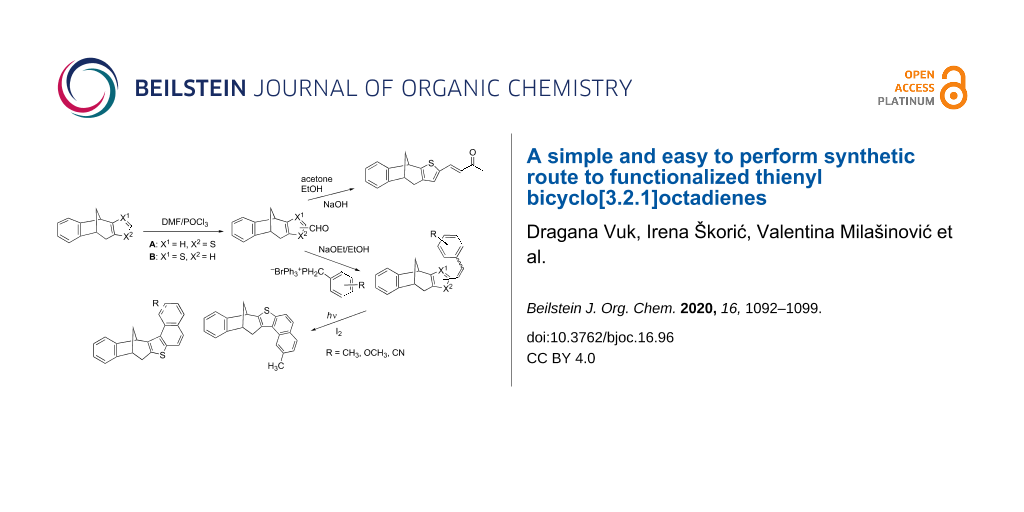
Abstract
In order to prepare novel polycyclic derivatives of bicyclo[3.2.1]octadiene systems fused with a thiophene ring, photochemical cyclization and aldol condensation reactions were carried out. The starting substrates were easily obtained by a Vilsmeier–Haack reaction of bicyclo[3.2.1]octadiene thiophene derivatives with dimethylformamide. From the obtained carbaldehydes, novel methyl, methoxy, and cyano-substituted styryl thienobenzobicyclo[3.2.1]octadiene derivatives were synthesized through Wittig reactions and subjected to photochemical cyclization, in terms of obtaining the new annulated structures. As part of this study, the aldol reaction of the starting 2-substituted carbaldehyde with acetone was also performed, which produced the thieno-fused benzobicyclo[3.2.1]octadiene compound with an extended conjugation.

Graphical Abstract
Introduction
The bicyclo[3.2.1]octane skeleton has become the subject of intensive research in recent years [1-3]. Its presence in numerous biologically active natural compounds (Figure 1) [4-7], their strenuous isolation procedures from plants, as well as their complicated multistage synthesis due to the complexity of their structure, encouraged us to develop a simple one-step synthetic procedure based on a photochemical methodology [8-21]. By using a simple photochemical procedure, it was possible to obtain a whole library of novel bicyclo[3.2.1]octadiene derivatives, available for further functionalization, which could enable the easier investigation of the relationship between structure and biological activity. During our previous investigation a series of functionalized compounds with a benzobicyclo[3.2.1]octadiene skeleton was prepared, among which some showed cholinesterase inhibitory properties (Figure 2) [2,3].
![[1860-5397-16-96-1]](/bjoc/content/figures/1860-5397-16-96-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Known biologically active bicyclo[3.2.1]octenes/octadienes.
Figure 1: Known biologically active bicyclo[3.2.1]octenes/octadienes.
![[1860-5397-16-96-2]](/bjoc/content/figures/1860-5397-16-96-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Previously prepared bicyclo[3.2.1]octenes/octadienes with cholinesterase inhibitory properties.
Figure 2: Previously prepared bicyclo[3.2.1]octenes/octadienes with cholinesterase inhibitory properties.
