Abstract
The preparation of large rylenes often needs the use of solubilizing groups along the rylene backbone, and all the substituents of the terrylenes and quaterrylenes were introduced before creating the rylene skeleton. In this work, we successfully synthesized 2,5,10,13-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)terrylene (TB4) by using an iridium-catalyzed direct borylation of C–H bonds in terrylene in 56% yield. The product is soluble in common organic solvents and could be purified without column chromatography. Single crystal X-ray diffraction analysis revealed that the terrylene core is not disturbed by the substituents and is perfectly flat. The photophysical properties of TB4 are also unchanged by the substituents because the carbon atoms at 2,5,10,13-positions have less coefficients on its HOMO and LUMO, estimated by theoretical calculations. Finally, the same borylation reaction was applied for quaterrylene, resulting in the formation of soluble tetra-borylated quaterrylene despite a low yield. The post modification of rylenes enables us to prepare their borylated products as versatile units after creating the rylene skeletons.

Graphical Abstract
Introduction
Compared with fruitful researches of oligorylene-bisimides for organic devices and single molecular spectroscopy [1-6], genuine oligorylenes have been sporadically investigated mainly because of their synthetic difficulty and low solubility (Figure 1) [7]. To be applied to electronic or photovoltaic components, the scalable synthesis of pure soluble compounds is essential. Recently, facile preparation methods of terrylene [8] and quaterrylene [9] were reported, after 50 years from the first reports, respectively [10,11]. Friedel–Crafts ring condensation reaction of 3-(1-naphthyl)perylene with AlCl3 reproducibly provided a pure terrylene [8]. Scholl reaction using a superacid catalyst in combination with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as oxidant provides a scalable preparation of quaterrylene [9], but unfortunately the low solubility prevents 1H NMR characterization. Larger oligorylenes have not been synthesized without the use of solubilizing groups along the rylene backbone.
![[1860-5397-16-58-1]](/bjoc/content/figures/1860-5397-16-58-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of a) oligorylene-bisimides, b) oligorylenes, c) bay-bridging oligorylenes, d) bay-alkoxy oligorylenes, and e) tetra-tert-butyl oligorylenes.
Figure 1: Chemical structures of a) oligorylene-bisimides, b) oligorylenes, c) bay-bridging oligorylenes, d) ...
For instance, bay-bridging perylenes with long alkyl chains [12-15] and bay-alkoxy-substitution [16-18] enable us to prepare longer rylenes (Figure 1c and 1d). However, tying the bay regions with carbon or nitrogen atoms causes bowing of the oligorylene backbone without disrupting the planarity of the aromatic backbone to give a flat banana-shaped molecule [15]. On the other hand, the bay-alkoxy-substitution is estimated to twist the core, which changes the original characteristics of rylenes but eventually helps to enhance the solubility of the macromolecules [7]. In order to avoid such deformation of the molecular skeleton, the solubilizing groups should be attained at the terminal positions. In this regard, the tetra-tert-butyl series of rylenes made it possible to study their intrinsic physical properties as a function of molecular length (Figure 1e) [19,20], while the low solubility of tert-butylpentarylene indicates that even such bulky group had reached its limit to interrupt π-stacking. The synthesis of higher oligorylenes would require more robust solubilizing groups.
All the substituents of the terrylenes and quaterrylenes were installed before creating the rylene skeleton and no derivatives have been produced by the post modification probably because their low solubility hampers such strategy. Therefore, the substituents on the rylene skeleton have been quite limited because they must suffer the harsh conditions of the rylene skeleton preparation.
Basic reactivity of rylenes with electrophiles is predicted from perylene in which the most reactive sites are the 3,4,9,10-positions [21]. On the other hand, the regioselectivity of the iridium (Ir)-catalyzed direct C–H borylation [22-25] gives 2,5,8,11-tetraborylated perylene [26]. The regioselectivity of the perylene borylation is determined by the steric factors rather than by the electron distribution in the arene and this regioselectivity complements that of electrophilic substitutions [26].
In light of the above, we plan to perform the Ir-catalyzed direct borylation of C–H bonds in terrylene because 1) the regioselectivity of the reaction is unique and should be remarkably high, thus terrylene will be borylated at the 2, 5, 10, and 13 positions, which don’t deform the rylene cores, 2) the product obtained is soluble in common organic solvents so that the separation and characterization would be readily performed, and 3) the resultant pure soluble terrylene derivative as a versatile unit can be functionalized by using conventional metal-catalyzed coupling reactions.
Results and Discussion
According to the literature [8], terrylene was prepared from a 3-(1-naphthyl)perylene by using 8 equivalents of AlCl3 in chlorobenzene at 80 °C for 4 h. The crude product was purified by Soxhlet extraction followed by recrystallization from toluene. After these simple manipulations, the pure terrylene was isolated. Note that the terrylene is slightly soluble in halogenated solvents, hence a 1H NMR spectrum in CDCl3 at room temperature can be measured.
