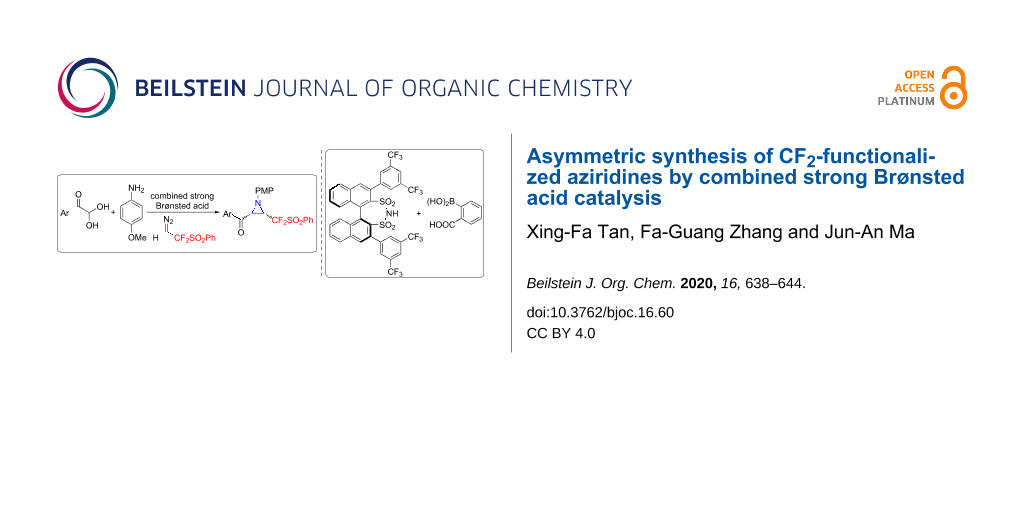
Abstract
A diastereo- and enantioselective approach to access chiral CF2-functionalized aziridines from difluorodiazoethyl phenyl sulfone (PhSO2CF2CHN2) and in situ-formed aldimines is described. This multicomponent reaction is enabled by a combined strong Brønsted acid catalytic platform consisting of a chiral disulfonimide and 2-carboxyphenylboronic acid. The optical purity of the obtained CF2-substituted aziridines could be further improved by a practical dissolution–filtration procedure.

Graphical Abstract
Introduction
Chiral aziridines are prevalently found in natural products and artificially made bioactive molecules, thus receiving significant attention in the past decades [1-6]. Among them, the introduction of fluorine or fluoroalkyl groups into three-membered N-heterocycles has emerged as an attractive direction due to the unique fluorine effect in pharmaceuticals and biology [7-11]. In this context, it is not surprising that the syntheses of trifluoromethylaziridines have been pursued from versatile precursors [12-25]. However, catalytic asymmetric approaches to chiral CF3-functionalized aziridines have only been reported by Cahard in 2012, who utilized trifluorodiazoethane (CF3CHN2) as the nucleophile to react with aldimines catalyzed by chiral phosphoric acid (Scheme 1a) [26]. In comparison, there is a significant dearth of available synthetic approaches to CF2-functionalized aziridines, particularly in a stereocontrolled manner. Indeed, a handful of reported methods document the employment of difluoromethylimines, difluoromethyl phenyl sulfone, and difluoromethyl vinyl sulfonium salts as the fluorinating partner en route to various CF2-substituted aziridines [27-31], and a general protocol to chiral CF2-aziridines remains an unsolved challenge. Thus, herein we report a diastereo- and enantioselective aza-Darzens reaction between in situ-generated aldimines and our recently developed difluorodiazo reagent PhSO2CF2CHN2 acting as the difluorinated nucleophile [32-35], providing access to a variety of chiral CF2-fuctionalized aziridines under mild conditions (Scheme 1b). The key to this multicomponent transformation hinges upon the discovery of a combined strong Brønsted acid system comprised of a chiral disulfonimide and 2-carboxyphenylboronic acid.
![[1860-5397-16-60-i1]](/bjoc/content/inline/1860-5397-16-60-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of chiral aziridines from fluorinated diazo reagents.
Scheme 1: Preparation of chiral aziridines from fluorinated diazo reagents.
