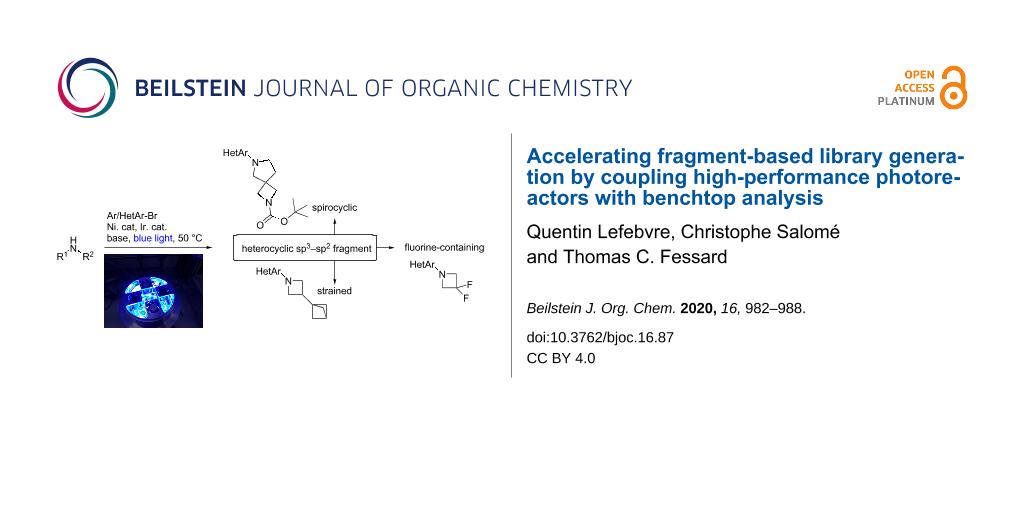
Abstract
Herein we report a workflow coupling photoredox-nickel dual-catalyzed N-arylation reactions to benchtop analysis for the efficient generation of fragment-based libraries. Technological advances in photoreactor design facilitated reliable and reproducible experimentation. Knowledge on the reactivity under previously reported reaction conditions of spirocyclic and strained heterocyclic building blocks, viewed as chemistry informers, could thus be rapidly accessed, identifying privileged or challenging scaffolds and paving the way for further exploration.

Graphical Abstract
Introduction
Heterocyclic sp2–sp3 fragments are sought-after in fragment-based libraries, as they offer improved pharmacokinetic properties over more classical, “flat” scaffolds [1]. Specifically, bicyclo- and spiroalkanes are sp3-rich (more 3D-like), conformationally-restricted and strained building blocks that are under-represented in screening collections. These are of high value to medicinal chemists to provide defined exit vectors and facilitate rational lead evolution [2-7]. Thus, we were interested in developing reliable synthetic methods and technologies to apply metal-catalyzed cross-coupling reactions to such scaffolds, which often exhibit different reactivity from unstrained substrates [8]. N-Arylation of spirocyclic amines would be a very efficient strategy for the modular synthesis of heterocyclic sp2–sp3 fragments, but their lack of stability to strongly basic conditions and heating might be an issue in palladium-catalyzed cross-coupling reactions [9]. Recently, milder cross-coupling methods using more abundant and affordable metals such as nickel, in combination with photochemistry or electrochemistry, significantly improved accessibility to N-arylated complex amines [10-12].
Results and Discussion
As we initiated a high-throughput program of fragment-based library generation and after shortcomings using more classical cross-coupling conditions, we turned our attention to the photoredox-nickel dual-catalysis strategy recently reported by MacMillan and Buchwald [10]. The reported conditions use lower temperatures and a mild base. Inspired by work at Merck on chemical informers, we decided to exploit our library of spirocyclic amines and strained scaffolds to generate a focused list of potential products [13]. We wanted to perform parallel reactions on small scale to gain further knowledge on the reactivity of these building blocks for future applications. Reaction times had to be short, and analysis, work-up and purification had to be streamlined. To comply with these project specifications, and because of the relative high cost of starting amines, the reported conditions of dual-catalyzed N-arylation were slightly altered: a smaller excess of amine was used, and dimethyl sulfoxide (DMSO) was chosen as solvent instead of dimethylformamide (DMF) or dimethylacetamide (DMA) to improve work-up and purification. The workflow was devised to ensure maximal productivity, using parallel experimentation coupled to benchtop analysis tools, namely thin-layer chromatography–mass spectrometry (TLC–MS, purchased from https://www.advion.com/rsc-product-note/direct-mass-analysis-of-tlc-plates/) and low-field nuclear magnetic resonance (NMR, purchased from https://www.nanalysis.com/) analysis (Scheme 1).