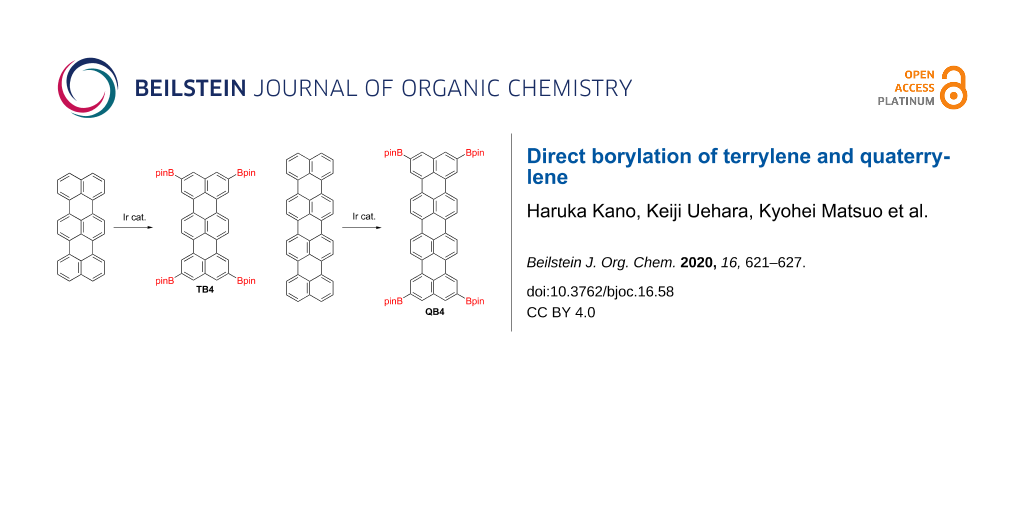
The reaction of terrylene (0.16 mmol), bis(pinacolato)diboron (8 equiv), [Ir(OMe)cod]2 (10 mol %) and di-tert-butylbipyridyl (20 mol %) in 1,4-dioxane (5 mL) under Ar at 105 °C for 30 h gave a deep orange solution (Scheme 1). Matrix assisted laser desorption/ionization (MALDI) mass spectrometry of the reaction mixture detected a single parent ion peak at m/z = 880.4687 (calcd for C54H60B4O8 = 880.4660 [M]+), making us expect its selective tetra-substitution with high yield. To isolate a pure product, the solvent was removed, then the reaction mixture was passed through a silica pad with CH2Cl2, followed by reprecipitation from MeOH. The yield of 2,5,10,13-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)terrylene (TB4) was 56%. Since insoluble materials and polar byproducts can be removed by short silica gel and the formation of tri-, bis- and mono-substituted compounds is negligible, TB4 can be purified without column chromatography.
![[1860-5397-16-58-i1]](/bjoc/content/inline/1860-5397-16-58-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2,5,10,13-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)terrylene (TB4): (a) (Bpin)2 (8 equiv), [Ir(OMe)cod]2 (10 mol %), di-tert-butylbipyridyl (20 mol %), 1,4-dioxane, at 105 °C, 30 h, yield 56%. Bpin = 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl.
Scheme 1: Synthesis of 2,5,10,13-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)terrylene (TB4): (a) (B...
The 1H NMR spectrum of TB4 in CDCl3 exhibited a highly symmetric D2h signal pattern that consists of three singlet peaks at 8.58, 8.37 and 8.23 ppm due to aromatic protons. The structure of TB4 was unambiguously revealed by single crystal X-ray diffraction analysis (Figure 2). The crystals suitable for X-ray diffraction were obtained by vapor diffusion of hexane into a solution of TB4 in CH2Cl2. The terrylene core is perfectly planar and forms a face-to-face stacking columnar structure with an interplanar distance of 3.40 Å along the a axis. The BO2 units are slightly tilted from the aromatic plane (6.7 and 20.3°).
![[1860-5397-16-58-2]](/bjoc/content/figures/1860-5397-16-58-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (Top) Single crystal X-ray structure of TB4. The thermal ellipsoids are scaled at 50% probability. (Bottom) Packing diagram of TB4. The solvent molecules are omitted for clarity.
Figure 2: (Top) Single crystal X-ray structure of TB4. The thermal ellipsoids are scaled at 50% probability. ...
TB4 in toluene absorbs UV–vis light with an absorption maximum at 566 nm, and emits fluorescence at 576 nm with a quantum yield of ΦF = 0.86 at 298 K (Figure 3). Both peaks are slightly red-shifted relative to those of intact terrylene (λabs = 560 nm and λem = 571 nm with ΦF = 0.82 in toluene).
![[1860-5397-16-58-3]](/bjoc/content/figures/1860-5397-16-58-3.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 3: UV–vis absorption and fluorescence spectra of terrylene (black) and TB4 (red) in toluene. λex = 489 nm. Fluorescence intensity reflects the absolute fluorescence quantum yields.