Results and Discussion
We commenced the desired one-pot transformation by conducting the model reaction between phenylglyoxal monohydrate (1a), 4-methoxyaniline (2a), and PhSO2CF2CHN2 (3, Ps-DFA). Initial screenings were focused on the evaluation of various chiral phosphoric acids that have proven effective in similar aza-Darzens reactions of diazo esters and trifluorodiazoethane [36-39]. Unfortunately, these endeavors resulted in either no conversion or no enantioselectivity at all. As arylboronic acids have been harnessed to enhance the Brønsted acidity in asymmetric organocatalysis in combination with chiral diols or chiral aminoalcohols [40-44], we envisioned that the simultaneous use of arylboronic acids and chiral Brønsted acids may bring about a complementary catalytic platform. Encouragingly, the targeted CF2-functionalized aziridine 4a was obtained in up to 51% ee and high diastereoselectivity, albeit in a low yield (Table 1, entries 1 and 2). The difficulty in further improving the conversions might be ascribed to the limited Brønsted acidity of chiral phosphoric acids. Bearing this in mind, we then turned our attention to chiral disulfonimides developed by List, which have been established as a unique type of stronger Brønsted acids [45]. Putting it into practice, a range of BINOL-derived disulfonimides was used as the chiral additive in combination with 2-carboxyphenylboronic acid (COOH-BA) in the model reaction (Table 1, entries 3–8). We were pleased to find that CDSI-4 gave the most promising result in terms of both yield and enantioselectivity (64% isolated yield with 73% ee, Table 1, entry 6). An examination on various arylboronic acids, solvent, temperature, and catalyst loadings resulted in no obvious improvement (Table 1, entries 9–15). Among them, the highest yield of 4a was observed (81%, Table 1, entry 12), albeit with slightly reduced ee value. This enhancement in catalytic activity could be attributed to the increased Brønsted acidity when the strong electron-withdrawing trifluoromethyl group was placed on the benzene ring of the arylboronic acid. Removing the boronic acid from the reaction system leads to a dramatic decrease in both yield and enantiocontrol (Table 1, entry 16).
Table 1: Representative screening results of the asymmetric aziridination reaction of PhSO2CF2CHN2.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-60-i4.svg?max-width=637&scale=1.0)
|
||||
| entry | arylboronic acid (mol %) | chiral Brønsted acid (mol %) | yield of 4a (%)b | ee (%) of 4a and dr of crude mixturec |
| 1 | COOH-BA (8) | CPA-1 (5) | 24 | 51, 13:1 |
| 2 | COOH-BA (8) | CPA-2 (5) | 28 | 25, 11:1 |
| 3 | COOH-BA (8) | CDSI-1 (5) | 21 | 41, 19:1 |
| 4 | COOH-BA (8) | CDSI-2 (5) | 50 | 41, 9:1 |
| 5d | COOH-BA (8) | CDSI-3 (5) | 16 | 60, 5:1 |
| 6d | COOH-BA (8) | CDSI-4 (5) | 64 | 73, 13:1 |
| 7 | COOH-BA (8) | CDSI-5 (5) | 34 | 33, 10:1 |
| 8 | COOH-BA (8) | CDSI-6 (5) | 47 | 52, 9:1 |
| 9 | OH-BA (8) | CDSI-4 (5) | 63 | 68, 28:1 |
| 10d | SO3H-BA (8) | CDSI-4 (5) | 62 | 66, 16:1 |
| 11d | NO2-BA (8) | CDSI-4 (5) | 45 | 62, 16:1 |
| 12 | CF3-COOH-BA (8) | CDSI-4 (5) | 81 | 67, 8:1 |
| 13e | COOH-BA (8) | CDSI-4 (5) | 60 | 47, 5:1 |
| 14f | COOH-BA (8) | CDSI-4 (5) | trace | n.d. |
| 15d | COOH-BA (8) | CDSI-4 (10) | 65 | 70, 12:1 |
| 16d | – | CDSI-4 (5) | 10 | 60, >20:1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-60-i5.svg?max-width=637&scale=1.18182)
aGeneral reaction conditions: 1a (8 mg, 0.05 mmol, 1.0 equiv), 2a (7 mg, 0.055 mmol), arylboronic acid (0.004 mmol), and Na2SO4 (40 mg) was stirred in toluene (1 mL) at rt for 30 min, then the chiral Brønsted acid (0.0025 mmol) and 3 (18 mg, 0.075 mmol) were added and the mixture was reacted at rt for 12 hours unless otherwise noted; byield of isolated product 4a was given for entries labelled with d; hexafluorobenzene was used as an internal standard to determine the yield in other cases; cee of 4a was determined by chiral HPLC analysis, and the dr of the crude reaction mixture was probed by 19F NMR analysis; d0.3 mmol scale of reaction was conducted: 1a (46 mg, 0.3 mmol, 1.0 equiv), 2a (41 mg, 0.33 mmol), arylboronic acid (0.024 mmol), and Na2SO4 (200 mg) was stirred in toluene (2 mL) at rt for 30 min, then the chiral Brønsted acid (0.015 mmol) and 3 (105 mg, 0.45 mmol) were added and the mixture was reacted at rt for 12–24 hours; eCH2Cl2 was used as the solvent; freaction was operated at 0 °C.