![[1860-5397-16-87-i1]](/bjoc/content/inline/1860-5397-16-87-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-day workflow for fragment-based library generation. QC: Quality control. MPLC: Medium-pressure liquid chromatography.
Scheme 1: One-day workflow for fragment-based library generation. QC: Quality control. MPLC: Medium-pressure ...
To ensure reproducibility of the reactions, we used a custom-made, high intensity photoreactor which enables accurate control of irradiation and heat [14,15]. Indeed, homogeneous irradiation and temperature in screening decreases the risk of false negatives triggered by high variability of reaction conditions compared to a single vial reactor. Active water cooling protects the LEDs from overheating, while additional air cooling with bespoke fans prevents the vessels to heat over 50 °C. Each vessel is placed at equal distance of a 35 W high power blue LED (Figure 1).
![[1860-5397-16-87-1]](/bjoc/content/figures/1860-5397-16-87-1.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Top: high-power blue LED photoreactors. Bottom, from left to right: photoreactor OFF, ON, and ON through protective yellow filter.
Figure 1: Top: high-power blue LED photoreactors. Bottom, from left to right: photoreactor OFF, ON, and ON th...
Reaction monitoring by TLC–MS enabled rapid and precise assessment of the success of each parallel reaction. As seen in Figure 2, a single TLC plate reporting five different reactions could be analyzed in five minutes, and the identity of the major product confirmed by mass.
![[1860-5397-16-87-2]](/bjoc/content/figures/1860-5397-16-87-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: TLC–MS equipment: analysis of 5 reactions in 5 minutes.
Figure 2: TLC–MS equipment: analysis of 5 reactions in 5 minutes.
Finally, after work-up and automated purification, a preliminary quality control (QC) could be performed using a benchtop 60 MHz NMR machine (Figure 3). Structural information was obtained to corroborate mass spectrometry data, an important information when dealing with strained bicycles or spirocycles, which could be prone to skeletal rearrangement. If diagnostic peaks of solvent impurities coming from the column eluents were observed, the purified material was dried further. Thus, costly and time-consuming measurements using a high-field NMR machine could be avoided: Only the final QC was performed on a 400 MHz NMR machine.
![[1860-5397-16-87-3]](/bjoc/content/figures/1860-5397-16-87-3.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Pre-QC validation by 60 MHz benchtop NMR.
Figure 3: Pre-QC validation by 60 MHz benchtop NMR.
In 4 days of experimentation, 29 combinations of bicycloalkylamines or spirocyclic amines with (hetero)aryl bromides were probed using this workflow, and the results are depicted in Scheme 2 and Scheme 3. To provide attractive heterocyclic sp2–sp3 fragments, we mainly focused on coupling 3-bromopyridine to five different classes of complex amines: bicyclopentanamines, azetidines, spiroazetidines, spiropyrrolidines and spiropiperidines. Functional group tolerance was probed by using functionalized building blocks bearing esters, carbamates, alcohols, tertiary amines, ethers, sulfones and fluorine atoms (Scheme 2 and Scheme 3).
![[1860-5397-16-87-i2]](/bjoc/content/inline/1860-5397-16-87-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the fragment-based library generation: BCP-amines and azetidines. See Supporting Information File 1 for experimental details.
Scheme 2: Scope of the fragment-based library generation: BCP-amines and azetidines. See Supporting Information File 1 for experimental de...
![[1860-5397-16-87-i3]](/bjoc/content/inline/1860-5397-16-87-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the fragment-based library generation: pyrrolidines, piperidines and morpholines. See Supporting Information File 1 for experimental details.
Scheme 3: Scope of the fragment-based library generation: pyrrolidines, piperidines and morpholines. See Supporting Information File 1 for...
Out of the 15 reactions showing product formation by TLC–MS, 7 gave the product in sufficient purity after one round of purification, while 8 were contaminated with solvent impurities, or obtained in too low yields for appropriate NMR analysis. Fourteen did not give the expected product, often showing low consumption of the aryl bromide. This corresponds to a success rate of 24% for product purification and isolation, and 52% for product formation. Although our focused library is not as functional-group-dense as the chemistry informer library used by Merck, these results are in line with previous findings on C–N cross-coupling conditions. In these works, nickel-photoredox-catalyzed cross-couplings were the most successful with success rates up to 50% [10]. This was an improvement from non-photocatalyzed conditions, where success rates of 11–33% were observed for palladium-catalyzed cross-coupling reactions, and ranged from 6% to 50% for copper-catalyzed cross-coupling reactions [13].