Figure 3: UV–vis absorption and fluorescence spectra of terrylene (black) and TB4 (red) in toluene. λex = 489...
We employed density functional theory (DFT) and time-dependent (TD)-DFT calculations, both of them at the B3LYP/6-31G(d) level and with Gaussian 09, to make these electronic features understandable (Figure 4) [27]. The HOMO, HOMO–1, LUMO, and LUMO+1 for terrylene are nondegenerative and the coefficient distribution in these four frontier MOs seem to be delocalized over the whole aromatic core. The highest band of terrylene at 553 nm is mainly caused by transition from HOMO (−4.59 eV) to LUMO (−2.30 eV) (oscillator strength, f = 0.76). Although the frontier MOs for TB4 are overall destabilized due to the smaller electronegativity of boron, the impact of substituents to the HOMO and LUMO distributions is negligible because of the existence of node at 2,5,10,13-positions. The longest band of TB4 at 563 nm comprises the transition from equally destabilized HOMO (−4.29 eV) to LUMO (−1.99 eV) (oscillator strength, f = 0.86). The transition energies and oscillator strengths simulated by TD-DFT calculations showed a good agreement with the observed absorption spectra.
![[1860-5397-16-58-4]](/bjoc/content/figures/1860-5397-16-58-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: MO diagrams of terrylene and TB4 based on calculations at the B3LYP/6-31G(d).
Figure 4: MO diagrams of terrylene and TB4 based on calculations at the B3LYP/6-31G(d).
Taking the successful result of the terrylene borylation, next we tried to perform the same reaction to quaterrylene (Scheme 2). The quaterrylene was prepared by the oxidative condensation reaction of perylene with TfOH and DDQ [9]. However, the crude product was not completely purified by Soxhlet extraction and by crystallization in our hands. The borylation reaction of hardly soluble crude quaterrylene gave a deep green suspension. MALDI mass spectrometry of the reaction mixture detected an ion peak at m/z = 1004.5018 (calcd for C64H64B4O8 = 1004.5003 [M]+), indicating that the mixture contains tetrasubstituted quaterrylene. To isolate the product, the combination of silica gel column chromatography and gel permeation chromatography techniques, followed by reprecipitation from MeOH gave a 2,5,12,15-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)quaterrylene (QB4) in 0.4% yield. The poor solubility of starting material prevented a smooth reaction.
![[1860-5397-16-58-i2]](/bjoc/content/inline/1860-5397-16-58-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2,5,12,15-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)quaterrylene (QB4): (a) (Bpin)2 (12 equiv), [Ir(OMe)cod]2 (20 mol %), di-tert-butylbipyridyl (40 mol %), 1,4-dioxane, at 105 °C, 38 h, yield 0.4%.
Scheme 2: Synthesis of 2,5,12,15-tetrakis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)quaterrylene (QB4): (a)...
It is difficult to observe accurate characteristics of quaterrylene in solution owing to its remarkable insolubility. The much improved solubility of QB4 enables 1H NMR measurement in CDCl3, which exhibits a highly symmetric D2h signal pattern that consists of two singlet peaks at 8.63 and 8.25 ppm, and two doublet peaks at 8.44 and 8.32 ppm for aromatic protons.
The UV–visible absorption spectrum of QB4 in toluene is shown in Figure 5. Compared with tert-butylquaterrylene in dioxane [19], a slightly red-shifted absorption peak at 668 nm is observed. Interestingly, QB4 emits only weak fluorescence as was described for tetra-tert-butylquaterrylene [20].
![[1860-5397-16-58-5]](/bjoc/content/figures/1860-5397-16-58-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: UV–vis absorption spectrum of QB4 in toluene.
Figure 5: UV–vis absorption spectrum of QB4 in toluene.
The coefficient distribution of four frontier MOs of quaterrylenes basically exhibits the same symmetric pattern with terrylenes (Figure 6). The longest band of QB4 at 676 nm comprises the transition from high energy level of HOMO (−4.13 eV) to LUMO (−2.26 eV) (oscillator strength, f = 1.41). The simulated absorption showed a good agreement with the observed spectra.
![[1860-5397-16-58-6]](/bjoc/content/figures/1860-5397-16-58-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: MO diagrams of terrylene and QB4 based on calculations at the B3LYP/6-31G(d).
Figure 6: MO diagrams of terrylene and QB4 based on calculations at the B3LYP/6-31G(d).
The calculated HOMO levels of –4.29 eV for TB4 and –4.13 eV for QB4 are pretty high among electron-rich compounds. Thus we tested the stability of TB4 and QB4 under air by means of UV–vis absorption spectroscopy. Surprisingly almost no spectral changes for both compounds were observed after 12 h, probably because the most reactive sites are protected by the steric hinderance of Bpin moieties.