The challenge to further improve the enantioselectivity promoted us to search for other practical solutions. Considering the poor solubility of 4a in organic solvents, a dissolution–filtration process with isopropanol was found to be workable for increasing the final ee value. This simple procedure could afford 4a with excellent enantiopurity as a single diastereoisomer (>99% ee, >50:1 dr, Scheme 2). By the aid of the developed one-pot aza-Darzens reaction and dissolution–filtration operation, a series of optically-pure CF2-aziridines 4b–h were furnished in moderate overall yields with uniformly excellent ee and dr values, including alkyl or halogen-substituted phenyl and 2-naphthyl ketones (Scheme 2). Unfortunately, phenylglyoxal monohydrates bearing strong electron-withdrawing groups were not compatible with the current conditions. X-ray analysis of aziridine 4a confirmed the absolute configuration of the chiral centers, pointing at a cis-aziridination process [46].
![[1860-5397-16-60-i2]](/bjoc/content/inline/1860-5397-16-60-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of chiral CF2-substituted aziridines from PhSO2CF2CHN2. General reaction conditions: Aryl glyoxal monohydrate (1, 0.3 mmol), 2a (41 mg, 0.33 mmol), COOH-BA (4 mg, 0.024 mmol), and Na2SO4 (200 mg) were stirred in toluene (2 mL) at rt for 30 min, then CDSI-4 (12 mg, 0.015 mmol) and Ps-DFA 3 (105 mg, 0.45 mmol) were added and the mixture was reacted at rt for 24 hours unless otherwise annotated. The yields are those of isolated products, and the dr was determined by 19F NMR analysis of the crude mixture. The results in parentheses are those of isolated products after the dissolution–filtration process: The corresponding CF2-functionalized aziridine 4 was dissolved in isopropanol (0.05–0.2 mL/mg) with the help of ultrasound, followed by filtration, and the obtained solution was concentrated to give 4 with increased ee and dr values. a0.006 mmol of COOH-BA was employed. bThe reaction was operated at 45 °C for 24 h.
Scheme 2: Substrate scope of chiral CF2-substituted aziridines from PhSO2CF2CHN2. General reaction conditions...
Scaled-up experiments with model substrate 1a also proved to be feasible, delivering the chiral CF2-aziridine 4a with comparable results (Scheme 3a). The 4-methoxyphenyl group of 4a was cleaved smoothly with ceric ammonium nitrate, giving the free aziridine 5a in 81% yield while maintaining the ee value. The reduction of the carbonyl moiety with either NaBH4 or LiAlH4 produced hydroxy-substituted CF2-functionalized aziridine 5b in excellent yield with exclusive diastereoselectivity [47]. Furthermore, the ring-opening of 4a under acidic conditions underwent well and gave rise to CF2-functionalized α-chloro-β-amino ketone 5c in 89% yield with >99% ee and >50:1 dr (confirmed by X-ray spectroscopy) [46].
![[1860-5397-16-60-i3]](/bjoc/content/inline/1860-5397-16-60-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scale-up experiment to 4a and further synthetic transformations.
Scheme 3: Scale-up experiment to 4a and further synthetic transformations.
Conclusion
In summary, an array of chiral CF2-functionalized aziridines was constructed from in situ-formed aldimines and difluorodiazoetyl phenyl sulfone under mild conditions by a combined strong Brønsted acid system consisting of chiral disulfonimide and 2-carboxyphenylboronic acid. The optical purity of the obtained CF2-substituted aziridines could be further improved by a practical dissolution–filtration procedure. Substrate expansion and mechanistic investigation are underway and will be reported in due course.