The aim of this study was to prepare novel thiophene bicyclo[3.2.1]octadiene derivatives with a structure convenient for the introduction of new functional groups. Further on, the study aimed at expanding the compound library and at creating preconditions for further biological investigations. This work represents a rational continuation of the research [17], previously done on similar furobicyclo[3.2.1]octadiene compounds. The previous study included the synthesis of aldehyde 03, which was via the corresponding styryl derivatives converted to the annulated products 04–07. These compounds were of particular importance due to their rigid methano-bridged junction of two aromatic units (Scheme 1). The idea herein was to prepare thienyl analogues of the annulated furyl derivatives, as substrates suitable for biological testing and/or new precursors for further functionalization.
![[1860-5397-16-96-i1]](/bjoc/content/inline/1860-5397-16-96-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of annulated furobenzobicyclo[3.2.1]octadiene compounds.
Scheme 1: Synthesis of annulated furobenzobicyclo[3.2.1]octadiene compounds.
Results and Discussion
As starting precursors two bicyclic thiophene derivatives 1' and 2', with different position of the sulfur in thiophene moiety, were selected. The compounds 1' and 2' were prepared according to the previously reported one-step photochemical methodology [15], and subjected to the Vilsmeier–Haack reaction (Scheme 2, Scheme 3), respectively. After chromatographic purification, the aldehydes 1 and 2 were obtained in very good yields (1: 79%; 2: 68%), and subsequently used as novel starting substrates for further addition/condensation reactions.
![[1860-5397-16-96-i2]](/bjoc/content/inline/1860-5397-16-96-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of annulated thiophenebicyclo[3.2.1]octadiene compounds 8-10.
Scheme 2: Synthesis of annulated thiophenebicyclo[3.2.1]octadiene compounds 8-10.
![[1860-5397-16-96-i3]](/bjoc/content/inline/1860-5397-16-96-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The Wittig reaction of the prepared aldehydes with the corresponding triphenylphosphonium salts provided five new styryl derivatives 3–7 as mixtures of cis and trans-isomers. The isomers of compounds 3 and 4 were separated by column chromatography and completely spectroscopically characterized, while in the case of compounds 5–7, only trans-isomers were obtained. Figure 3 presents parts of the 1H NMR spectra of the trans-isomers 3–6 as representative examples. The detailed analysis of all new compounds' NMR spectra can be found in Supporting Information File 1. The 1H NMR spectra of the presented examples confirmed the conservation of the bicyclic core. Six proton pattern, characteristic for these bicyclic systems, were clearly visible, with similar shifts in all cases, due to the only slight impact of the substituents on the phenyl moiety. The most significant difference was related to the protons of the methoxy group, which were shifted upfield as expected. Also a slight impact of para-substituents on the chemical shifts of the aromatic protons could be observed, with the proton chemical shift shifting upfield.
![[1860-5397-16-96-3]](/bjoc/content/figures/1860-5397-16-96-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: 1H NMR spectra (CDCl3) for the trans-isomers 3–6.
Figure 3: 1H NMR spectra (CDCl3) for the trans-isomers 3–6.
The comparison of the UV spectra of the cis- and trans-isomers of compound 3 (Figure 4) showed the expected bathochromic and hyperchromic shifts of the trans-isomers, due to the planarity of the structure.
![[1860-5397-16-96-4]](/bjoc/content/figures/1860-5397-16-96-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV spectra in ethanol (95%) of the cis- and trans-isomers of compound 3.
Figure 4: UV spectra in ethanol (95%) of the cis- and trans-isomers of compound 3.
Further, the separated isomers of 3–7 were irradiated and the reaction course followed by UV spectroscopy. In all cases, the longest wavelength absorption band gradually disappeared upon irradiation. Based on previous research, it was assumed, that the preliminary process could be a photoisomerization, which could be accompanied by a photochemical annulation. The photolysis spectra of compound's 3 isomers are shown in Figure 5, as representative examples.
![[1860-5397-16-96-5]](/bjoc/content/figures/1860-5397-16-96-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Photolysis spectra of cis-3 (a) and trans-3 (b) in ethanol (95%).
Figure 5: Photolysis spectra of cis-3 (a) and trans-3 (b) in ethanol (95%).