Finally, to demonstrate the utility of the borylated oligorylenes, the Suzuki–Miyaura cross-coupling reaction of TB4 under the standard conditions was performed (Scheme 3). Coupling of TB4 and 2-bromomesitylene with Pd(PPh3)4, Cs2CO3 and CsF in a mixture of toluene/DMF furnished 2,5,10,13-tetramesitylterrylene (TM4) in 58% yield. TM4 was successfully isolated through a silica gel pad and by reprecipitation. The structure of TM4 was characterized by mass spectrometry and 1H and 13C NMR spectroscopy. High-resolution MALDI mass spectrometry detected the parent ion peak at m/z = 848.4377 (calcd. for C66H56 = 848.4377 [M]+). The 1H NMR spectrum of TM4 in CDCl3 reveals only a single set of signals that consists of four singlet peaks for aromatic protons at 8.21, 8.02, 7.44, and 7.03 ppm.
![[1860-5397-16-58-i3]](/bjoc/content/inline/1860-5397-16-58-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Suzuki–Miyaura cross-coupling reaction of TB4 with 2-bromomesitylene. (a) 2-bromomesitylene (8 equiv), Pd(PPh3)4 (40mol %), Cs2CO3 (40 equiv), CsF (40 equiv), toluene/DMF (1:1), at 110 °C, 24 h, yield 58%.
Scheme 3: Suzuki–Miyaura cross-coupling reaction of TB4 with 2-bromomesitylene. (a) 2-bromomesitylene (8 equi...
Conclusion
In this work, we synthesized soluble terrylene and quaterrylene derivatives by using an Ir-catalyzed borylation reaction. Our results provide a highly potential route to various functional oligorylene derivatives. In fact, TB4 could be transformed into the tetraarylterrylene TM4 in good yield by the conventional cross-coupling reaction. TB4 exhibits face-to-face interaction in the crystals, resulting in the 1D columnar structure as a whole, which is expected to have higher carrier mobility. Recently, oligo-perinaphthalenes, namely oligorylenes, are of great interest as good model compounds of armchair graphene nanoribbons (AGNRs), since AGNRs with a width of N = 3p + 2 are predicted to be metallic with a small bandgap [28-30], and the narrowest AGNR is polyrylene (5-AGNR, p = 1) [31,32]. Many potential applications await techniques to prepare large scale quantities of functionalized oligorylenes. We believe that our approach opens fruitful oligorylene researches that find many uses in organic devices.
Experimental
1H NMR (500 MHz) and 13C NMR (126 MHz) spectra were recorded with a JEOL JNM-ECX500 spectrometer at 20 °C by using tetramethylsilane as an internal standard. High-resolution mass spectra (HRMS) were measured with the matrix-assisted laser desorption/ionization-time-of-flight (MALDI–TOF) method on a JEOL SpiralTOF/JMS-S3000 spectrometer. X-ray crystallographic data were recorded at 103 K with a BRUKER-APEXII X-ray diffractometer using Mo Kα radiation equipped with a large area CCD detector. UV–vis absorption spectra were measured with a JASCO UV/Vis/NIR spectrophotometer V-570, and fluorescence spectra were measured with a JASCO PL spectrofluorometer FP-6600. Fluorescence quantum yields were measured on a HAMAMATSU Absolute PL Quantum Yield Measurement System C9920-02G. TLC and gravity column chromatography were performed on Art. 5554 (Merck KGaA) plates and silica gel 60N (Kanto Chemical), respectively. All solvents and chemicals were of reagent-grade quality, obtained commercially, and used without further purification. Terrylene and quaterrylene were prepared according to the literature [8,9]. For spectral measurements, spectral-grade toluene was purchased from Nacalai Tesque.
Synthesis of TB4. [Ir(OMe)(cod)]2 (10.6 mg, 16 μmol), di-tert-butylbipyridyl (8.6 mg, 32 μmol), terrylene (60 mg, 0.16 mmol) and (Bpin)2 (325 mg, 1.3 mmol) were added to a flask and dissolved in 1,4-dioxane (5 mL) under Ar. After bubbled with Ar for 10 min, the mixture was heated at 105 °C for 30 h and concentrated. Extracted with CH2Cl2 and washed with H2O, dried over Na2SO4 and concentrated. The residue was dissolved with CH2Cl2 and passed through a pad of silica gel and reprecipitated with MeOH to afford TB4 (78 mg, 56%). 1H NMR (500 MHz, CDCl3) δ 8.58 (s, 4H), 8.37 (s, 4H), 8.23 (s, 4H), 1.44 (s, 48H) ppm; 13C NMR (126 MHz, CDCl3) δ 136.60, 133.49, 131.50, 130.71, 130.53, 129.88, 127.02, 126.04, 121.61, 84.61, 25.28 ppm; HRMS (Spiral MALDI) m/z: [M]+ calcd for C54H60B4O8, 880.4686; found, 880.4687; UV–vis (toluene): λmax (ε [104 M−1 cm−1]) = 489 (1.5), 524 (4.1) and 566 (7.2) nm; fluorescence (toluene): λmax (λex = 489 nm) = 576 and 622 nm.
Crystallographic data have been deposited with Cambridge Crystallographic Data Centre: Deposition number CCDC-1981465. Copies of the data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html.