Experimental
General procedure for the preparation of chiral CF2-functionalized aziridines 4: To a 25 mL Schlenk tube equipped with a stirring bar were added 2,2-dihydroxy-1-arylethan-1-one (1, 0.3 mmol, 1 equiv), 4-methoxyaniline (2a, 40.6 mg, 0.33 mmol), 2-boronobenzoic acid (COOH-BA, 3.98 mg, 0.024 mmol), anhydrous Na2SO4 (200 mg) and toluene (1 mL) at room temperature under an argon atmosphere. After reacting for 30 minutes at room temperature, ((2-diazo-1,1-difluoroethyl)sulfonyl)benzene (Ps-DFA 3, 104.5 mg, 77.4 uL, 0.45 mmol) was added with a micro syringe and CDSI-4 (12.3 mg, 0.015 mmol) in toluene (1 mL) was added dropwise. The reaction was allowed to stir for 24 hours at room temperature under an argon atmosphere until the consumption of substrates was completed (as monitored by TLC). The reaction mixture was quenched with saturated aq NaHCO3 and extracted with ethyl acetate three times. The combined organic layer was washed with water and brine, and then dried over anhydrous Na2SO4, filtered and evaporated under vacuum. The residue was purified by neutral alumina column chromatography (eluting with dichloromethane/petroleum ether) to give CF2-substituted aziridine 4. The enantiomeric excess was determined by chiral HPLC analysis. See Supporting Information File 1 for the dissolution–filtration procedure for each compound.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization, NMR spectra of all new compounds, and HPLC traces. | ||
| Format: PDF | Size: 3.1 MB | Download |
| Supporting Information File 2: X-ray data for compound 4a. | ||
| Format: CIF | Size: 17.6 KB | Download |
| Supporting Information File 3: X-ray data for compound 5c. | ||
| Format: CIF | Size: 278.0 KB | Download |
References
-
Tanner, D. Angew. Chem., Int. Ed. Engl. 1994, 33, 599–619. doi:10.1002/anie.199405991
Return to citation in text: [1] -
Sweeney, J. B. Chem. Soc. Rev. 2002, 31, 247–258. doi:10.1039/b006015l
Return to citation in text: [1] -
Singh, G. S.; D'hooghe, M.; De Kimpe, N. Chem. Rev. 2007, 107, 2080–2135. doi:10.1021/cr0680033
Return to citation in text: [1] -
Sweeney, J. Eur. J. Org. Chem. 2009, 4911–4919. doi:10.1002/ejoc.200900211
Return to citation in text: [1] -
Stanković, S.; D'hooghe, M.; Catak, S.; Eum, H.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N.; Ha, H.-J. Chem. Soc. Rev. 2012, 41, 643–665. doi:10.1039/c1cs15140a
Return to citation in text: [1] -
de los Santos, J. M.; Ochoa de Retana, A. M.; Martínez de Marigorta, E.; Vicario, J.; Palacios, F. ChemCatChem 2018, 10, 5092–5114. doi:10.1002/cctc.201801026
Return to citation in text: [1] -
Muller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v
Return to citation in text: [1] -
Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788
Return to citation in text: [1] -
Rong, J.; Ni, C.; Hu, J. Asian J. Org. Chem. 2017, 6, 139–152. doi:10.1002/ajoc.201600509
Return to citation in text: [1] -
Meyer, F. Chem. Commun. 2016, 52, 3077–3094. doi:10.1039/c5cc09414c
Return to citation in text: [1] -
Dolfen, J.; De Kimpe, N.; D’hooghe, M. Synlett 2016, 27, 1486–1510. doi:10.1055/s-0035-1561379
Return to citation in text: [1] -
