Abstract
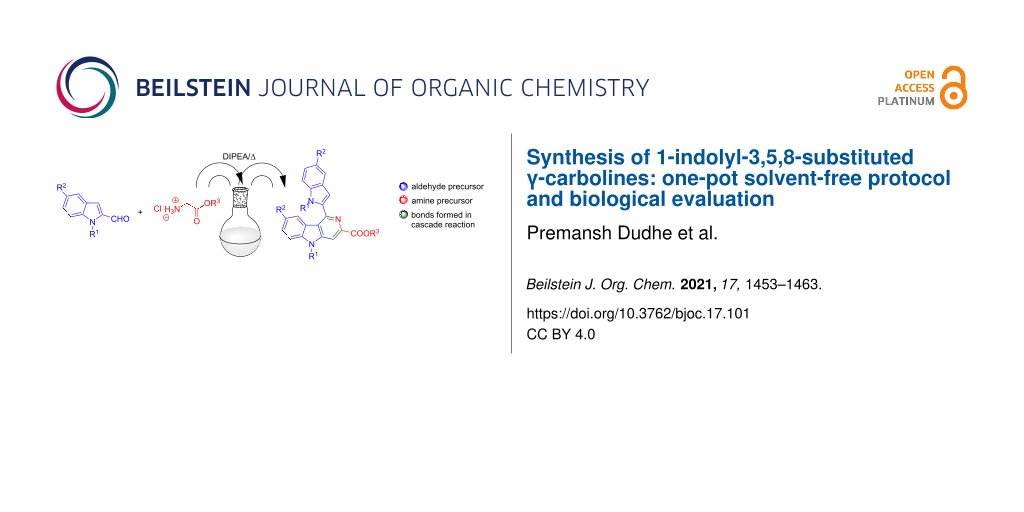
1,5-Disubstituted indole-2-carboxaldehyde derivatives 1a–h and glycine alkyl esters 2a–c are shown to undergo a novel cascade imination-heterocylization in the presence of the organic base DIPEA to provide 1-indolyl-3,5,8-substituted γ-carbolines 3aa–ea in good yields. The γ-carbolines are fluorescent and exhibit anticancer activities against cervical, lung, breast, skin, and kidney cancer cells.

Graphical Abstract
Introduction
Carbolines are privileged aza-heterocycles found in the core of several natural and synthetic compounds and are known for their biological applications. Among the four different isomers, 9H-pyrido[3,4-b]indole (β-carboline) is the most naturally abundant, present for instance, in the alkaloid harmine, a well-known selective inhibitor of monoamine oxidase-A (MAO-A) [1]. On the contrary, 5H-pyrido[4,3-b]indoles (γ-carbolines) are comparatively less examined, although these heterocycles have shown promising biological activities in preclinical and clinical studies (Figure 1) [2-6].
![[1860-5397-17-101-1]](/bjoc/content/figures/1860-5397-17-101-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of compounds containing the γ-carboline core.
Figure 1: Selected examples of compounds containing the γ-carboline core.
The pyrimidine-γ-carboline alkaloid ingenine B (isolated from an Indonesian sponge) exhibits a pronounced cytotoxicity against a murine lymphoma cell line [7] and several isocanthine analogs are effective against cervical cancer (HeLa cells) [8]. Moreover, γ-carbolines are known for their well-established DNA intercalating [9] and anti-Alzheimer [10] properties.
The classical Graebe–Ullmann synthesis [11] of γ-carbolines, one of the very early protocols in the domain, is based on the thermal decomposition of N-pyridylbenzotriazoles. Later, the reaction conditions were modified to make this reaction more versatile and operationally simple such as by the use of microwave irradiation [12]. Meanwhile, the Fischer indole synthesis was successfully extended for the synthesis of significant biologically active tetrahydro-γ-carboline derivatives [13,14]. A systematic assessment of the above Graebe–Ullmann and Fischer synthesis protocols revealed that these reactions are associated with i) low product yield, ii) limited scope including the use of a very specific set of substrates, and iii) involvement of extreme thermal conditions with the use of corrosive reagents. Much later, Larock and co-workers developed a Pd/Cu-catalyzed imino-annulation of internal alkynes [15], which paved the way for transition-metal-catalyzed cyclizations as easy access to these scaffolds. Notably, the gold-catalyzed tandem cycloisomerization/Pictet–Spengler cyclization of 2-(4-aminobut-1-yn-1-yl)aniline [16], the Ru and Rh-catalyzed [2 + 2 + 2] cycloadditions of yne-ynamides [17], and the Pd-catalyzed tandem coupling-cyclization [18] are significant works in the area (Scheme 1). However, the use of toxic and expensive metal catalysts has limited their development as environment-friendly synthetic protocols. More recently, an acid-catalyzed cyclization of α-indol-2-ylmethyl TosMIC (tosylmethyl isocyanide) derivatives to synthesize heterocycles [19] has been thoroughly studied (Scheme 1), including the synthesis of the carboline alkaloid ingenine B [20]. The iodine-catalyzed [3 + 3] cycloaddition of indolyl alcohol to enaminones [21] and the thiourea-catalyzed iso-Pictet–Spengler reaction of isotryptamine with aldehydes [22], are some noteworthy contributions to the field.