Figure 6 presents the UV spectra of products' 3–7 trans-isomers. All the isomers showed an absorption maxima between 300–400 nm. It can be noticed that p-substituents (trans-3, trans-4, and trans-7) enabled a higher value of molar extinction coefficients, in comparison to o-substituted compounds (trans-5 and trans-6). The cyano-substituted compound trans-6 showed the largest bathochromic shift, in regards to the methyl and methoxy-substituted compounds (trans-3–5, trans-7).
![[1860-5397-16-96-6]](/bjoc/content/figures/1860-5397-16-96-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: UV spectra in ethanol (95%) of the trans-isomers of compounds 3–7.
Figure 6: UV spectra in ethanol (95%) of the trans-isomers of compounds 3–7.
The structure of trans-6 was also confirmed by X-ray analysis (Figure 7). The compound crystallized in the ![[Graphic 1]](/bjoc/content/inline/1860-5397-16-96-i6.svg?max-width=637&scale=1.18182) space group, with the molecular symmetry Ci. The crystal packing is presented in Figure 8.
space group, with the molecular symmetry Ci. The crystal packing is presented in Figure 8.
![[1860-5397-16-96-7]](/bjoc/content/figures/1860-5397-16-96-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Molecular structure of compound trans-6. Displacement ellipsoids are drawn for the probability of 30% and hydrogen atoms are shown as spheres of arbitrary radii.
Figure 7: Molecular structure of compound trans-6. Displacement ellipsoids are drawn for the probability of 3...
![[1860-5397-16-96-8]](/bjoc/content/figures/1860-5397-16-96-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Crystal packing of trans-6. (a) Chain parallel to [100] and (b) chain parallel to [010].
Figure 8: Crystal packing of trans-6. (a) Chain parallel to [100] and (b) chain parallel to [010].
The next synthesis step involved the preparation of the annulated bicyclo[3.2.1]octadiene derivatives by irradiating the toluene solution of compound's 3–7 mixture of cis- and trans-isomers in the presence of iodine (Scheme 2 and Scheme 3). The electrocyclization reactions were successfully implemented in most cases and photoproducts 8–11 were obtained in moderate yields. The only exception was the cyano derivative 6 which was proven to be non-reactive, since the reaction mixture showed solely the presence of the initial cis- and trans-isomers.
Figure 9 presents the 1H NMR spectra of photoproducts 8 and 9, in comparison to the spectra of the starting aldehyde 1. The effect of the substituent could be seen through a shift of the aromatic singlet, which is, in the case of methoxy-substituted derivative 9, shifted downfield, due to the electronic and anisotropic effect of the methoxy group.
![[1860-5397-16-96-9]](/bjoc/content/figures/1860-5397-16-96-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: 1H NMR spectra (CDCl3) of compounds 1, 8, and 9.
Figure 9: 1H NMR spectra (CDCl3) of compounds 1, 8, and 9.
In continuation of the study herein presented, the aldol condensation reaction of the bicyclo[3.2.1]octadiene aldehyde 1 and acetone was conducted (Scheme 4). After purification of the reaction mixture the product 12 was obtained. The aim of this experiment was to obtain a system with an extended conjugation of the heteroaromatic moiety under mild conditions, while leaving the bicyclic skeleton preserved.
![[1860-5397-16-96-i4]](/bjoc/content/inline/1860-5397-16-96-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The UV spectrum of the aldol product 12, in comparison to the starting aldehyde 1, showed the expected red shift, under the prolonged conjugation in product 12 (Figure 10). Contrary to the results obtained on the styryl analogs 3–7, the preliminary irradiation experiments of compound 12 indicated its lower photoreactivity, as it was shown by only a slight decrease of the absorption band (Figure 11).
![[1860-5397-16-96-10]](/bjoc/content/figures/1860-5397-16-96-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: UV spectra of compounds 1 and 12 in ethanol (95%).
Figure 10: UV spectra of compounds 1 and 12 in ethanol (95%).
