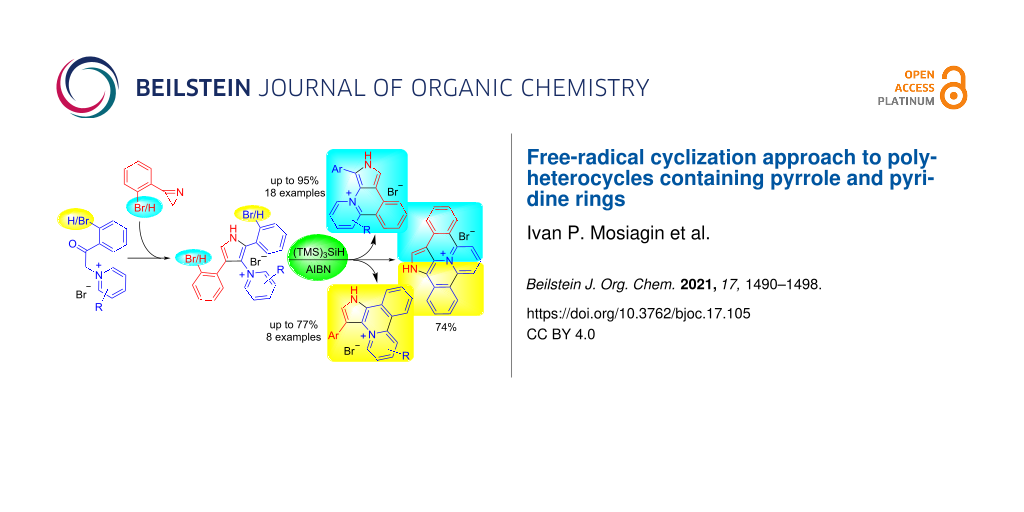
Abstract
A wide range of derivatives with new pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline skeleton was synthesized by free-radical intramolecular cyclization of o-bromophenyl-substituted pyrrolylpyridinium salts using the (TMS)3SiH/AIBN system. The cyclization provides generally good yields of pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline hydrobromides having no additional radical-sensitive substituents. The free bases can be obtained from the synthesized hydrobromides in quantitative yield by basification at room temperature. The selectivity control of intramolecular arylation was achieved by replacing the halogen: the use of 1-(2-(ortho-bromophenyl)-4-(ortho-iodophenyl)pyrrol-3-yl)pyridinium bromide makes it possible to obtain a monocyclization product, and the bicyclization product from the dibromo derivative. The procedure is also applicable to obtain 3-arylpyrido[2,1-a]pyrrolo[3,2-c]isoquinoline derivatives including 2-unsubstituted skeletons that are inaccessible via Pd-catalyzed cyclization.

Graphical Abstract
Introduction
Polycyclic heteroaromatic molecules, which have a tunable electronic structure and excellent self-assembling properties, are highly desirable in materials science, especially in post-silicon electronics [1-4]. Though the remarkable progress in the synthesis of the polycyclic heteroarenes during the last two decades, further development of the existing methodologies is still required for broadening of the scope of polycyclic heteroaromatics available for various applications. Fused polyheteroarenes, containing a pyrrole moiety, are widely present in natural products and pharmacologically important agents and have a significant importance in the development of new materials useful for bioimaging applications and chemosensor systems [3-13]. Pyrroles with vicinal o-bromophenyl and heteroaryl substituents, which are readily accessible via reactions of the corresponding 2H-azirines and phenacylcycloiminium ylides [14-19], are excellent precursors for various fused aza-heteroaromatics via metal-catalyzed arylation [17-19]. We have recently successfully applied this approach to synthesize the new luminescent heterocyclic system pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline A [17], which turned out to be useful for bioimaging [13] and as a ligand for the preparation of luminescent metal complexes, Au(I) [20], Ir(III) [21] and Eu(III) [22] (Scheme 1). According to the calculations, the isomeric pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline system B (Scheme 1) should have no less interesting photophysical properties [17], than skeleton A but its synthesis is still a challenge. In particular, attempts to assemble this framework using Pd-catalyzed intramolecular arylation of compounds C' (X = Br, I) failed [17]. In this work, we report an effective method for the assembly of pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline and related frameworks via a free radical cyclization of pyrrolylpyridinium salts.
![[1860-5397-17-105-i1]](/bjoc/content/inline/1860-5397-17-105-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of heterocycles A and B.
Scheme 1: Retrosynthetic analysis of heterocycles A and B.