Much information could be obtained from this screening. Electron-deficient aryl bromides led to better yields than neutral and electron-rich partners, as observed in previous reports on photochemically- or electrochemically-mediated nickel-catalyzed cross-couplings. Electron-deficient aniline products are less prone to oxidative decomposition. BCP-amines were viable coupling partners but gave the corresponding products 1a–e in poor purities. Simple azetidines partook the reaction to give 2a–c while azaspiro[3,3]heptane analogues were unreactive. The higher reactivity of azaspiro[3,4]octane example 3d highlights the distinct reactivity of azaspiro[3,3]heptanes. On one hand, a rationalization of this observation could be traced back to the first step of the catalytic cycle. Reduction of the nickel(II) pre-catalyst to nickel(0) is believed to occur by β-hydride elimination on a sacrificial amount of amine [10]. The strained character of azaspiro[3,3]heptane might prevent this event. On the other hand, pyramidalization at nitrogen is much more important in azetidines [16-18]. This might lower the oxidation potential of the arylated product and promote oxidative decomposition. Several spirocyclic compounds with larger ring sizes could be coupled to 3-bromopyridine to give products 4c, 4d, 5a and 5b, provided that steric hindrance was not too high (see 5c). Surprisingly, oxetanes appear to be incompatible with the reaction conditions (see 4a, 4f, 5d and 5f), despite a successful report on their decarboxylative arylation under nickel-photoredox-catalyzed conditions [19]. As this report and MacMillan’s report use more basic, but less nucleophilic bases than DABCO, namely Barton’s base and MTBE, the issue might come from ring opening [20,21].
Conclusion
In summary, we have developed a new workflow allowing to efficiently perform photoredox-nickel dual-catalyzed reactions in parallel for fragment-based library generation. We have screened 29 combinations of complex spirocyclic amines and (hetero)aryl halides to get an insight into the specific reactivity of these building blocks. The workflow combined a powerful photoreactor with precise and reproducible conditions control with benchtop analytical tools such as TLC–MS and low-field NMR. This enabled the identification of privileged spirocyclic scaffolds compatible with this chemistry, and challenging substrates requiring additional investigations. The knowledge obtained within this four-day experimentation period could be built-upon in-house and allowed the efficient synthesis of original heterocyclic sp2–sp3 fragments.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Lovering, F.; Bikker, J.; Humblet, C. J. Med. Chem. 2009, 52, 6752–6756. doi:10.1021/jm901241e
Return to citation in text: [1] -
Carreira, E. M.; Fessard, T. C. Chem. Rev. 2014, 114, 8257–8322. doi:10.1021/cr500127b
Return to citation in text: [1] -
Guérot, C.; Tchitchanov, B. H.; Knust, H.; Carreira, E. M. Org. Lett. 2011, 13, 780–783. doi:10.1021/ol103050c
Return to citation in text: [1] -
Burkhard, J. A.; Guérot, C.; Knust, H.; Rogers-Evans, M.; Carreira, E. M. Org. Lett. 2010, 12, 1944–1947. doi:10.1021/ol1003302
Return to citation in text: [1] -
Burkhard, J. A.; Wagner, B.; Fischer, H.; Schuler, F.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 3524–3527. doi:10.1002/anie.200907108