![[1860-5397-16-96-11]](/bjoc/content/figures/1860-5397-16-96-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Photolysis spectra of compound 12 in ethanol (95%).
Figure 11: Photolysis spectra of compound 12 in ethanol (95%).
As previously emphasized, the prepared products 3–7 and 12, due to the presence of a double bond in their structure, could serve as potential starting precursors for further functionalization. These functionalizations, beside the addition reaction, could involve photooxygenation reactions (Scheme 5) [1,22-25], previously studied in our laboratory. These reactions could result in a completely new spectrum of products, with preserved bicyclo[3.2.1.]octadiene skeleton, crucial for biological testing.
![[1860-5397-16-96-i5]](/bjoc/content/inline/1860-5397-16-96-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Possible outcomes of future photocatalytic oxygenation reactions of new benzobicyclo[3.2.1.]octadienes.
Scheme 5: Possible outcomes of future photocatalytic oxygenation reactions of new benzobicyclo[3.2.1.]octadie...
Conclusion
From the two starting thiophene derivatives 1 and 2, ten novel products 3–12 have been prepared by a simple and low-cost procedure, paving the way to new researches, some of which could be directed toward inclusion of new heterocycles. Due to their indicative structure the prepared compounds 1–12 are candidates for biological assays. The novel styryl derivatives 3–7 and 12 could also find application in further research for functionalization of the bicyclo[3.2.1.]octadiene core.
Supporting Information
| Supporting Information File 1: Experimental details, copies of spectra and X-ray crystallographic data. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Vuk, D.; Horváth, O.; Škorić, I. Catalysts 2019, 9, 304. doi:10.3390/catal9040304
Return to citation in text: [1] [2] -
Šagud, I.; Škorić, I.; Vuk, D.; Ratković, A.; Burčul, F. Turk. J. Chem. 2019, 43, 1170–1182. doi:10.3906/kim-1903-74
Return to citation in text: [1] [2] -
Ratković, A.; Pavlović, K.; Barić, D.; Marinić, Ž.; Grgičević, I.; Škorić, I. J. Mol. Struct. 2020, 1200, 127149. doi:10.1016/j.molstruc.2019.127149
Return to citation in text: [1] [2] -
Huang, Y.-L.; Tsai, W.-J.; Shen, C.-C.; Chen, C.-C. J. Nat. Prod. 2005, 68, 217–220. doi:10.1021/np049686p
Return to citation in text: [1] -
Winkelmann, K.; Heilmann, J.; Zerbe, O.; Rali, T.; Sticher, O. Helv. Chim. Acta 2001, 84, 3380–3392. doi:10.1002/1522-2675(20011114)84:11<3380::aid-hlca3380>3.0.co;2-o
Return to citation in text: [1] -
Cuesta-Rubio, O.; Padron, A.; Castro, H. V.; Pizza, C.; Rastrelli, L. J. Nat. Prod. 2001, 64, 973–975. doi:10.1021/np0100035
Return to citation in text: [1] -
Presset, M.; Coquerel, Y.; Rodriguez, J. Chem. Rev. 2013, 113, 525–595. doi:10.1021/cr200364p
Return to citation in text: [1] -
Šindler-Kulyk, M.; Špoljarić, L.; Marinić, Ž. Heterocycles 1989, 29, 679–682. doi:10.3987/com-89-4629
Return to citation in text: [1] -
ŠindlerKulyk, M.; Tomšić, S.; Marinić, Ž.; Metelko, B. Recl. Trav. Chim. Pays-Bas 1995, 114, 476–479. doi:10.1002/recl.19951141109
Return to citation in text: [1] -