Katagiri, T.; Ihara, H.; Takahashi, M.; Kashino, S.; Furuhashi, K.; Uneyama, K. Tetrahedron: Asymmetry 1997, 8, 2933–2937. doi:10.1016/s0957-4166(97)00320-0
Return to citation in text: [1] -
Colantoni, D.; Fioravanti, S.; Pellacani, L.; Tardella, P. A. Org. Lett. 2004, 6, 197–200. doi:10.1021/ol0361554
Return to citation in text: [1] -
Maeda, R.; Ooyama, K.; Anno, R.; Shiosaki, M.; Azema, T.; Hanamoto, T. Org. Lett. 2010, 12, 2548–2550. doi:10.1021/ol100768s
Return to citation in text: [1] -
Künzi, S. A.; Morandi, B.; Carreira, E. M. Org. Lett. 2012, 14, 1900–1901. doi:10.1021/ol300539e
Return to citation in text: [1] -
Yang, Y.; Huang, Y.; Qing, F.-L. Tetrahedron Lett. 2013, 54, 3826–3830. doi:10.1016/j.tetlet.2013.05.048
Return to citation in text: [1] -
Dolfen, J.; Kenis, S.; Van Hecke, K.; De Kimpe, N.; D'hooghe, M. Chem. – Eur. J. 2014, 20, 10650–10653. doi:10.1002/chem.201304759
Return to citation in text: [1] -
Wang, F.; Zhu, N.; Chen, P.; Ye, J.; Liu, G. Angew. Chem., Int. Ed. 2015, 54, 9356–9360. doi:10.1002/anie.201503412
Return to citation in text: [1] -
Duan, Y.; Zhou, B.; Lin, J.-H.; Xiao, J.-C. Chem. Commun. 2015, 51, 13127–13130. doi:10.1039/c5cc04991a
Return to citation in text: [1] -
Huang, Y.-J.; Qiao, B.; Zhang, F.-G.; Ma, J.-A. Tetrahedron 2018, 74, 3791–3796. doi:10.1016/j.tet.2018.05.033
Return to citation in text: [1] -
Mészáros, Á.; Székely, A.; Stirling, A.; Novák, Z. Angew. Chem., Int. Ed. 2018, 57, 6643–6647. doi:10.1002/anie.201802347
Return to citation in text: [1] -
Thi, H. D.; Thuy, G. L. N.; Catak, S.; Van Speybroeck, V.; Nguyen, T. V.; D’hooghe, M. Synthesis 2018, 50, 1439–1456. doi:10.1055/s-0036-1591537
Return to citation in text: [1] -
Wu, K.; Zhou, C.-Y.; Che, C.-M. Org. Lett. 2019, 21, 85–89. doi:10.1021/acs.orglett.8b03514
Return to citation in text: [1] -
Chai, Z.; Bouillon, J.-P.; Cahard, D. Chem. Commun. 2012, 48, 9471–9473. doi:10.1039/c2cc35246j
Return to citation in text: [1] -
Mae, M.; Matsuura, M.; Amii, H.; Uneyama, K. Tetrahedron Lett. 2002, 43, 2069–2072. doi:10.1016/s0040-4039(02)00205-8
Return to citation in text: [1] -
Palacios, F.; Ochoa de Retana, A. M.; Alonso, J. M. J. Org. Chem. 2006, 71, 6141–6148. doi:10.1021/jo060865g
Return to citation in text: [1] -
Liu, J.; Hu, J. Chem. – Eur. J. 2010, 16, 11443–11454. doi:10.1002/chem.201000893
Return to citation in text: [1] -
Kurosato, F.; Ishikawa, T.; Yamada, Y.; Hanamoto, T. Synlett 2015, 26, 1827–1830. doi:10.1055/s-0034-1381008
Return to citation in text: [1] -
Zhang, W.; Wang, X.; Zhu, B.; Zhu, D.; Han, J.; Wzorek, A.; Sato, A.; Soloshonok, V. A.; Zhou, J.; Pan, Y. Adv. Synth. Catal. 2017, 359, 4267–4273. doi:10.1002/adsc.201701066
Return to citation in text: [1] -
Zeng, J.-L.; Chen, Z.; Zhang, F.-G.; Ma, J.-A. Org. Lett. 2018, 20, 4562–4565. doi:10.1021/acs.orglett.8b01854
Return to citation in text: [1] -
Peng, X.; Xiao, M.-Y.; Zeng, J.-L.; Zhang, F.-G.; Ma, J.-A. Org. Lett. 2019, 21, 4808–4811. doi:10.1021/acs.orglett.9b01697
Return to citation in text: [1] -
Zeng, J.-L.; Zhang, Y.; Zheng, M.-M.; Zhang, Z.-Q.; Xue, X.-S.; Zhang, F.-G.; Ma, J.-A. Org. Lett. 2019, 21, 8244–8249. doi:10.1021/acs.orglett.9b02989
Return to citation in text: [1] -