![[1860-5397-17-101-i1]](/bjoc/content/inline/1860-5397-17-101-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthetic strategy of present work in comparison with previous reports.
Scheme 1: The synthetic strategy of present work in comparison with previous reports.
A cascade or domino reaction is an interesting approach for the design of efficient one-step transformations for complex molecule synthesis [23,24]. Employing domino reactions to simplify cumbersome industrial processes can afford complex pharmaceutical products in an economical and environment-friendly manner [25]. Easy workup procedures and single-step purification reduce the efforts in the synthesis of complex molecular architectures. Therefore, cascade reactions are essential in synthetic organic chemistry, even with moderate yields [26]. Recently, such reactions have claimed their much deserving place in drug design and natural product synthesis [27]. In the literature, there is only a limited number of direct synthetic procedures to prepare γ-carbolines till date [28], and this gives a cutting edge advantage to our new protocol wherein a solvent and metal-free direct access to the γ-carboline core from substituted indole-2-aldehyes and glycine ester salts has been discovered.
Results and Discussion
Optimization of reaction conditions and chemical synthesis
The base-catalyzed imination of aromatic aldehydes is a valuable method in organic synthesis to synthesize a variety of heterocyclic building blocks [29]. Among all the reported iminoesters, alkyl N-arylideneglycinates have attracted much attention in recent years. For instance, the metal-catalyzed asymmetric [3 + 2] cycloaddition of ethyl N-benzylideneglycinates with electron-deficient alkenes has been reported to yield substituted pyrrolidines [30].
Recently, we reported the synthesis of substituted pyrrole-2-aldehydes to 5-azaindole transformation during a base-catalyzed imination reaction [31]. However, we envisioned that our methodology might be strategically applied towards the synthesis of substituted γ-carbolines as a C-3 nucleophilic attack is more favored in indoles than in pyrroles. Herein, we report an interesting observation for conversion of substituted indole-2-aldehydes 1 to 1-indolyl-3,5,8-substituted γ-carbolines 3 by a cascade imination-heterocylization pathway when treated with the salt glycine methyl ester hydrochloride (2a) in the presence of a base.
Earlier in the literature, it was reported that 1H-indole-2-carbaldehyde derivatives underwent condensation with N-arylideneglycinate to form pyrimidoindole derivatives [32]. However, when 1-methyl-1H-indole-2-carbaldehyde (1a) and glycine methyl ester hydrochloride salt (2a) were reacted in the presence of DIPEA (Hünig’s base) at room temperature in a non-polar solvent such as toluene, only marginal amounts of the corresponding imine were observed, that could not be isolated (Table 1, entry 1). When the reaction mixture was further heated to reflux for 16 h, only traces of 1-indolyl 3,5,8-substituted γ-carboline 3aa were formed that were still insufficient for complete characterization. Intending to improve the yield of 3aa, we screened various solvents, non-nucleophilic organic bases such as triethylamine and DBU, and several inorganic bases like K2CO3, Cs2CO3, and NaH (Table 1, entries 2–6).
Table 1: Optimization of the reaction conditions for the transformation of 1-methylindole-2-carbaldeyde (1a) to γ-carboline 3aa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-101-i4.svg?max-width=637&scale=1.0)
|
||||||
| entry | mmol of 1a (conc.) | mmol of 2a (conc.) | base (3.5 equiv, no of mmol, conc.) |
solvent
(5 mL) |
temp. | yield |
|---|---|---|---|---|---|---|
| 1 |
0.62 mmol
(0.12 M) |
0.31 mmol
(0.06 M) |
DIPEA (1.09 mmol, 0.21 M) | toluenea | rt to reflux | trace |
| 2 |
0.62 mmol
(0.12 M) |
0.31 mmol
(0.06 M) |
Et3N (1.09 mmol, 0.21 M) | toluenea | rt to reflux | no product |
| 3 |
0.62 mmol
(0.12 M) |
0.31 mmol
(0.06 M) |
DBU (1.09 mmol, 0.21 M) | toluenea | rt to reflux | trace |
| 4 |
0.62 mmol
(0.12 M) |
0.31 mmol
(0.06 M) |
K2CO3 (1.09 mmol, 0.21 M) | Et2Oa | rt to reflux | no product |
| 5 |
0.62 mmol
(0.12 M) |
0.31 mmol
(0.06 M) |
Cs2CO3 (1.09 mmol, 0.21 M) | DMFa | rt to reflux | no product |
| 6 |
0.62 mmol
(0.12 M) |
0.31 mmol
(0.06 M) |
NaH (1.09 mmol, 0.21 M) | THFa | rt to reflux | no product |
| 7 | 0.62 mmol | 0.31 mmol | DIPEAb | – | 120 °C | 70% |
aReactions were monitored by TLC for 3 h at room temperature followed by reflux for 16 h in the appropriate solvent; bsolvent-free reaction carried out in a 25 mL borosilicate sealed tube in a preheated oil bath in an air atmosphere at 120 °C.
After systematic screening of several reaction conditions, we found that heating at 120 °C of a neat mixture consisting of 1-methyl-1H-indole-2-carbaldehyde (1a, 2.0 equiv), glycine methyl ester HCl salt (2a, 1.0 equiv), and DIPEA (3.5 equiv) in a sealed tube for 6 h, led to the isolation of γ-carboline 3aa in 70% yield (Table 1, entry 7). The product 3aa was subsequently characterized by various spectroscopic techniques.