Results and Discussion
Although the intramolecular free-radical cyclization is widely used for the synthesis of heteroaromatic compounds [23-26], only a few examples of the successful use of radical cyclization for the synthesis of pyrrole-containing fused heteroaromatics are known in the literature [23-30]. We initiated our study with the attempts to carry out the cyclization of 4-(2-bromophenyl)pyrrole 1a under standard radical cyclization conditions (azobisisobutyronitrile (AIBN)/Bu3SnH), which previously had been successfully used for the cyclization of 2-bromophenyl-substituted pyrroles into 7-oxa-2a1-azabenzo[b]-cyclopenta[pq]pleiadenes [30]. However, an attempt to use AIBN/Bu3SnH for the cyclization of pyrrole 1a to compound 3a only led to a tarring of the reaction mixture, regardless of the temperature (70–110 °C, MeCN) and the protocol for mixing the reagents (Scheme 2). Luckily, 4-(2-iodophenyl)pyrrole 2 gave under these conditions target heterocyclic skeleton 3a in 50% yield (Scheme 2). Salt 3a was characterized by 1H, 13C NMR and HRMS, its structure was also confirmed by single-crystal X-ray diffraction analysis (Figure 1).
![[1860-5397-17-105-1]](/bjoc/content/figures/1860-5397-17-105-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of compound 3a, displacement parameters are drawn at 50%.
Figure 1: Molecular structure of compound 3a, displacement parameters are drawn at 50%.
Since iodo-substituted compounds are more expensive and less accessible than bromo-substituted analogs, we tried to accomplish the cyclization of pyridinium salt 1a using another radical mediator, tris(trimethylsilyl)silane (TTMSS) [31,32], which, moreover, is much less toxic than tributylstannane. Fortunately, free-radical cyclization with TTMSS gave salt 3a in 74% yield from 4-(2-bromophenyl)pyrrole 1a, that is comparable with the yield for the 4-(2-iodophenyl)pyrrole 2 cyclization. Protection of pyrrole nitrogen with benzyl, methyl or acetyl PG did not improve the yield of the cyclization product 3a (Scheme 2). Acetylated compound N-Ac-1a gives deacetylated product 3a. The use of 1,1'-azobis(cyclohexanecarbonitrile) or Et3B as radical reaction initiators instead of AIBN was unsuccessful.
![[1860-5397-17-105-i2]](/bjoc/content/inline/1860-5397-17-105-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Free-radical cyclization of N-protected and N-unprotected pyrroles 1a and 2.
Scheme 2: Free-radical cyclization of N-protected and N-unprotected pyrroles 1a and 2.
Therefore, the reaction conditions for the free-radical cyclization of salt 1a were further optimized by variation of the mediator/initiator ratio, temperature, reaction time, concentration of 1a, and reagent mixing procedure (Table 1). The optimal conditions found (entry 8, Table 1), characterized by the use of minimum amounts of reagents, were used to evaluate the substrate scope for the synthesis of compounds 3 (Table 2). The developed protocol doesn’t require column chromatography for purification of target compounds 3 and can be performed in a gram scale.
Table 1: Optimization of the reaction conditions for the cyclization of 1a with TTMSS/AIBN in MeCN.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-105-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | TTMSS/AIBN, equiv |
T, °C,
(time, h) |
Concentration
of 1a, mg/mL |
Reagent mixing procedurea | Conversionb of 1a to 3a, % |
| 1 | 3/3 | 100 (12) | 0.7 | A | 20 |
| 2 | 3/3 | 100 (12) | 10 | A | 100 |
| 3 | 3/3 | 100 (12) | 7 | A | 100 |
| 4 | 2/3 | 100 (12) | 7 | B | 100 |
| 5 | 2/2 | 100 (6) | 10 | C | 100 |
| 6 | 1.5/3 | 100 (5) | 10 | C | 100 |
| 7 | 1.7/2.2 | 85 (5) | 10 | C | 100 |
| 8 | 1.5/2 | 75 (25) | 10 | C | 100 |
aMethod A: slow addition of the TTMSS/AIBN mixture through a syringe pump during all the reaction time; method B: slow addition of AIBN to the 1a/TTMSS mixture; method C: heating of premixed mixture. bConversion was determined by 1H NMR.
Pyridinium bromides 1a–l,n–w and iodide 1m were prepared by the reaction of 3-(2-bromophenyl)-2H-azirine (4a) with substituted N-phenacyl pyridinium salts 5a–w according to the published method (Table 2) [14]. The free-radical cyclization tolerates 4-F and 4-Cl substituents in the 2-phenyl ring (Table 2, entries 2,3), whereas the reaction of compound 1d, with a 4-Br substituent in the 2-phenyl ring, led to the formation of a complex mixture of products because of competitive radical reactions. Methyl and methoxy substituents in the 2-phenyl ring (Table 2, entries 5–9) did not affect the cyclization, whereas the presence of 3- or 4-NO2-substituents (Table 2, entries 10 and 11) led to tarring of the reaction mixtures probably due to participation of the NO2 group in side radical reactions. The fact that this result is not related to the electron-withdrawing nature of this substituent follows from the fact that another strong EWG at the position 4, such as cyano, did not interfere with the reaction, and the product was obtained in good yield. (Table 2, entry 12). It is important that the reaction of iodide 1m with 2-pyridyl substituent gave a good yield of the product 3m, which could be used for the preparation of bidentate ligand for metal complexes. Reaction of salts 1 with electron-donating para-substituents (Me, MeO, Me2N) in the pyridine ring (1n–p,s) and aryl substituents (1q,r) afforded the corresponding compounds 3 in 51–82% yield (Table 2, entries 14–19). Heating of isoquinolinium salt 1t with the TTMSS/AIBN system, disappointingly, led to intensive tarring without the formation of the cyclization product in even trace amounts. Salts with electron-withdrawing para-substituents in the pyridine ring (1u–w) do not cyclize under used reaction conditions. The reaction of 2H-pyrido[2,1-a]pyrrolo[3,4-c]isoquinolin-4-ium bromides 3 with aq KOH at room temperature gave quantitatively the corresponding bases 6 (Table 2).
