. doi:10.1021/jo00079a024 |
| 1. | Millar, J. G. Annu. Rev. Entomol. 2000, 45, 575–604. doi:10.1146/annurev.ento.45.1.575 |
| 6. | Yasui, H.; Fujiwara-Tsujii, N.; Yasuda, T.; Fukaya, M.; Kiriyama, S.; Nakano, A.; Watanabe, T.; Mori, K. Appl. Entomol. Zool. 2019, 54, 109–114. doi:10.1007/s13355-018-0600-x |
| 7. | Mori, K. Tetrahedron 2018, 74, 1444–1448. doi:10.1016/j.tet.2018.01.052 |
| 25. | Keinan, E.; Sinha, S. C.; Sinha-Bagchi, A.; Zhi-Min, W.; Xiu-Lian, Z.; Sharpless, K. B. Tetrahedron Lett. 1992, 33, 6411–6414. doi:10.1016/s0040-4039(00)79002-2 |
| 26. | Sinha-Bagchi, A.; Sinha, S. C.; Keinan, E. Tetrahedron: Asymmetry 1995, 6, 2889–2892. doi:10.1016/0957-4166(95)00405-x |
| 5. | Xu, T.; Yasui, H.; Teale, S. A.; Fujiwara-Tsujii, N.; Wickham, J. D.; Fukaya, M.; Hansen, L.; Kiriyama, S.; Hao, D.; Nakano, A.; Zhang, L.; Watanabe, T.; Tokoro, M.; Millar, J. G. Sci. Rep. 2017, 7, 7330. doi:10.1038/s41598-017-07520-1 |
| 27. | Hu, S.; Jayaraman, S.; Oehlschlager, A. C. J. Org. Chem. 1999, 64, 3719–3721. doi:10.1021/jo9820871 |
| 3. | Haynes, K. F.; Gemeno, C.; Yeargan, K. V.; Millar, J. G.; Johnson, K. M. Chemoecology 2002, 12, 99–105. doi:10.1007/s00049-002-8332-2 |
| 4. | Olsen, C. A.; Kristensen, A. S.; Strømgaard, K. Angew. Chem., Int. Ed. 2011, 50, 11296–11311. doi:10.1002/anie.201101599 |
| 18. | Paolucci, C.; Mazzini, C.; Fava, A. J. Org. Chem. 1995, 60, 169–175. doi:10.1021/jo00106a030 |
| 2. | Haynes, K. F.; Yeargan, K. V.; Millar, J. G.; Chastain, B. B. J. Chem. Ecol. 1996, 22, 75–89. doi:10.1007/bf02040201 |
| 19. | Rossiter, B. E.; Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1981, 103, 464–465. doi:10.1021/ja00392a038 |
| 20. | Sato, T.; Itoh, T.; Fujisawa, T. Tetrahedron Lett. 1987, 28, 5677–5680. doi:10.1016/s0040-4039(00)96811-4 |
| 21. | Mori, K.; Ebata, T. Tetrahedron Lett. 1981, 22, 4281–4282. doi:10.1016/s0040-4039(01)82934-8 |
| 22. | Mori, K.; Ebata, T. Tetrahedron 1986, 42, 3471–3478. doi:10.1016/s0040-4020(01)87314-2 |
| 23. | Marczak, S.; Masnyk, M.; Wicha, J. Liebigs Ann. Chem. 1990, 345–347. doi:10.1002/jlac.199019900165 |
| 24. | Li, L. H.; Wang, D.; Chan, T. H. Tetrahedron Lett. 1997, 38, 101–104. doi:10.1016/s0040-4039(96)02226-5 |
| 11. | Gries, G.; Gries, R.; Khaskin, G.; Slessor, K. N.; Grant, G. G.; Liška, J.; Kapitola, P. Naturwissenschaften 1996, 83, 382–385. doi:10.1007/bf01142006 |
| 14. | Mori, K.; Takigawa, T.; Matsui, M. Tetrahedron Lett. 1976, 17, 3953–3956. doi:10.1016/s0040-4039(00)92545-0 |
| 15. | Mori, K.; Takigawa, T.; Matsui, M. Tetrahedron 1979, 35, 833–837. doi:10.1016/0040-4020(79)80102-7 |
| 16. | Masaki, Y.; Serizawa, Y.; Nagata, K.; Oda, H.; Nagashima, H.; Kaji, K. Tetrahedron Lett. 1986, 27, 231–234. doi:10.1016/s0040-4039(00)83984-2 |