With the initial success at hand, the reaction was found to be equally effective with various glycine alkyl ester HCl salts 2a–c but failed to result in the formation of 1-indolyl-3-cyano-5-methyl γ-carboline derivative 3ad when 2-aminoacetonitrile 2d was utilized as the condensation component. Then, a range of 1- and 1,5-disubstituted indole-2-carboxaldehyde derivatives 1a–h was synthesized (for details see Supporting Information File 1) to evaluate the scope of the reaction further.
Indole-2-carbaldehyde derivatives with electron-donating substituents in the 1-position of the indole ring system 1a–e were transformed in moderate to good yields into their corresponding γ-carboline derivatives 3aa–ac and 3ba–ea due to an enhanced C-3 nucleophilicity of the indole nucleus (Scheme 2). The formation of the corresponding γ-carboline products was confirmed unequivocally by single-crystal X-ray diffraction analysis of 3ac (Figure 2).
![[1860-5397-17-101-i2]](/bjoc/content/inline/1860-5397-17-101-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Series of synthesized 1-indolyl-3,5,8-substituted γ-carboline 3aa–ac, 3ba-ea and 1-indolyl-1,2-dihydro-3,5-substituted γ-carboline 3ga derivatives.
Scheme 2: Series of synthesized 1-indolyl-3,5,8-substituted γ-carboline 3aa–ac, 3ba-ea and 1-indolyl-1,2-dihy...
![[1860-5397-17-101-2]](/bjoc/content/figures/1860-5397-17-101-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Single-crystal XRD structure of 3ac (CCDC: 1897787).
Figure 2: Single-crystal XRD structure of 3ac (CCDC: 1897787).
The presence of two substituents in the 1,5-position of indole-2-carbaldehyde substrates such as 1e (5-methoxy-1-methyl-1H-indole-2-carbaldehyde) and 1f (1-methyl-5-phenyl-1H-indole-2-carbaldehyde) influenced the outcome of the heterocylization reaction in different ways. For instance, 1-methyl-5-methoxy-substituted compound 1e was successfully transformed into 1-indolyl-3-carbomethoxy-5-methyl-8-methoxy γ-carboline (3ea) in 72% yield, whereas the 1-methoxy-5-phenyl-substituted indolecarbaldehyde 1f remained unreacted under the optimized reaction conditions and did not yield the expected 5-methyl-1-(1-methyl-5-phenyl-1H-indol-2-yl)-3-carbomethoxy-8-phenyl γ-carboline derivative 3fa. The reason for this remains unclear but is likely due to the electron-withdrawing nature of the phenyl substituent at the 5-position in substrate 1f. Indole substrates with a weak electron-withdrawing substituent in the 1-position such as N-tosyl in 1-tosyl-1H-indole-2-carbaldehyde (1g) did not affect the reaction course. The substrate was smoothly transformed into the corresponding 1-indolyl-3,5-disubstituted 1,2-dihydro-γ-carboline derivative 3ga instead of completely aromatized γ-carboline when heated at 120 °C with glycine methyl ester hydrochloride and DIPEA for 8 h in a sealed tube. However, electron-withdrawing 1-substituents such as an N-Boc group in 1-tert-butyloxycarbonyl-1H-indole-2-carbaldehyde (1h), impeded the conversion to γ-carboline 3ha (structure not shown) due to probable decomposition and decrease in nucleophilicity at the 3-position in substrate 1h.
Plausible mechanism for the formation of γ-carbolines
The probable mechanistic explanation (Scheme 3) for the formation of γ-carboline derivatives 3aa–ac and 3ba–ea involves the initial formation of trans-iminoester 4 from the N-protected indole-2-carboxaldehydes 1a–e and 1g, and glycine alkyl esters 2a–c. The Hünig’s base, DIPEA, helps abstract the active methylene proton from iminoester 4 to generate enolate ion 5, which undergoes nucleophilic addition with another molecule of aldehyde 1 to furnish the iminoalcohol intermediate 6. The iminoalcohol 6 undergoes dehydration under the reaction conditions to give E-imine/Z-enamine 7a or Z-imine/E-enamine 7c intermediates irreversibly, which plays a decisive role in determining ring closure either via path a or path b.
![[1860-5397-17-101-i3]](/bjoc/content/inline/1860-5397-17-101-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible mechanism for the formation of 1,2-dihydro-γ-carboline derivative 3ga and 1-indolyl-3,5,8-substituted γ-carbolines 3aa–ac and 3ba–ea.
Scheme 3: Plausible mechanism for the formation of 1,2-dihydro-γ-carboline derivative 3ga and 1-indolyl-3,5,8...