Synthesis of QB4. [Ir(OMe)(cod)]2 (26.5 mg, 0.04 mmol), di-tert-butylbipyridyl (21.5 mg, 0.08 mmol), quaterrylene (100 mg, 0.20 mmol) and (Bpin)2 (609 mg, 2.4 mmol) was added to a flask and dissolved in 1,4-dioxane (2 mL) under Ar. After bubbled with Ar for 10 min, the mixture was heated at 105 °C for 38 h and concentrated. Extracted with CH2Cl2 and washed with H2O, dried over Na2SO4 and concentrated. The residue was dissolved with CH2Cl2 and passed through a pad of silica gel. The combination of silica gel column chromatography and gel permeation column chromatography, followed by reprecipitation with MeOH afforded QB4 (0.7 mg, 0.4%). 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 4H), 8.44 (d, J = 8.5 Hz, 4H), 8.32 (d, J = 8.5 Hz, 4H), 8.25 (s, 4H), 1.44 (s, 48H) ppm; HRMS (Spiral MALDI) m/z: [M]+ calcd for C64H64B4O8, 1004.5003; found, 1004.5018; UV–vis (toluene): λmax = 565, 610 and 668 nm.
Synthesis of TM4. TB4 (8.8 mg, 0.01 mmol), 2-bromomesitylene (7.2 μL, 0.08 mmol), Pd(PPh3)4 (4.6 mg, 4 μmol), Cs2CO3 (130 mg, 0.4 mmol) and CsF (61 mg, 0.4 mmol) were added to a flask. The atmosphere was exchanged by applying vacuum and backfilling with Ar (this process was conducted three times). Toluene (0.3 mL) and DMF (0.3 mL) were added to the flask and the resulting mixture was stirred at 110 °C for 24 h. Upon cooling to room temperature, the mixture was extracted with Et2O, washed with H2O, and evaporated. The residue was dissolved with CH2Cl2 and passed through a pad of silica gel, followed by reprecipitation with MeOH to afford TM4 (4.9 mg, 58%). 1H NMR (500 MHz, CDCl3) δ 8.21 (s, 4H), 8.02 (s, 4H), 7.44 (s, 4H), 7.03 (s, 4H), 2.38 (s, 12H), 2.16 (s, 24H) ppm; 13C NMR (126 MHz, CDCl3) δ 139.83, 138.83, 137.13, 136.34, 135.49, 131.51, 130.83, 129.90, 128.35, 127.90, 126.22, 122.10, 121.59, 21.26, 21.15 ppm; HRMS (Spiral MALDI) m/z: [M]+ calcd for C66H56, 848.4377; found, 848.4377; UV–vis (toluene): λmax (ε [104 M−1 cm−1]) = 493 (1.8), 528 (4.9) and 572 (8.6) nm; fluorescence (toluene): λmax (λex = 489 nm) = 583, 629 and 682 nm.
References
-
Lee, S. K.; Zu, Y.; Herrmann, A.; Geerts, Y.; Müllen, K.; Bard, A. J. J. Am. Chem. Soc. 1999, 121, 3513–3520. doi:10.1021/ja984188m
Return to citation in text: [1] -
Langhals, H.; Christian, S.; Hofer, A. J. Org. Chem. 2013, 78, 9883–9891. doi:10.1021/jo401597u
Return to citation in text: [1] -
Langhals, H.; Zgela, D.; Lüling, R. J. Org. Chem. 2015, 80, 12146–12150. doi:10.1021/acs.joc.5b02092
Return to citation in text: [1] -
Liu, C.; Zhang, S.; Li, J.; Wei, J.; Müllen, K.; Yin, M. Angew. Chem., Int. Ed. 2019, 58, 1638–1642. doi:10.1002/anie.201810541
Return to citation in text: [1] -
Yuan, Z.; Lee, S.-L.; Chen, L.; Li, C.; Mali, K. S.; De Feyter, S.; Müllen, K. Chem. – Eur. J. 2013, 19, 11842–11846. doi:10.1002/chem.201302086
Return to citation in text: [1] -
Tam-Chang, S.-W.; Seo, W.; Iverson, I. K.; Casey, S. M. Angew. Chem., Int. Ed. 2003, 42, 897–900. doi:10.1002/anie.200390236
Return to citation in text: [1] -
Markiewicz, J. T.; Wudl, F. ACS Appl. Mater. Interfaces 2015, 7, 28063–28085. doi:10.1021/acsami.5b02243
Return to citation in text: [1] [2] -