| 11. | Gries, G.; Gries, R.; Khaskin, G.; Slessor, K. N.; Grant, G. G.; Liška, J.; Kapitola, P. Naturwissenschaften 1996, 83, 382–385. doi:10.1007/bf01142006 |
| 12. | Gries, R.; Khaskin, G.; Schaefer, P. W.; Hahn, R.; Gotoh, T.; Gries, G. J. Chem. Ecol. 2005, 31, 49–62. doi:10.1007/s10886-005-0973-5 |
| 17. | Achmatowicz, O., Jr.; Sadownik, A. J. Carbohydr. Chem. 1985, 4, 435–440. doi:10.1080/07328308508070193 |
| 10. | Bierl, B. A.; Beroza, M.; Collier, C. W. Science 1970, 170, 87–89. doi:10.1126/science.170.3953.87 |
| 8. | Nilsson, S.; Sjöberg, J.; Amundin, M.; Hartmann, C.; Buettner, A.; Laska, M. PLoS One 2014, 9, e112694. doi:10.1371/journal.pone.0112694 |
| 9. | Mori, K.; Osada, K.; Amaike, M. Tetrahedron: Asymmetry 2015, 26, 861–867. doi:10.1016/j.tetasy.2015.06.011 |
| 13. | Iwaki, S.; Marumo, S.; Saito, T.; Yamada, M.; Katagiri, K. J. Am. Chem. Soc. 1974, 96, 7842–7844. doi:10.1021/ja00832a055 |
| 29. | Pinnelli, G. R.; Terrado, M.; Hillier, N. K.; Lance, D. R.; Plettner, E. Eur. J. Org. Chem. 2019, 6807–6821. doi:10.1002/ejoc.201901164 |
| 28. | Klosowski, D. W.; Martin, S. F. Org. Lett. 2018, 20, 1269–1271. doi:10.1021/acs.orglett.7b03911 |
| 29. | Pinnelli, G. R.; Terrado, M.; Hillier, N. K.; Lance, D. R.; Plettner, E. Eur. J. Org. Chem. 2019, 6807–6821. doi:10.1002/ejoc.201901164 |
| 37. | Drop, A.; Wojtasek, H.; Frąckowiak-Wojtasek, B. Tetrahedron Lett. 2017, 58, 1453–1455. doi:10.1016/j.tetlet.2017.02.072 |
| 37. | Drop, A.; Wojtasek, H.; Frąckowiak-Wojtasek, B. Tetrahedron Lett. 2017, 58, 1453–1455. doi:10.1016/j.tetlet.2017.02.072 |
| 35. | Maycock, C. D.; Ventura, M. R. Tetrahedron: Asymmetry 2012, 23, 1262–1271. doi:10.1016/j.tetasy.2012.08.008 |
| 36. | Barros, M. T.; Burke, A. J.; Maycock, C. D. Tetrahedron Lett. 1999, 40, 1583–1586. doi:10.1016/s0040-4039(98)02650-1 |
| 32. | Dixon, D. J.; Foster, A. C.; Ley, S. V. Can. J. Chem. 2001, 79, 1668–1680. doi:10.1139/v01-144 |
| 32. | Dixon, D. J.; Foster, A. C.; Ley, S. V. Can. J. Chem. 2001, 79, 1668–1680. doi:10.1139/v01-144 |
| 33. | Carter, C. F.; Baxendale, I. R.; O'Brien, M.; Pavey, J. B. J.; Ley, S. V. Org. Biomol. Chem. 2009, 7, 4594–4597. doi:10.1039/b917289k |
| 34. | Carter, C. F.; Baxendale, I. R.; Pavey, J. B. J.; Ley, S. V. Org. Biomol. Chem. 2010, 8, 1588–1595. doi:10.1039/b924309g |
| 29. | Pinnelli, G. R.; Terrado, M.; Hillier, N. K.; Lance, D. R.; Plettner, E. Eur. J. Org. Chem. 2019, 6807–6821. doi:10.1002/ejoc.201901164 |
| 26. | Sinha-Bagchi, A.; Sinha, S. C.; Keinan, E. Tetrahedron: Asymmetry 1995, 6, 2889–2892. doi:10.1016/0957-4166(95)00405-x |
| 30. | Koumbis, A. E.; Chronopoulos, D. D. Tetrahedron Lett. 2005, 46, 4353–4355. doi:10.1016/j.tetlet.2005.04.081 |
| 31. | Kim, S.-G. Synthesis 2009, 2418–2422. doi:10.1055/s-0029-1216855 |
© 2020 Drop et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)