In path a, the protonation of the imine nitrogen in 7a by the conjugate acid (+ BH) leads to an electrophilic aromatic substitution at the 3-position of the indole unit to form a carbon–carbon bond in the intermediate 8. A further proton abstraction in 8 by the base then gives the 1-indolyl-3,5-substituted 1,2-dihydro-γ-carboline intermediate 9a or 3ga. In path b, the intermediate 7a cyclizes via a thermal 6π-electrocyclic reaction of the conjugated triene system to form the 1-indolyl-3,5-substituted 1,9β-dihydro-γ-carboline 9b, that may also undergo a [1,5]-sigmatropic hydrogen shift, to reinstall aromaticity of the indole ring, leading to the formation of 9a. In situ oxidation of intermediates 9a or 9b, probably from the dissolved oxygen present in the reaction mixture, leads to the formation of 1-indolyl-3,5,8-substituted γ-carbolines 3aa–ac and 3ba–ea. We successfully isolated and characterized the 1,2-dihydro γ-carboline derivative 3ga, which again verifies the proposed mechanism. During the formation of carbolines, the substrates, 1a–e and 1g were exclusively transformed to γ-carbolines or the 1,2-dihydro-γ-carboline 9a, and no traces of any β-carboline product were observed, which proves that the heterocyclization reaction is highly regiospecific.
Optical properties of γ-carbolines
Interestingly, the γ-carboline derivatives were found to be highly fluorescent under UV light irradiation. A systematic literature survey revealed that the structural core of carbolines had been widely exploited for the development of organic fluorescent entities, and in general, their UV absorbance ranges between 340 to 380 nm. For a deeper insight into the optical properties of the novel substituted γ-carbolines, absorption and emission studies were carried out in different organic solvents (Figure 3). The representative γ-carboline derivative tert-butyl-5-methyl-1-(1-methyl-1H-indol-2-yl)-5H-pyrido[4,3-b]indole-3-carboxylate (3ac) revealed similar absorption features with a shift in absorption maximum in different solvents. The highest absorption maximum (λmax) was observed at 230 nm for 3ac in DMSO (Figure 3, left side).
![[1860-5397-17-101-3]](/bjoc/content/figures/1860-5397-17-101-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis absorption (left side) and emission (right side) spectra of 3ac measured in different solvents.
Figure 3: UV–vis absorption (left side) and emission (right side) spectra of 3ac measured in different solven...
The fluorescence studies carried out for 3ac in four different solvents revealed that the emission maxima shifted bathochromically by almost 40 nm upon changing the solvent polarity, for instance, from non-polar hexane to moderately polar dichloromethane and then highly polar DMSO (Table 2, Figure 3). The fluorescence quenching of 3ac in methanol is attributed to the partial protonation of the carboline unit's nitrogen atoms facilitated by polar-protic solvents [33]. The fluorescence lifetimes were measured by time-correlated single-photon counting (TCSPC) experiments. The average fluorescence lifetime of compound 3ac was found to be 8.35 ns and 4.73 ns in DMSO and DCM, respectively (Table 2, Figure 4).
![[1860-5397-17-101-4]](/bjoc/content/figures/1860-5397-17-101-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fluorescence decay profile of 3ac in DMSO (left side; λex 360 nm) and 10−5 M solutions of compound 3ac in four different solvents in a UV chamber (right side).
Figure 4: Fluorescence decay profile of 3ac in DMSO (left side; λex 360 nm) and 10−5 M solutions of compound ...
Biological evaluation of γ-carbolines as anticancer agents
A panel of carboline derivatives 3ac, 3bc, 3ca, and 3ga, along with a standard drug, doxorubicin, were screened for their cytotoxicity against various cancer lines (Figure 5, Table 3 and Figure S2, in Supporting Information File 1) such as MCF-7 (breast cancer), HeLa (cervical cancer), HEK293 (human embryonic kidney cells), A431 (skin cancer), A549 (lung cancer), and macrophage or immune cell line (RAW 264.7). The cancer cells were treated with increasing concentrations of the carboline derivatives 3ac, 3bc, 3ca, 3ga and doxorubicin (0.1 μM, 0.25 μM, 0.5 μM, 1 μM, 2.5 μM, 5 μM, 10 μM, 25 μM, 50 μM, 100 μM) and incubated for 48 h. Half-maximal inhibitory studies show that the γ-carbolines are highly toxic to cancer cells at micromolar concentrations similar to doxorubicin, whereas they are non-cytotoxic (Figure 6) to human macrophages or immune cells.
![[1860-5397-17-101-5]](/bjoc/content/figures/1860-5397-17-101-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Dose–response curves for (A) γ-carbolines 3ac, 3bc, 3ca, 3ga in the breast cancer cell line, MCF7 and (B) doxorubicin against the panel of tested cancer cell lines.
Figure 5: Dose–response curves for (A) γ-carbolines 3ac, 3bc, 3ca, 3ga in the breast cancer cell line, MCF7 a...
Table 3: IC50 values of γ-carbolines 3ac, 3bc, 3ca, 3ga and doxorubicin against various cancer cell lines.
| compound | IC50 (µM) in cancer cell lines | ||||
|---|---|---|---|---|---|
| γ-carboline | MCF7 | A431 | A549 | HEK293 | HeLa |
| 3ac | 5.59 | 4.89 | 4.76 | 2.29 | 4.89 |
| 3bc | 7.07 | 9.18 | 5.53 | 7.14 | 8.15 |
| 3ca | 2.99 | 4.47 | 5.27 | 6.73 | 1.30 |
| 3ga | 3.71 | 3.57 | 5.05 | 4.98 | 1.07 |
| doxorubicin | 5.92 | 4.68 | 2.09 | 8.84 | 7.77 |
![[1860-5397-17-101-6]](/bjoc/content/figures/1860-5397-17-101-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Dose–response curve of γ-carbolines 3ac, 3bc, 3ca, 3ga in macrophage cell line, RAW264.7.