Zhang, Z.-Q.; Zheng, M.-M.; Xue, X.-S.; Marek, I.; Zhang, F.-G.; Ma, J.-A. Angew. Chem., Int. Ed. 2019, 58, 18191–18196. doi:10.1002/anie.201911701
Return to citation in text: [1] -
Zhang, Y.; Wang, J. Chem. Commun. 2009, 5350–5361. doi:10.1039/b908378b
Return to citation in text: [1] -
Zhang, Y.; Lu, Z.; Wulff, W. D. Synlett 2009, 2715–2739. doi:10.1055/s-0029-1217979
Return to citation in text: [1] -
Pellissier, H. Adv. Synth. Catal. 2014, 356, 1899–1935. doi:10.1002/adsc.201400312
Return to citation in text: [1] -
Liu, Y. Curr. Org. Chem. 2016, 20, 19–28. doi:10.2174/1385272819666150810225618
Return to citation in text: [1] -
Yamamoto, H.; Futatsugi, K. Angew. Chem., Int. Ed. 2005, 44, 1924–1942. doi:10.1002/anie.200460394
Return to citation in text: [1] -
Dimitrijević, E.; Taylor, M. S. ACS Catal. 2013, 3, 945–962. doi:10.1021/cs4000848
Return to citation in text: [1] -
Hashimoto, T.; Gálvez, A. O.; Maruoka, K. J. Am. Chem. Soc. 2013, 135, 17667–17670. doi:10.1021/ja407764u
Return to citation in text: [1] -
Huang, B.; He, Y.; Levin, M. D.; Coelho, J. A. S.; Bergman, R. G.; Toste, F. D. Adv. Synth. Catal. 2020, 362, 295–301. doi:10.1002/adsc.201900816
Return to citation in text: [1] -
Zhang, Q.-X.; Li, Y.; Wang, J.; Yang, C.; Liu, C.-J.; Li, X.; Cheng, J.-P. Angew. Chem., Int. Ed. 2020, 59, 4550–4556. doi:10.1002/anie.201915226
Return to citation in text: [1] -
James, T.; van Gemmeren, M.; List, B. Chem. Rev. 2015, 115, 9388–9409. doi:10.1021/acs.chemrev.5b00128
Return to citation in text: [1] -
CCDC 1983642 (4a) and 1983643 (5c) contain the supplementary crystallographic data. The crystallographic data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif. It should be mentioned that the stereochemistry of 3-CF2-aziridines in this reaction (2R,3S) are opposite to that of 3-CF3-aziridines (2S,3R) in Cahard’s report ([26]). We hypothesized that the enantiodetermining step in our reaction might be controlled through an in situ formed chiral boronate complex from chiral disulfonimide and 2-carboxyphenylboronic acid. Further efforts to improve the level of stereoselectivity and detailed mechanistic elucidation are still undergoing in our lab.
Return to citation in text: [1] [2] -
The stereochemistry of compound 5b was determined based on the 1H NMR, 19F NMR, and 2D NOE analysis. In the 2D NOE spectrum, the correlation between HO (4.83 ppm) and HC–N (2.7 ppm) was observed, whereas no correlation between HC–O (3.7 ppm) and HC–CF2 (3.02 ppm) was found.
Return to citation in text: [1]
| 1. | Tanner, D. Angew. Chem., Int. Ed. Engl. 1994, 33, 599–619. doi:10.1002/anie.199405991 |
| 2. | Sweeney, J. B. Chem. Soc. Rev. 2002, 31, 247–258. doi:10.1039/b006015l |
| 3. | Singh, G. S.; D'hooghe, M.; De Kimpe, N. Chem. Rev. 2007, 107, 2080–2135. doi:10.1021/cr0680033 |
| 4. | Sweeney, J. Eur. J. Org. Chem. 2009, 4911–4919. doi:10.1002/ejoc.200900211 |
| 5. | Stanković, S.; D'hooghe, M.; Catak, S.; Eum, H.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N.; Ha, H.-J. Chem. Soc. Rev. 2012, 41, 643–665. doi:10.1039/c1cs15140a |
| 6. | de los Santos, J. M.; Ochoa de Retana, A. M.; Martínez de Marigorta, E.; Vicario, J.; Palacios, F. ChemCatChem 2018, 10, 5092–5114. doi:10.1002/cctc.201801026 |