Šindler-Kulyk, M.; Škorić, I.; Tomšić, S.; Marinić, Ž.; Mrvoš-Sermek, D. Heterocycles 1999, 51, 1355–1369. doi:10.3987/com-99-8502
Return to citation in text: [1] -
Škorić, I.; Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. Heterocycles 2001, 55, 1889–1896. doi:10.3987/com-01-9314
Return to citation in text: [1] -
Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2003, 68, 7524–7527. doi:10.1021/jo0346454
Return to citation in text: [1] -
Škorić, I.; Basarić, N.; Marinić, Ž.; Višnjevac, A.; KojićProdić, B.; ŠindlerKulyk, M. Chem. – Eur. J. 2005, 11, 543–551. doi:10.1002/chem.200401005
Return to citation in text: [1] -
Škorić, I.; Flegar, I.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2006, 62, 7396–7407. doi:10.1016/j.tet.2006.05.034
Return to citation in text: [1] -
Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062
Return to citation in text: [1] [2] -
Škorić, I.; Šmehil, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. J. Photochem. Photobiol., A 2009, 207, 190–196. doi:10.1016/j.jphotochem.2009.07.008
Return to citation in text: [1] -
Kikaš, I.; Škorić, I.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9405–9414. doi:10.1016/j.tet.2010.09.093
Return to citation in text: [1] [2] -
Škorić, I.; Kikaš, I.; Kovács, M.; Fodor, L.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Horváth, O. J. Org. Chem. 2011, 76, 8641–8657. doi:10.1021/jo200691x
Return to citation in text: [1] -
Vuk, D.; Potroško, D.; Šindler-Kulyk, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Škorić, I. J. Mol. Struct. 2013, 1051, 1–14. doi:10.1016/j.molstruc.2013.07.052
Return to citation in text: [1] -
Vuk, D.; Marinić, Ž.; Škorić, I. Croat. Chem. Acta 2014, 87, 465–473. doi:10.5562/cca2454
Return to citation in text: [1] -
Šagud, I.; Levačić, M.; Marinić, Ž.; Škorić, I. Eur. J. Org. Chem. 2017, 3787–3794. doi:10.1002/ejoc.201700481
Return to citation in text: [1] -
Kikaš, I.; Horváth, O.; Škorić, I. Tetrahedron Lett. 2011, 52, 6255–6259. doi:10.1016/j.tetlet.2011.09.076
Return to citation in text: [1] -
Kikaš, I.; Horváth, O.; Škorić, I. J. Mol. Struct. 2013, 1034, 62–68. doi:10.1016/j.molstruc.2012.09.005
Return to citation in text: [1] -
Vuk, D.; Kikaš, I.; Molčanov, K.; Horváth, O.; Škorić, I. J. Mol. Struct. 2014, 1063, 83–91. doi:10.1016/j.molstruc.2014.01.055
Return to citation in text: [1] -
Vuk, D.; Horváth, O.; Marinić, Ž.; Škorić, I. J. Mol. Struct. 2016, 1107, 70–76. doi:10.1016/j.molstruc.2015.11.036
Return to citation in text: [1]
| 1. | Vuk, D.; Horváth, O.; Škorić, I. Catalysts 2019, 9, 304. doi:10.3390/catal9040304 |
| 2. | Šagud, I.; Škorić, I.; Vuk, D.; Ratković, A.; Burčul, F. Turk. J. Chem. 2019, 43, 1170–1182. doi:10.3906/kim-1903-74 |
| 3. | Ratković, A.; Pavlović, K.; Barić, D.; Marinić, Ž.; Grgičević, I.; Škorić, I. J. Mol. Struct. 2020, 1200, 127149. doi:10.1016/j.molstruc.2019.127149 |
| 17. | Kikaš, I.; Škorić, I.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9405–9414. doi:10.1016/j.tet.2010.09.093 |
| 2. | Šagud, I.; Škorić, I.; Vuk, D.; Ratković, A.; Burčul, F. Turk. J. Chem. 2019, 43, 1170–1182. doi:10.3906/kim-1903-74 |
| 3. | Ratković, A.; Pavlović, K.; Barić, D.; Marinić, Ž.; Grgičević, I.; Škorić, I. J. Mol. Struct. 2020, 1200, 127149. doi:10.1016/j.molstruc.2019.127149 |