Figure 6: Dose–response curve of γ-carbolines 3ac, 3bc, 3ca, 3ga in macrophage cell line, RAW264.7.
At last, to evaluate cell uptake of the novel γ-carboline for fluorescence imaging, live-cell imaging experiments were performed. In brief, HeLa cells were incubated with carboline 3ac (10 μM and 100 nM), and the cellular uptake was examined using confocal microscopy (λex = 405 nm; λem = 420–470 nm). Compound 3ac showed excellent cytosolic uptake in cancer cells when incubated at a 10 μM concentration (Figure 7), whereas only little uptake was observed at a concentration of 100 nM (Figure S3, Supporting Information File 1).
![[1860-5397-17-101-7]](/bjoc/content/figures/1860-5397-17-101-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Laser scanning confocal microscopy studies (λex = 405 nm; collection range = 420–470 nm) for uptake of carboline derivative 3ac in HeLa cells. a) Confocal fluorescent image of HeLa cells after 3 h incubation with 10 μM concentration of 3ac (20-fold magnification, 2-fold zoom); b) DIC image of HeLa cells; c) overlay of (a) and (b) indicating the distribution of 3ac in the cytoplasm with distinct cell nucleus.
Figure 7: Laser scanning confocal microscopy studies (λex = 405 nm; collection range = 420–470 nm) for uptake...
Conclusion
In summary, we have developed an operationally simple one-pot synthetic protocol for the synthesis of highly substituted γ-carboline derivatives. The metal- and solvent-free method provides direct access to complex molecular structures in good yield from inexpensive substrates. The optical and biological evaluations carried out for representative γ-carbolines revealed promising photophysical and anticancer properties of the core framework for developing novel theranostic applications to diagnose and treat cancer in the future.
Experimental
General methods and materials
All reactions were carried out in oven-dried glassware with magnetic stirring. Starting materials and other reagents were obtained from a commercial supplier and used without further purification. NMR spectra were recorded on an Avance III 400 Ascend Bruker spectrometer. CDCl3 and D2O were used as NMR solvents. Chemical shifts (δ) were reported as part per million (ppm), and TMS was used as an internal reference. High-resolution mass spectra were recorded using a Bruker Daltonik High-Performance LC-MS (electrospray ionization quadrupole time-of-flight) spectrometer. X-ray structure analysis was carried out at a Bruker KAPPA APEXII single crystal X-ray diffractometer. Melting points (mp) are uncorrected and were measured on a Veego melting point apparatus (capillary method). Analytical thin-layer chromatography (TLC) was carried out on silica gel plates (silica gel 60 F254 aluminum supported plates), and the spots were visualized with a UV lamp (254 nm and 365 nm) or using chemical staining with Brady’s reagent, KMnO4, ninhydrin, iodine, and bromocresol. Column chromatography was performed using silica gel (100–200 mesh or 230–400 mesh) and neutral alumina (175 mesh). DMF, DCM, DMA, toluene, and acetonitrile were dried using CaH2 and distilled over flame-dried 4 Å molecular sieves. THF and Et2O were dried over Na/benzophenone and stored over flame-dried 4 Å molecular sieves under an inert atmosphere prior to use. Organic bases, including DIPEA, Et3N, and DBU, were stored over anhydrous KOH pellets.
In vitro cytotoxicity studies
Cytotoxicity analysis in cancer and macrophage cells
Cancer (MCF7, A431, A549, HEK293 or HeLa cell lines) or RAW264.7 cells were seeded in a 96-well plate (4,200 cells/well) and allowed to form a monolayer for a period of 48 h. Old medium was replaced with fresh medium (0.2 mL) containing an increasing concentration of γ-carboline derivatives 3ac, 3bc, 3ca, 3ga and doxorubicin (0.1 μM, 0.25 μM, 0.5 μM, 1 μM, 2.5 μM, 5 μM, 10 μM, 25 μM, 50 μM, 100 μM) and incubated for 48 h or 3 h, respectively. The medium in each well was discarded, and the cells were rinsed with PBS (3 × 0.2 mL) followed by treatment with 0.5% crystal violet (0.05 mL) for 20 minutes at room temperature. The cells were rinsed with PBS (3 × 0.2 mL) and methanol (0.20 mL) was added to each well followed by incubation for 20 minutes. The absorbance from each well proportional to the live cell was measured using a Synergy H1 multimode plate reader (BioTek Instruments, Inc., Winooski, VT, USA) at an excitation and emission wavelength of 530 nm and 590 nm, respectively.
Dose–response curves were obtained from a plot of the semi-logarithmic [conc] vs the intensity of the fluorescence emission, and the IC50 (concentration at which 50% of the enzymatic activity is inhibited) was calculated for the carboline derivatives or doxorubicin using GraphPad Prism, version 7.02 for Windows (GraphPad Software, San Diego, CA).
HeLa cell uptake study of γ-carboline 3ac
A live-cell imaging experiment was performed with HeLa cells. The HeLa cells were placed in a 4-well confocal dish (cell count ≈ 100 cells per well) and incubated for 48 h at 37 °C under 5% CO2. After 3 h of incubation with carboline derivative 3ac (10 nM, 100 nM, 1 μM, 10 μM, and 100 μM), the cellular uptake and distribution was monitored by using confocal microscopy (λex = 405 nm; λem range = 420–470 nm).