| 27. | Mae, M.; Matsuura, M.; Amii, H.; Uneyama, K. Tetrahedron Lett. 2002, 43, 2069–2072. doi:10.1016/s0040-4039(02)00205-8 |
| 28. | Palacios, F.; Ochoa de Retana, A. M.; Alonso, J. M. J. Org. Chem. 2006, 71, 6141–6148. doi:10.1021/jo060865g |
| 29. | Liu, J.; Hu, J. Chem. – Eur. J. 2010, 16, 11443–11454. doi:10.1002/chem.201000893 |
| 30. | Kurosato, F.; Ishikawa, T.; Yamada, Y.; Hanamoto, T. Synlett 2015, 26, 1827–1830. doi:10.1055/s-0034-1381008 |
| 31. | Zhang, W.; Wang, X.; Zhu, B.; Zhu, D.; Han, J.; Wzorek, A.; Sato, A.; Soloshonok, V. A.; Zhou, J.; Pan, Y. Adv. Synth. Catal. 2017, 359, 4267–4273. doi:10.1002/adsc.201701066 |
| 26. | Chai, Z.; Bouillon, J.-P.; Cahard, D. Chem. Commun. 2012, 48, 9471–9473. doi:10.1039/c2cc35246j |
| 12. | Meyer, F. Chem. Commun. 2016, 52, 3077–3094. doi:10.1039/c5cc09414c |
| 13. | Dolfen, J.; De Kimpe, N.; D’hooghe, M. Synlett 2016, 27, 1486–1510. doi:10.1055/s-0035-1561379 |
| 14. | Katagiri, T.; Ihara, H.; Takahashi, M.; Kashino, S.; Furuhashi, K.; Uneyama, K. Tetrahedron: Asymmetry 1997, 8, 2933–2937. doi:10.1016/s0957-4166(97)00320-0 |
| 15. | Colantoni, D.; Fioravanti, S.; Pellacani, L.; Tardella, P. A. Org. Lett. 2004, 6, 197–200. doi:10.1021/ol0361554 |
| 16. | Maeda, R.; Ooyama, K.; Anno, R.; Shiosaki, M.; Azema, T.; Hanamoto, T. Org. Lett. 2010, 12, 2548–2550. doi:10.1021/ol100768s |
| 17. | Künzi, S. A.; Morandi, B.; Carreira, E. M. Org. Lett. 2012, 14, 1900–1901. doi:10.1021/ol300539e |
| 18. | Yang, Y.; Huang, Y.; Qing, F.-L. Tetrahedron Lett. 2013, 54, 3826–3830. doi:10.1016/j.tetlet.2013.05.048 |
| 19. | Dolfen, J.; Kenis, S.; Van Hecke, K.; De Kimpe, N.; D'hooghe, M. Chem. – Eur. J. 2014, 20, 10650–10653. doi:10.1002/chem.201304759 |
| 20. | Wang, F.; Zhu, N.; Chen, P.; Ye, J.; Liu, G. Angew. Chem., Int. Ed. 2015, 54, 9356–9360. doi:10.1002/anie.201503412 |
| 21. | Duan, Y.; Zhou, B.; Lin, J.-H.; Xiao, J.-C. Chem. Commun. 2015, 51, 13127–13130. doi:10.1039/c5cc04991a |
| 22. | Huang, Y.-J.; Qiao, B.; Zhang, F.-G.; Ma, J.-A. Tetrahedron 2018, 74, 3791–3796. doi:10.1016/j.tet.2018.05.033 |
| 23. | Mészáros, Á.; Székely, A.; Stirling, A.; Novák, Z. Angew. Chem., Int. Ed. 2018, 57, 6643–6647. doi:10.1002/anie.201802347 |
| 24. | Thi, H. D.; Thuy, G. L. N.; Catak, S.; Van Speybroeck, V.; Nguyen, T. V.; D’hooghe, M. Synthesis 2018, 50, 1439–1456. doi:10.1055/s-0036-1591537 |
| 25. | Wu, K.; Zhou, C.-Y.; Che, C.-M. Org. Lett. 2019, 21, 85–89. doi:10.1021/acs.orglett.8b03514 |
| 26. | Chai, Z.; Bouillon, J.-P.; Cahard, D. Chem. Commun. 2012, 48, 9471–9473. doi:10.1039/c2cc35246j |
| 7. | Muller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 8. | Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1–PR43. doi:10.1021/cr800221v |
| 9. | Nie, J.; Guo, H.-C.; Cahard, D.; Ma, J.-A. Chem. Rev. 2011, 111, 455–529. doi:10.1021/cr100166a |
| 10. | Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788 |
| 11. | Rong, J.; Ni, C.; Hu, J. Asian J. Org. Chem. 2017, 6, 139–152. doi:10.1002/ajoc.201600509 |
| 45. | James, T.; van Gemmeren, M.; List, B. Chem. Rev. 2015, 115, 9388–9409. doi:10.1021/acs.chemrev.5b00128 |