Avlasevich, Y.; Kohl, C.; Müllen, K. J. Mater. Chem. 2006, 16, 1053–1057. doi:10.1039/b516264e
Return to citation in text: [1] [2] [3] [4] -
Thamatam, R.; Skraba, S. L.; Johnson, R. P. Chem. Commun. 2013, 49, 9122–9124. doi:10.1039/c3cc46270f
Return to citation in text: [1] [2] [3] [4] -
Clar, E.; Kelly, W.; Laird, R. M. Monatsh. Chem. 1956, 87, 391–398. doi:10.1007/bf00902634
Return to citation in text: [1] -
Ott, R.; Zeschko, E.; Zinke, A. Monatsh. Chem. 1963, 94, 51–62. doi:10.1007/bf00900219
Return to citation in text: [1] -
Li, Y.; Gao, J.; Di Motta, S.; Negri, F.; Wang, Z. J. Am. Chem. Soc. 2010, 132, 4208–4213. doi:10.1021/ja100276x
Return to citation in text: [1] -
Qi, Q.; Burrezo, P. M.; Phan, H.; Herng, T. S.; Gopalakrishna, T. Y.; Zeng, W.; Ding, J.; Casado, J.; Wu, J. Chem. – Eur. J. 2017, 23, 7595–7606. doi:10.1002/chem.201701102
Return to citation in text: [1] -
Zeng, W.; Hong, Y.; Medina Rivero, S.; Kim, J.; Zafra, J. L.; Phan, H.; Gopalakrishna, T. Y.; Herng, T. S.; Ding, J.; Casado, J.; Kim, D.; Wu, J. Chem. – Eur. J. 2018, 24, 4944–4951. doi:10.1002/chem.201706041
Return to citation in text: [1] -
Zeng, W.; Phan, H.; Herng, T. S.; Gopalakrishna, T. Y.; Aratani, N.; Zeng, Z.; Yamada, H.; Ding, J.; Wu, J. Chem 2017, 2, 81–92. doi:10.1016/j.chempr.2016.12.001
Return to citation in text: [1] [2] -
Geerts, Y.; Quante, H.; Platz, H.; Mahrt, R.; Hopmeier, M.; Böhm, A.; Müllen, K. J. Mater. Chem. 1998, 8, 2357–2369. doi:10.1039/a804337j
Return to citation in text: [1] -
Former, C.; Becker, S.; Grimsdale, A. C.; Müllen, K. Macromolecules 2002, 35, 1576–1582. doi:10.1021/ma011724d
Return to citation in text: [1] -
Miletić, T.; Fermi, A.; Papadakis, I.; Orfanos, I.; Karampitsos, N.; Avramopoulos, A.; Demitri, N.; De Leo, F.; Pope, S. J. A.; Papadopoulos, M. G.; Couris, S.; Bonifazi, D. Helv. Chim. Acta 2017, 100, e1700192. doi:10.1002/hlca.201700192
Return to citation in text: [1] -
Bohnen, A.; Koch, K.-H.; Lüttke, W.; Müllen, K. Angew. Chem., Int. Ed. Engl. 1990, 29, 525–527. doi:10.1002/anie.199005251
Return to citation in text: [1] [2] -
Koch, K.-H.; Müllen, K. Chem. Ber. 1991, 124, 2091–2100. doi:10.1002/cber.19911240935
Return to citation in text: [1] [2] -
Matsumoto, A.; Suzuki, M.; Hayashi, H.; Kuzuhara, D.; Yuasa, J.; Kawai, T.; Aratani, N.; Yamada, H. Chem. – Eur. J. 2016, 22, 14462–14466. doi:10.1002/chem.201602188
Return to citation in text: [1] -
Ishiyama, T.; Miyaura, N. J. Organomet. Chem. 2003, 680, 3–11. doi:10.1016/s0022-328x(03)00176-1
Return to citation in text: [1] -
Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R., III. Science 2002, 295, 305–308. doi:10.1126/science.1067074
Return to citation in text: [1] -
Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390–391. doi:10.1021/ja0173019
Return to citation in text: [1] -
Ishiyama, T.; Takagi, J.; Hartwig, J. F.; Miyaura, N. Angew. Chem., Int. Ed. 2002, 41, 3056. doi:10.1002/1521-3773(20020816)41:16<3056::aid-anie3056>3.0.co;2-#
Return to citation in text: [1] -
Coventry, D. N.; Batsanov, A. S.; Goeta, A. E.; Howard, J. A. K.; Marder, T. B.; Perutz, R. N. Chem. Commun. 2005, 2172–2174. doi:10.1039/b501778e
Return to citation in text: [1] [2] -
Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2010.