Supporting Information
| Supporting Information File 1: Copies of 1H, 13C NMR spectra of 1a–h, 3aa–ac, 3ba–bc, 3da, 3ea, 3ga, 12a–b, 12e–f, 12i, 14d, 14g and 15, UV calibration curves in different organic solvents for γ-carboline 3ac, and single-crystal XRD data of 3ac. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Bioorg. Med. Chem. 2011, 19, 134–144. doi:10.1016/j.bmc.2010.11.041
Return to citation in text: [1] -
Alekseyev, R. S.; Kurkin, A. V.; Yurovskaya, M. A. Khim. Geterotsikl. Soedin. 2009, 45, 889–925. doi:10.1007/s10593-009-0373-9
Return to citation in text: [1] -
Pontecorvo, M. J.; Devoussr, M. D.; Navitsky, M.; Lu, M.; Salloway, S.; Schaerf, F. W.; Jennings, D.; Arora, A. K.; McGeehan, A.; Lim, N. C.; Xiong, H.; Joshi, A. D.; Siderowf, A.; Mintun, M. A. Brain 2017, 140, 748–763. doi:10.1093/brain/aww334
Return to citation in text: [1] -
Snyder, J. K.; Wei, G. W.; Strosberg, A. D.; Kota, S.; Takahashi, V. Small Molecule Inhibitors of Hepatitis C Virus. Int. Pat. Appl. WO 2011/056630 A2, Oct 27, 2010.
Return to citation in text: [1] -
Guerquin-Kern, J.-L.; Coppey, M.; Carrez, D.; Brunet, A.-C.; Nguyen, C. H.; Rivalle, C.; Slodzian, G.; Croisy, A. Microsc. Res. Tech. 1997, 36, 287–295. doi:10.1002/(sici)1097-0029(19970215)36:4<287::aid-jemt6>3.0.co;2-j
Return to citation in text: [1] -
Ran, X.; Zhao, Y.; Liu, L.; Bai, L.; Yang, C.-Y.; Zhou, B.; Meagher, J. L.; Chinnaswamy, K.; Stuckey, J. A.; Wang, S. J. Med. Chem. 2015, 58, 4927–4939. doi:10.1021/acs.jmedchem.5b00613
Return to citation in text: [1] -
Ibrahim, S. R. M.; Mohamed, G. A.; Zayed, M. F.; Sayed, H. M. Drug Res. (Stuttgart, Ger.) 2015, 65, 361–365. doi:10.1055/s-0034-1384577
Return to citation in text: [1] -
Naik, P. N.; Khan, A.; Kusurkar, R. S. Tetrahedron 2013, 69, 10733–10738. doi:10.1016/j.tet.2013.10.054
Return to citation in text: [1] -
Jia, T.; Wang, J.; Guo, P.; Yu, J. Org. Biomol. Chem. 2015, 13, 1234–1242. doi:10.1039/c4ob01905a
Return to citation in text: [1] -
Otto, R.; Penzis, R.; Gaube, F.; Winckler, T.; Appenroth, D.; Fleck, C.; Tränkle, C.; Lehmann, J.; Enzensperger, C. Eur. J. Med. Chem. 2014, 87, 63–70. doi:10.1016/j.ejmech.2014.09.048
Return to citation in text: [1] -
Shuvalov, V. Y.; Shestakov, A. N.; Kulakova, L. A.; Kuratova, A. K.; Vorontsova, M. A.; Sagitullina, G. P. Khim. Geterotsikl. Soedin. 2019, 55, 844–850. doi:10.1007/s10593-019-02547-w
Return to citation in text: [1] -
Alekseev, R. S.; Kurkin, A. V.; Yurovskaya, M. A. Khim. Geterotsikl. Soedin. 2012, 48, 1235–1250. doi:10.1007/s10593-012-1127-7
Return to citation in text: [1] -
Harbert, C. A.; Plattner, J. J.; Welch, W. M.; Weissman, A.; Koe, B. K. J. Med. Chem. 1980, 23, 635–643. doi:10.1021/jm00180a011
Return to citation in text: [1] -
Butler, K. V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A. P. J. Am. Chem. Soc. 2010, 132, 10842–10846. doi:10.1021/ja102758v
Return to citation in text: [1] -
Zhang, H.; Larock, R. C. Tetrahedron Lett. 2002, 43, 1359–1362. doi:10.1016/s0040-4039(02)00005-9
Return to citation in text: [1] -
Subba Reddy, B. V.; Swain, M.; Reddy, S. M.; Yadav, J. S.; Sridhar, B. J. Org. Chem. 2012, 77, 11355–11361. doi:10.1021/jo302068e
Return to citation in text: [1] -
Nissen, F.; Richard, V.; Alayrac, C.; Witulski, B. Chem. Commun. 2011, 47, 6656–6658. doi:10.1039/c1cc11298h
Return to citation in text: [1] -
Wang, T.-T.; Zhang, D.; Liao, W.-W. Chem. Commun. 2018, 54, 2048–2051. doi:10.1039/c8cc00040a
Return to citation in text: [1] -