| 47. | The stereochemistry of compound 5b was determined based on the 1H NMR, 19F NMR, and 2D NOE analysis. In the 2D NOE spectrum, the correlation between HO (4.83 ppm) and HC–N (2.7 ppm) was observed, whereas no correlation between HC–O (3.7 ppm) and HC–CF2 (3.02 ppm) was found. |
| 40. | Yamamoto, H.; Futatsugi, K. Angew. Chem., Int. Ed. 2005, 44, 1924–1942. doi:10.1002/anie.200460394 |
| 41. | Dimitrijević, E.; Taylor, M. S. ACS Catal. 2013, 3, 945–962. doi:10.1021/cs4000848 |
| 42. | Hashimoto, T.; Gálvez, A. O.; Maruoka, K. J. Am. Chem. Soc. 2013, 135, 17667–17670. doi:10.1021/ja407764u |
| 43. | Huang, B.; He, Y.; Levin, M. D.; Coelho, J. A. S.; Bergman, R. G.; Toste, F. D. Adv. Synth. Catal. 2020, 362, 295–301. doi:10.1002/adsc.201900816 |
| 44. | Zhang, Q.-X.; Li, Y.; Wang, J.; Yang, C.; Liu, C.-J.; Li, X.; Cheng, J.-P. Angew. Chem., Int. Ed. 2020, 59, 4550–4556. doi:10.1002/anie.201915226 |
| 46. | CCDC 1983642 (4a) and 1983643 (5c) contain the supplementary crystallographic data. The crystallographic data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif. It should be mentioned that the stereochemistry of 3-CF2-aziridines in this reaction (2R,3S) are opposite to that of 3-CF3-aziridines (2S,3R) in Cahard’s report ([26]). We hypothesized that the enantiodetermining step in our reaction might be controlled through an in situ formed chiral boronate complex from chiral disulfonimide and 2-carboxyphenylboronic acid. Further efforts to improve the level of stereoselectivity and detailed mechanistic elucidation are still undergoing in our lab. |
| 36. | Zhang, Y.; Wang, J. Chem. Commun. 2009, 5350–5361. doi:10.1039/b908378b |
| 37. | Zhang, Y.; Lu, Z.; Wulff, W. D. Synlett 2009, 2715–2739. doi:10.1055/s-0029-1217979 |
| 38. | Pellissier, H. Adv. Synth. Catal. 2014, 356, 1899–1935. doi:10.1002/adsc.201400312 |
| 39. | Liu, Y. Curr. Org. Chem. 2016, 20, 19–28. doi:10.2174/1385272819666150810225618 |
| 32. | Zeng, J.-L.; Chen, Z.; Zhang, F.-G.; Ma, J.-A. Org. Lett. 2018, 20, 4562–4565. doi:10.1021/acs.orglett.8b01854 |
| 33. | Peng, X.; Xiao, M.-Y.; Zeng, J.-L.; Zhang, F.-G.; Ma, J.-A. Org. Lett. 2019, 21, 4808–4811. doi:10.1021/acs.orglett.9b01697 |
| 34. | Zeng, J.-L.; Zhang, Y.; Zheng, M.-M.; Zhang, Z.-Q.; Xue, X.-S.; Zhang, F.-G.; Ma, J.-A. Org. Lett. 2019, 21, 8244–8249. doi:10.1021/acs.orglett.9b02989 |
| 35. | Zhang, Z.-Q.; Zheng, M.-M.; Xue, X.-S.; Marek, I.; Zhang, F.-G.; Ma, J.-A. Angew. Chem., Int. Ed. 2019, 58, 18191–18196. doi:10.1002/anie.201911701 |
| 46. | CCDC 1983642 (4a) and 1983643 (5c) contain the supplementary crystallographic data. The crystallographic data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif. It should be mentioned that the stereochemistry of 3-CF2-aziridines in this reaction (2R,3S) are opposite to that of 3-CF3-aziridines (2S,3R) in Cahard’s report ([26]). We hypothesized that the enantiodetermining step in our reaction might be controlled through an in situ formed chiral boronate complex from chiral disulfonimide and 2-carboxyphenylboronic acid. Further efforts to improve the level of stereoselectivity and detailed mechanistic elucidation are still undergoing in our lab. |
© 2020 Tan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)