Return to citation in text: [1] -
Li, D. B.; Rogers-Evans, M.; Carreira, E. M. Org. Lett. 2011, 13, 6134–6136. doi:10.1021/ol2025313
Return to citation in text: [1] -
Li, D. B.; Rogers-Evans, M.; Carreira, E. M. Org. Lett. 2013, 15, 4766–4769. doi:10.1021/ol402127b
Return to citation in text: [1] -
Bollenbach, M.; Lecroq, W.; Wagner, P.; Fessard, T.; Schmitt, M.; Salomé, C. Chem. Commun. 2019, 55, 1623–1626. doi:10.1039/c8cc09110b
Return to citation in text: [1] -
Burkhard, J.; Carreira, E. M. Org. Lett. 2008, 10, 3525–3526. doi:10.1021/ol801293f
Return to citation in text: [1] -
Corcoran, E. B.; Pirnot, M. T.; Lin, S.; Dreher, S. D.; DiRocco, D. A.; Davies, I. W.; Buchwald, S. L.; MacMillan, D. W. C. Science 2016, 353, 279–283. doi:10.1126/science.aag0209
Return to citation in text: [1] [2] [3] [4] -
Li, C.; Kawamata, Y.; Nakamura, H.; Vantourout, J. C.; Liu, Z.; Hou, Q.; Bao, D.; Starr, J. T.; Chen, J.; Yan, M.; Baran, P. S. Angew. Chem., Int. Ed. 2017, 56, 13088–13093. doi:10.1002/anie.201707906
Return to citation in text: [1] -
Lim, C.-H.; Kudisch, M.; Liu, B.; Miyake, G. M. J. Am. Chem. Soc. 2018, 140, 7667–7673. doi:10.1021/jacs.8b03744
Return to citation in text: [1] -
Kutchukian, P. S.; Dropinski, J. F.; Dykstra, K. D.; Li, B.; DiRocco, D. A.; Streckfuss, E. C.; Campeau, L.-C.; Cernak, T.; Vachal, P.; Davies, I. W.; Krska, S. W.; Dreher, S. D. Chem. Sci. 2016, 7, 2604–2613. doi:10.1039/c5sc04751j
Return to citation in text: [1] [2] -
Jelier, B. J.; Tripet, P. F.; Pietrasiak, E.; Franzoni, I.; Jeschke, G.; Togni, A. Angew. Chem., Int. Ed. 2018, 57, 13784–13789. doi:10.1002/anie.201806296
Return to citation in text: [1] -
Rössler, S. L.; Jelier, B. J.; Tripet, P. F.; Shemet, A.; Jeschke, G.; Togni, A.; Carreira, E. M. Angew. Chem., Int. Ed. 2019, 58, 526–531. doi:10.1002/anie.201810261
Return to citation in text: [1] -
Revilla-López, G.; Warren, J. G.; Torras, J.; Jiménez, A. I.; Cativiela, C.; Alemán, C. Biopolymers 2012, 98, 98–110. doi:10.1002/bip.21716
Return to citation in text: [1] -
Schnitzer, T.; Möhler, J. S.; Wennemers, H. Chem. Sci. 2020, 11, 1943–1947. doi:10.1039/c9sc05410c
Return to citation in text: [1] -
Grimm, J. B.; English, B. P.; Chen, J.; Slaughter, J. P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J. J.; Normanno, D.; Singer, R. H.; Lionnet, T.; Lavis, L. D. Nat. Methods 2015, 12, 244–250. doi:10.1038/nmeth.3256
Return to citation in text: [1] -
Kolahdouzan, K.; Khalaf, R.; Grandner, J. M.; Chen, Y.; Terrett, J. A.; Huestis, M. P. ACS Catal. 2020, 10, 405–411. doi:10.1021/acscatal.9b03596
Return to citation in text: [1] -
Baidya, M.; Mayr, H. Chem. Commun. 2008, 1792–1794. doi:10.1039/b801811a
Return to citation in text: [1] -
Maji, B.; Stephenson, D. S.; Mayr, H. ChemCatChem 2012, 4, 993–999. doi:10.1002/cctc.201200143
Return to citation in text: [1]
| 1. | Lovering, F.; Bikker, J.; Humblet, C. J. Med. Chem. 2009, 52, 6752–6756. doi:10.1021/jm901241e |
| 10. | Corcoran, E. B.; Pirnot, M. T.; Lin, S.; Dreher, S. D.; DiRocco, D. A.; Davies, I. W.; Buchwald, S. L.; MacMillan, D. W. C. Science 2016, 353, 279–283. doi:10.1126/science.aag0209 |
| 11. | Li, C.; Kawamata, Y.; Nakamura, H.; Vantourout, J. C.; Liu, Z.; Hou, Q.; Bao, D.; Starr, J. T.; Chen, J.; Yan, M.; Baran, P. S. Angew. Chem., Int. Ed. 2017, 56, 13088–13093. doi:10.1002/anie.201707906 |