Gutiérrez, S.; Sucunza, D.; Vaquero, J. J. J. Org. Chem. 2018, 83, 6623–6632. doi:10.1021/acs.joc.8b00906
Return to citation in text: [1] -
Chepyshev, S. V.; Lujan-Montelongo, J. A.; Chao, A.; Fleming, F. F. Angew. Chem., Int. Ed. 2017, 56, 4310–4313. doi:10.1002/anie.201612574
Return to citation in text: [1] -
Hao, W.-J.; Wang, S.-Y.; Ji, S.-J. ACS Catal. 2013, 3, 2501–2504. doi:10.1021/cs400703u
Return to citation in text: [1] -
Lee, Y.; Klausen, R. S.; Jacobsen, E. N. Org. Lett. 2011, 13, 5564–5567. doi:10.1021/ol202300t
Return to citation in text: [1] -
Touré, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439–4486. doi:10.1021/cr800296p
Return to citation in text: [1] -
Held, F. E.; Guryev, A. A.; Fröhlich, T.; Hampel, F.; Kahnt, A.; Hutterer, C.; Steingruber, M.; Bahsi, H.; von Bojničić-Kninski, C.; Mattes, D. S.; Foertsch, T. C.; Nesterov-Mueller, A.; Marschall, M.; Tsogoeva, S. B. Nat. Commun. 2017, 8, 15071. doi:10.1038/ncomms15071
Return to citation in text: [1] -
Tietze, L. F.; Rackelmann, N. Pure Appl. Chem. 2004, 76, 1967–1983. doi:10.1351/pac200476111967
Return to citation in text: [1] -
Tietze, L. F.; Modi, A. Med. Res. Rev. 2000, 20, 304–322. doi:10.1002/1098-1128(200007)20:4<304::aid-med3>3.0.co;2-8
Return to citation in text: [1] -
Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. doi:10.1002/anie.200601872
Return to citation in text: [1] -
Dai, J.; Dan, W.; Zhang, Y.; Wang, J. Eur. J. Med. Chem. 2018, 157, 447–461. doi:10.1016/j.ejmech.2018.08.015
Return to citation in text: [1] -
Martin, S. F. Pure Appl. Chem. 2009, 81, 195–204. doi:10.1351/pac-con-08-07-03
Return to citation in text: [1] -
López-Pérez, A.; Adrio, J.; Carretero, J. C. Angew. Chem., Int. Ed. 2009, 48, 340–343. doi:10.1002/anie.200805063
Return to citation in text: [1] -
Dudhe, P.; Venkatasubbaiah, K.; Pathak, B.; Chelvam, V. Org. Biomol. Chem. 2020, 18, 1582–1587. doi:10.1039/c9ob02657f
Return to citation in text: [1] -
Das, T.; Kayet, A.; Mishra, R.; Singh, V. K. Chem. Commun. 2016, 52, 11231–11234. doi:10.1039/c6cc05378e
Return to citation in text: [1] -
Zhao, G.-J.; Liu, J.-Y.; Zhou, L.-C.; Han, K.-L. J. Phys. Chem. B 2007, 111, 8940–8945. doi:10.1021/jp0734530
Return to citation in text: [1]
| 1. | Reniers, J.; Robert, S.; Frederick, R.; Masereel, B.; Vincent, S.; Wouters, J. Bioorg. Med. Chem. 2011, 19, 134–144. doi:10.1016/j.bmc.2010.11.041 |
| 9. | Jia, T.; Wang, J.; Guo, P.; Yu, J. Org. Biomol. Chem. 2015, 13, 1234–1242. doi:10.1039/c4ob01905a |
| 20. | Chepyshev, S. V.; Lujan-Montelongo, J. A.; Chao, A.; Fleming, F. F. Angew. Chem., Int. Ed. 2017, 56, 4310–4313. doi:10.1002/anie.201612574 |
| 8. | Naik, P. N.; Khan, A.; Kusurkar, R. S. Tetrahedron 2013, 69, 10733–10738. doi:10.1016/j.tet.2013.10.054 |
| 21. | Hao, W.-J.; Wang, S.-Y.; Ji, S.-J. ACS Catal. 2013, 3, 2501–2504. doi:10.1021/cs400703u |
| 7. | Ibrahim, S. R. M.; Mohamed, G. A.; Zayed, M. F.; Sayed, H. M. Drug Res. (Stuttgart, Ger.) 2015, 65, 361–365. doi:10.1055/s-0034-1384577 |
| 18. | Wang, T.-T.; Zhang, D.; Liao, W.-W. Chem. Commun. 2018, 54, 2048–2051. doi:10.1039/c8cc00040a |
| 2. | Alekseyev, R. S.; Kurkin, A. V.; Yurovskaya, M. A. Khim. Geterotsikl. Soedin. 2009, 45, 889–925. doi:10.1007/s10593-009-0373-9 |
| 3. | Pontecorvo, M. J.; Devoussr, M. D.; Navitsky, M.; Lu, M.; Salloway, S.; Schaerf, F. W.; Jennings, D.; Arora, A. K.; McGeehan, A.; Lim, N. C.; Xiong, H.; Joshi, A. D.; Siderowf, A.; Mintun, M. A. Brain 2017, 140, 748–763. doi:10.1093/brain/aww334 |