| 8. | Šindler-Kulyk, M.; Špoljarić, L.; Marinić, Ž. Heterocycles 1989, 29, 679–682. doi:10.3987/com-89-4629 |
| 9. | ŠindlerKulyk, M.; Tomšić, S.; Marinić, Ž.; Metelko, B. Recl. Trav. Chim. Pays-Bas 1995, 114, 476–479. doi:10.1002/recl.19951141109 |
| 10. | Šindler-Kulyk, M.; Škorić, I.; Tomšić, S.; Marinić, Ž.; Mrvoš-Sermek, D. Heterocycles 1999, 51, 1355–1369. doi:10.3987/com-99-8502 |
| 11. | Škorić, I.; Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. Heterocycles 2001, 55, 1889–1896. doi:10.3987/com-01-9314 |
| 12. | Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2003, 68, 7524–7527. doi:10.1021/jo0346454 |
| 13. | Škorić, I.; Basarić, N.; Marinić, Ž.; Višnjevac, A.; KojićProdić, B.; ŠindlerKulyk, M. Chem. – Eur. J. 2005, 11, 543–551. doi:10.1002/chem.200401005 |
| 14. | Škorić, I.; Flegar, I.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2006, 62, 7396–7407. doi:10.1016/j.tet.2006.05.034 |
| 15. | Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062 |
| 16. | Škorić, I.; Šmehil, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. J. Photochem. Photobiol., A 2009, 207, 190–196. doi:10.1016/j.jphotochem.2009.07.008 |
| 17. | Kikaš, I.; Škorić, I.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9405–9414. doi:10.1016/j.tet.2010.09.093 |
| 18. | Škorić, I.; Kikaš, I.; Kovács, M.; Fodor, L.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Horváth, O. J. Org. Chem. 2011, 76, 8641–8657. doi:10.1021/jo200691x |
| 19. | Vuk, D.; Potroško, D.; Šindler-Kulyk, M.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Škorić, I. J. Mol. Struct. 2013, 1051, 1–14. doi:10.1016/j.molstruc.2013.07.052 |
| 20. | Vuk, D.; Marinić, Ž.; Škorić, I. Croat. Chem. Acta 2014, 87, 465–473. doi:10.5562/cca2454 |
| 21. | Šagud, I.; Levačić, M.; Marinić, Ž.; Škorić, I. Eur. J. Org. Chem. 2017, 3787–3794. doi:10.1002/ejoc.201700481 |
| 4. | Huang, Y.-L.; Tsai, W.-J.; Shen, C.-C.; Chen, C.-C. J. Nat. Prod. 2005, 68, 217–220. doi:10.1021/np049686p |
| 5. | Winkelmann, K.; Heilmann, J.; Zerbe, O.; Rali, T.; Sticher, O. Helv. Chim. Acta 2001, 84, 3380–3392. doi:10.1002/1522-2675(20011114)84:11<3380::aid-hlca3380>3.0.co;2-o |
| 6. | Cuesta-Rubio, O.; Padron, A.; Castro, H. V.; Pizza, C.; Rastrelli, L. J. Nat. Prod. 2001, 64, 973–975. doi:10.1021/np0100035 |
| 7. | Presset, M.; Coquerel, Y.; Rodriguez, J. Chem. Rev. 2013, 113, 525–595. doi:10.1021/cr200364p |
| 1. | Vuk, D.; Horváth, O.; Škorić, I. Catalysts 2019, 9, 304. doi:10.3390/catal9040304 |
| 22. | Kikaš, I.; Horváth, O.; Škorić, I. Tetrahedron Lett. 2011, 52, 6255–6259. doi:10.1016/j.tetlet.2011.09.076 |
| 23. | Kikaš, I.; Horváth, O.; Škorić, I. J. Mol. Struct. 2013, 1034, 62–68. doi:10.1016/j.molstruc.2012.09.005 |
| 24. | Vuk, D.; Kikaš, I.; Molčanov, K.; Horváth, O.; Škorić, I. J. Mol. Struct. 2014, 1063, 83–91. doi:10.1016/j.molstruc.2014.01.055 |
| 25. | Vuk, D.; Horváth, O.; Marinić, Ž.; Škorić, I. J. Mol. Struct. 2016, 1107, 70–76. doi:10.1016/j.molstruc.2015.11.036 |
| 15. | Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062 |
© 2020 Vuk et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)