| 12. | Lim, C.-H.; Kudisch, M.; Liu, B.; Miyake, G. M. J. Am. Chem. Soc. 2018, 140, 7667–7673. doi:10.1021/jacs.8b03744 |
| 9. | Burkhard, J.; Carreira, E. M. Org. Lett. 2008, 10, 3525–3526. doi:10.1021/ol801293f |
| 8. | Bollenbach, M.; Lecroq, W.; Wagner, P.; Fessard, T.; Schmitt, M.; Salomé, C. Chem. Commun. 2019, 55, 1623–1626. doi:10.1039/c8cc09110b |
| 19. | Kolahdouzan, K.; Khalaf, R.; Grandner, J. M.; Chen, Y.; Terrett, J. A.; Huestis, M. P. ACS Catal. 2020, 10, 405–411. doi:10.1021/acscatal.9b03596 |
| 2. | Carreira, E. M.; Fessard, T. C. Chem. Rev. 2014, 114, 8257–8322. doi:10.1021/cr500127b |
| 3. | Guérot, C.; Tchitchanov, B. H.; Knust, H.; Carreira, E. M. Org. Lett. 2011, 13, 780–783. doi:10.1021/ol103050c |
| 4. | Burkhard, J. A.; Guérot, C.; Knust, H.; Rogers-Evans, M.; Carreira, E. M. Org. Lett. 2010, 12, 1944–1947. doi:10.1021/ol1003302 |
| 5. | Burkhard, J. A.; Wagner, B.; Fischer, H.; Schuler, F.; Müller, K.; Carreira, E. M. Angew. Chem., Int. Ed. 2010, 49, 3524–3527. doi:10.1002/anie.200907108 |
| 6. | Li, D. B.; Rogers-Evans, M.; Carreira, E. M. Org. Lett. 2011, 13, 6134–6136. doi:10.1021/ol2025313 |
| 7. | Li, D. B.; Rogers-Evans, M.; Carreira, E. M. Org. Lett. 2013, 15, 4766–4769. doi:10.1021/ol402127b |
| 20. | Baidya, M.; Mayr, H. Chem. Commun. 2008, 1792–1794. doi:10.1039/b801811a |
| 21. | Maji, B.; Stephenson, D. S.; Mayr, H. ChemCatChem 2012, 4, 993–999. doi:10.1002/cctc.201200143 |
| 10. | Corcoran, E. B.; Pirnot, M. T.; Lin, S.; Dreher, S. D.; DiRocco, D. A.; Davies, I. W.; Buchwald, S. L.; MacMillan, D. W. C. Science 2016, 353, 279–283. doi:10.1126/science.aag0209 |
| 10. | Corcoran, E. B.; Pirnot, M. T.; Lin, S.; Dreher, S. D.; DiRocco, D. A.; Davies, I. W.; Buchwald, S. L.; MacMillan, D. W. C. Science 2016, 353, 279–283. doi:10.1126/science.aag0209 |
| 14. | Jelier, B. J.; Tripet, P. F.; Pietrasiak, E.; Franzoni, I.; Jeschke, G.; Togni, A. Angew. Chem., Int. Ed. 2018, 57, 13784–13789. doi:10.1002/anie.201806296 |
| 15. | Rössler, S. L.; Jelier, B. J.; Tripet, P. F.; Shemet, A.; Jeschke, G.; Togni, A.; Carreira, E. M. Angew. Chem., Int. Ed. 2019, 58, 526–531. doi:10.1002/anie.201810261 |
| 16. | Revilla-López, G.; Warren, J. G.; Torras, J.; Jiménez, A. I.; Cativiela, C.; Alemán, C. Biopolymers 2012, 98, 98–110. doi:10.1002/bip.21716 |
| 17. | Schnitzer, T.; Möhler, J. S.; Wennemers, H. Chem. Sci. 2020, 11, 1943–1947. doi:10.1039/c9sc05410c |
| 18. | Grimm, J. B.; English, B. P.; Chen, J.; Slaughter, J. P.; Zhang, Z.; Revyakin, A.; Patel, R.; Macklin, J. J.; Normanno, D.; Singer, R. H.; Lionnet, T.; Lavis, L. D. Nat. Methods 2015, 12, 244–250. doi:10.1038/nmeth.3256 |
| 13. | Kutchukian, P. S.; Dropinski, J. F.; Dykstra, K. D.; Li, B.; DiRocco, D. A.; Streckfuss, E. C.; Campeau, L.-C.; Cernak, T.; Vachal, P.; Davies, I. W.; Krska, S. W.; Dreher, S. D. Chem. Sci. 2016, 7, 2604–2613. doi:10.1039/c5sc04751j |
| 10. | Corcoran, E. B.; Pirnot, M. T.; Lin, S.; Dreher, S. D.; DiRocco, D. A.; Davies, I. W.; Buchwald, S. L.; MacMillan, D. W. C. Science 2016, 353, 279–283. doi:10.1126/science.aag0209 |
| 13. | Kutchukian, P. S.; Dropinski, J. F.; Dykstra, K. D.; Li, B.; DiRocco, D. A.; Streckfuss, E. C.; Campeau, L.-C.; Cernak, T.; Vachal, P.; Davies, I. W.; Krska, S. W.; Dreher, S. D. Chem. Sci. 2016, 7, 2604–2613. doi:10.1039/c5sc04751j |
© 2020 Lefebvre et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)