Return to citation in text: [1] -
Nakada, K.; Fujita, M.; Dresselhaus, G.; Dresselhaus, M. S. Phys. Rev. B 1996, 54, 17954–17961. doi:10.1103/physrevb.54.17954
Return to citation in text: [1] -
Son, Y.-W.; Cohen, M. L.; Louie, S. G. Phys. Rev. Lett. 2006, 97, 216803. doi:10.1103/physrevlett.97.216803
Return to citation in text: [1] -
Yang, L.; Park, C.-H.; Son, Y.-W.; Cohen, M. L.; Louie, S. G. Phys. Rev. Lett. 2007, 99, 186801. doi:10.1103/physrevlett.99.186801
Return to citation in text: [1] -
Zhang, H.; Lin, H.; Sun, K.; Chen, L.; Zagranyarski, Y.; Aghdassi, N.; Duhm, S.; Li, Q.; Zhong, D.; Li, Y.; Müllen, K.; Fuchs, H.; Chi, L. J. Am. Chem. Soc. 2015, 137, 4022–4025. doi:10.1021/ja511995r
Return to citation in text: [1] -
Cai, Z.; She, L.; He, Y.; Wu, L.; Cai, L.; Zhong, D. Macromol. Chem. Phys. 2017, 218, 1700155. doi:10.1002/macp.201700155
Return to citation in text: [1]
| 9. | Thamatam, R.; Skraba, S. L.; Johnson, R. P. Chem. Commun. 2013, 49, 9122–9124. doi:10.1039/c3cc46270f |
| 8. | Avlasevich, Y.; Kohl, C.; Müllen, K. J. Mater. Chem. 2006, 16, 1053–1057. doi:10.1039/b516264e |
| 1. | Lee, S. K.; Zu, Y.; Herrmann, A.; Geerts, Y.; Müllen, K.; Bard, A. J. J. Am. Chem. Soc. 1999, 121, 3513–3520. doi:10.1021/ja984188m |
| 2. | Langhals, H.; Christian, S.; Hofer, A. J. Org. Chem. 2013, 78, 9883–9891. doi:10.1021/jo401597u |
| 3. | Langhals, H.; Zgela, D.; Lüling, R. J. Org. Chem. 2015, 80, 12146–12150. doi:10.1021/acs.joc.5b02092 |
| 4. | Liu, C.; Zhang, S.; Li, J.; Wei, J.; Müllen, K.; Yin, M. Angew. Chem., Int. Ed. 2019, 58, 1638–1642. doi:10.1002/anie.201810541 |
| 5. | Yuan, Z.; Lee, S.-L.; Chen, L.; Li, C.; Mali, K. S.; De Feyter, S.; Müllen, K. Chem. – Eur. J. 2013, 19, 11842–11846. doi:10.1002/chem.201302086 |
| 6. | Tam-Chang, S.-W.; Seo, W.; Iverson, I. K.; Casey, S. M. Angew. Chem., Int. Ed. 2003, 42, 897–900. doi:10.1002/anie.200390236 |
| 10. | Clar, E.; Kelly, W.; Laird, R. M. Monatsh. Chem. 1956, 87, 391–398. doi:10.1007/bf00902634 |
| 11. | Ott, R.; Zeschko, E.; Zinke, A. Monatsh. Chem. 1963, 94, 51–62. doi:10.1007/bf00900219 |
| 26. | Coventry, D. N.; Batsanov, A. S.; Goeta, A. E.; Howard, J. A. K.; Marder, T. B.; Perutz, R. N. Chem. Commun. 2005, 2172–2174. doi:10.1039/b501778e |
| 9. | Thamatam, R.; Skraba, S. L.; Johnson, R. P. Chem. Commun. 2013, 49, 9122–9124. doi:10.1039/c3cc46270f |
| 26. | Coventry, D. N.; Batsanov, A. S.; Goeta, A. E.; Howard, J. A. K.; Marder, T. B.; Perutz, R. N. Chem. Commun. 2005, 2172–2174. doi:10.1039/b501778e |
| 8. | Avlasevich, Y.; Kohl, C.; Müllen, K. J. Mater. Chem. 2006, 16, 1053–1057. doi:10.1039/b516264e |
| 21. | Matsumoto, A.; Suzuki, M.; Hayashi, H.; Kuzuhara, D.; Yuasa, J.; Kawai, T.; Aratani, N.; Yamada, H. Chem. – Eur. J. 2016, 22, 14462–14466. doi:10.1002/chem.201602188 |
| 8. | Avlasevich, Y.; Kohl, C.; Müllen, K. J. Mater. Chem. 2006, 16, 1053–1057. doi:10.1039/b516264e |
| 9. | Thamatam, R.; Skraba, S. L.; Johnson, R. P. Chem. Commun. 2013, 49, 9122–9124. doi:10.1039/c3cc46270f |
| 7. | Markiewicz, J. T.; Wudl, F. ACS Appl. Mater. Interfaces 2015, 7, 28063–28085. doi:10.1021/acsami.5b02243 |
| 22. | Ishiyama, T.; Miyaura, N. J. Organomet. Chem. 2003, 680, 3–11. doi:10.1016/s0022-328x(03)00176-1 |
| 23. | Cho, J.-Y.; Tse, M. K.; Holmes, D.; Maleczka, R. E., Jr.; Smith, M. R., III. Science 2002, 295, 305–308. doi:10.1126/science.1067074 |