| 4. | Snyder, J. K.; Wei, G. W.; Strosberg, A. D.; Kota, S.; Takahashi, V. Small Molecule Inhibitors of Hepatitis C Virus. Int. Pat. Appl. WO 2011/056630 A2, Oct 27, 2010. |
| 5. | Guerquin-Kern, J.-L.; Coppey, M.; Carrez, D.; Brunet, A.-C.; Nguyen, C. H.; Rivalle, C.; Slodzian, G.; Croisy, A. Microsc. Res. Tech. 1997, 36, 287–295. doi:10.1002/(sici)1097-0029(19970215)36:4<287::aid-jemt6>3.0.co;2-j |
| 6. | Ran, X.; Zhao, Y.; Liu, L.; Bai, L.; Yang, C.-Y.; Zhou, B.; Meagher, J. L.; Chinnaswamy, K.; Stuckey, J. A.; Wang, S. J. Med. Chem. 2015, 58, 4927–4939. doi:10.1021/acs.jmedchem.5b00613 |
| 19. | Gutiérrez, S.; Sucunza, D.; Vaquero, J. J. J. Org. Chem. 2018, 83, 6623–6632. doi:10.1021/acs.joc.8b00906 |
| 13. | Harbert, C. A.; Plattner, J. J.; Welch, W. M.; Weissman, A.; Koe, B. K. J. Med. Chem. 1980, 23, 635–643. doi:10.1021/jm00180a011 |
| 14. | Butler, K. V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A. P. J. Am. Chem. Soc. 2010, 132, 10842–10846. doi:10.1021/ja102758v |
| 16. | Subba Reddy, B. V.; Swain, M.; Reddy, S. M.; Yadav, J. S.; Sridhar, B. J. Org. Chem. 2012, 77, 11355–11361. doi:10.1021/jo302068e |
| 12. | Alekseev, R. S.; Kurkin, A. V.; Yurovskaya, M. A. Khim. Geterotsikl. Soedin. 2012, 48, 1235–1250. doi:10.1007/s10593-012-1127-7 |
| 17. | Nissen, F.; Richard, V.; Alayrac, C.; Witulski, B. Chem. Commun. 2011, 47, 6656–6658. doi:10.1039/c1cc11298h |
| 11. | Shuvalov, V. Y.; Shestakov, A. N.; Kulakova, L. A.; Kuratova, A. K.; Vorontsova, M. A.; Sagitullina, G. P. Khim. Geterotsikl. Soedin. 2019, 55, 844–850. doi:10.1007/s10593-019-02547-w |
| 10. | Otto, R.; Penzis, R.; Gaube, F.; Winckler, T.; Appenroth, D.; Fleck, C.; Tränkle, C.; Lehmann, J.; Enzensperger, C. Eur. J. Med. Chem. 2014, 87, 63–70. doi:10.1016/j.ejmech.2014.09.048 |
| 15. | Zhang, H.; Larock, R. C. Tetrahedron Lett. 2002, 43, 1359–1362. doi:10.1016/s0040-4039(02)00005-9 |
| 25. | Tietze, L. F.; Rackelmann, N. Pure Appl. Chem. 2004, 76, 1967–1983. doi:10.1351/pac200476111967 |
| 22. | Lee, Y.; Klausen, R. S.; Jacobsen, E. N. Org. Lett. 2011, 13, 5564–5567. doi:10.1021/ol202300t |
| 23. | Touré, B. B.; Hall, D. G. Chem. Rev. 2009, 109, 4439–4486. doi:10.1021/cr800296p |
| 24. | Held, F. E.; Guryev, A. A.; Fröhlich, T.; Hampel, F.; Kahnt, A.; Hutterer, C.; Steingruber, M.; Bahsi, H.; von Bojničić-Kninski, C.; Mattes, D. S.; Foertsch, T. C.; Nesterov-Mueller, A.; Marschall, M.; Tsogoeva, S. B. Nat. Commun. 2017, 8, 15071. doi:10.1038/ncomms15071 |
| 32. | Das, T.; Kayet, A.; Mishra, R.; Singh, V. K. Chem. Commun. 2016, 52, 11231–11234. doi:10.1039/c6cc05378e |
| 33. | Zhao, G.-J.; Liu, J.-Y.; Zhou, L.-C.; Han, K.-L. J. Phys. Chem. B 2007, 111, 8940–8945. doi:10.1021/jp0734530 |
| 30. | López-Pérez, A.; Adrio, J.; Carretero, J. C. Angew. Chem., Int. Ed. 2009, 48, 340–343. doi:10.1002/anie.200805063 |
| 31. | Dudhe, P.; Venkatasubbaiah, K.; Pathak, B.; Chelvam, V. Org. Biomol. Chem. 2020, 18, 1582–1587. doi:10.1039/c9ob02657f |
| 28. | Dai, J.; Dan, W.; Zhang, Y.; Wang, J. Eur. J. Med. Chem. 2018, 157, 447–461. doi:10.1016/j.ejmech.2018.08.015 |
| 29. | Martin, S. F. Pure Appl. Chem. 2009, 81, 195–204. doi:10.1351/pac-con-08-07-03 |
| 26. | Tietze, L. F.; Modi, A. Med. Res. Rev. 2000, 20, 304–322. doi:10.1002/1098-1128(200007)20:4<304::aid-med3>3.0.co;2-8 |
| 27. | Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem., Int. Ed. 2006, 45, 7134–7186. doi:10.1002/anie.200601872 |
© 2021 Dudhe et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)