| 24. | Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390–391. doi:10.1021/ja0173019 |
| 25. | Ishiyama, T.; Takagi, J.; Hartwig, J. F.; Miyaura, N. Angew. Chem., Int. Ed. 2002, 41, 3056. doi:10.1002/1521-3773(20020816)41:16<3056::aid-anie3056>3.0.co;2-# |
| 16. | Geerts, Y.; Quante, H.; Platz, H.; Mahrt, R.; Hopmeier, M.; Böhm, A.; Müllen, K. J. Mater. Chem. 1998, 8, 2357–2369. doi:10.1039/a804337j |
| 17. | Former, C.; Becker, S.; Grimsdale, A. C.; Müllen, K. Macromolecules 2002, 35, 1576–1582. doi:10.1021/ma011724d |
| 18. | Miletić, T.; Fermi, A.; Papadakis, I.; Orfanos, I.; Karampitsos, N.; Avramopoulos, A.; Demitri, N.; De Leo, F.; Pope, S. J. A.; Papadopoulos, M. G.; Couris, S.; Bonifazi, D. Helv. Chim. Acta 2017, 100, e1700192. doi:10.1002/hlca.201700192 |
| 7. | Markiewicz, J. T.; Wudl, F. ACS Appl. Mater. Interfaces 2015, 7, 28063–28085. doi:10.1021/acsami.5b02243 |
| 28. | Nakada, K.; Fujita, M.; Dresselhaus, G.; Dresselhaus, M. S. Phys. Rev. B 1996, 54, 17954–17961. doi:10.1103/physrevb.54.17954 |
| 29. | Son, Y.-W.; Cohen, M. L.; Louie, S. G. Phys. Rev. Lett. 2006, 97, 216803. doi:10.1103/physrevlett.97.216803 |
| 30. | Yang, L.; Park, C.-H.; Son, Y.-W.; Cohen, M. L.; Louie, S. G. Phys. Rev. Lett. 2007, 99, 186801. doi:10.1103/physrevlett.99.186801 |
| 12. | Li, Y.; Gao, J.; Di Motta, S.; Negri, F.; Wang, Z. J. Am. Chem. Soc. 2010, 132, 4208–4213. doi:10.1021/ja100276x |
| 13. | Qi, Q.; Burrezo, P. M.; Phan, H.; Herng, T. S.; Gopalakrishna, T. Y.; Zeng, W.; Ding, J.; Casado, J.; Wu, J. Chem. – Eur. J. 2017, 23, 7595–7606. doi:10.1002/chem.201701102 |
| 14. | Zeng, W.; Hong, Y.; Medina Rivero, S.; Kim, J.; Zafra, J. L.; Phan, H.; Gopalakrishna, T. Y.; Herng, T. S.; Ding, J.; Casado, J.; Kim, D.; Wu, J. Chem. – Eur. J. 2018, 24, 4944–4951. doi:10.1002/chem.201706041 |
| 15. | Zeng, W.; Phan, H.; Herng, T. S.; Gopalakrishna, T. Y.; Aratani, N.; Zeng, Z.; Yamada, H.; Ding, J.; Wu, J. Chem 2017, 2, 81–92. doi:10.1016/j.chempr.2016.12.001 |
| 19. | Bohnen, A.; Koch, K.-H.; Lüttke, W.; Müllen, K. Angew. Chem., Int. Ed. Engl. 1990, 29, 525–527. doi:10.1002/anie.199005251 |
| 20. | Koch, K.-H.; Müllen, K. Chem. Ber. 1991, 124, 2091–2100. doi:10.1002/cber.19911240935 |
| 31. | Zhang, H.; Lin, H.; Sun, K.; Chen, L.; Zagranyarski, Y.; Aghdassi, N.; Duhm, S.; Li, Q.; Zhong, D.; Li, Y.; Müllen, K.; Fuchs, H.; Chi, L. J. Am. Chem. Soc. 2015, 137, 4022–4025. doi:10.1021/ja511995r |
| 32. | Cai, Z.; She, L.; He, Y.; Wu, L.; Cai, L.; Zhong, D. Macromol. Chem. Phys. 2017, 218, 1700155. doi:10.1002/macp.201700155 |
| 9. | Thamatam, R.; Skraba, S. L.; Johnson, R. P. Chem. Commun. 2013, 49, 9122–9124. doi:10.1039/c3cc46270f |
| 19. | Bohnen, A.; Koch, K.-H.; Lüttke, W.; Müllen, K. Angew. Chem., Int. Ed. Engl. 1990, 29, 525–527. doi:10.1002/anie.199005251 |
| 8. | Avlasevich, Y.; Kohl, C.; Müllen, K. J. Mater. Chem. 2006, 16, 1053–1057. doi:10.1039/b516264e |
| 15. | Zeng, W.; Phan, H.; Herng, T. S.; Gopalakrishna, T. Y.; Aratani, N.; Zeng, Z.; Yamada, H.; Ding, J.; Wu, J. Chem 2017, 2, 81–92. doi:10.1016/j.chempr.2016.12.001 |
| 20. | Koch, K.-H.; Müllen, K. Chem. Ber. 1991, 124, 2091–2100. doi:10.1002/cber.19911240935 |
© 2020 Kano et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)