Table 2: Synthesis of compounds 1, 3 and 6.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-105-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Ar | R1 | R2 = R3 | Yield of 1, % | Yield of 3, % |
| 1 | Ph | H | H | a, 81 | a, 74 |
| 2 | 4-FC6H4 | H | H | b, 72 | b, 60 |
| 3 | 4-ClC6H4 | H | H | c, 80 | c, 71 |
| 4 | 4-BrC6H4 | H | H | d, 79 | –a |
| 5 | 4-MeC6H4 | H | H | e, 76 | e, 58 |
| 6 | 4-MeOC6H4 | H | H | f, 83 | f, 70 |
| 7 | 2-MeOC6H4 | H | H | g, 63 | g, 63 |
| 8 | 3-MeOC6H4 | H | H | h, 68 | h, 74 |
| 9 | 2,4-(MeO)2C6H3 | H | H | i, 56 | i, 93 |
| 10 | 4-NO2C6H4 | H | H | j, 72 | –a |
| 11 | 3-NO2C6H4 | H | H | k, 65 | –a |
| 12 | 4-NCC6H4 | H | H | l, 50 | l, 73 |
| 13 | 2-Pyridylb | H | H | m, 89 | m, 92 |
| 14 | Ph | Me | H | n, 82 | n, 77 |
| 15 | Ph | MeO | H | o, 55 | o, 72 |
| 16 | Ph | Me2N | H | p, 51 | p, 85 |
| 17 | Ph | Ph | H | q, 80 | q, 53 |
| 18 | Ph | 4-MeOС6H4 | H | r, 61 | r, 71 |
| 19 | Ph | H | Me | s, 73 | s, 56 |
| 20 | Ph | R1 + R2 = (CH=CH)2; R3 = H | t, 40 | –a | |
| 21 | Ph | CO2Me | H | u, 52 | –a |
| 22 | Ph | CN | H | v, 64c | –a |
| 23 | Ph | Bz | H | w, 42c | –a |
aComplex mixture of unidentified products.b1m, 3m, 5m - iodides.
cNiBr2·3H2O was used as a base instead of Et3N.
Pyridopyrroloisoquinolinium bromide 8, functionalized with an alkoxycarbonyl group at the C1 position, was also synthesized by the developed approach, starting from azirine 4b (Scheme 3).
![[1860-5397-17-105-i3]](/bjoc/content/inline/1860-5397-17-105-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2H-pyrido[2,1-a]pyrrolo[3,4-c]isoquinolin-4-ium bromide 8.
Scheme 3: Synthesis of 2H-pyrido[2,1-a]pyrrolo[3,4-c]isoquinolin-4-ium bromide 8.
Iodo-substituted salt 2 should be more reactive towards cyclization as compared to its bromo-analogue, and this can be used to control the selectivity of intramolecular arylation. Thus compound 10 was obtained from iodo/bromo-substituted salt 9 using 1.5 equiv of TTMSS. Dibromo-substituted salt 12 under the same conditions with 4 equiv of TTMSS gave the product of the double arylation, new heterocyclic skeleton 13, 1H-dibenzo[b,g]pyrido[2,1,6-de]pyrrolo[2,3,4-ij]quinolizin-14-ium bromide (Scheme 4). The structure of 13 was confirmed by XRD-analysis (Figure 2). The reaction of bromide 13 with aq KOH at room temperature gave quantitatively the corresponding base 14. This new 24π-electron hexacyclic phenalenoid doped with two nitrogens is potentially useful material for organic field-effect transistors [1].
![[1860-5397-17-105-i4]](/bjoc/content/inline/1860-5397-17-105-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Free-radical cyclization of dihalogeno-substituted salts 9 and 12.
Scheme 4: Free-radical cyclization of dihalogeno-substituted salts 9 and 12.
![[1860-5397-17-105-2]](/bjoc/content/figures/1860-5397-17-105-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of compound 13, displacement parameters are drawn at 50% probability level.
Figure 2: Molecular structure of compound 13, displacement parameters are drawn at 50% probability level.
Bases 6 can be easily methylated and benzylated on the pyrrole nitrogen with formation of the corresponding salts N-Me-and N-Bn-3a in quantitative yield (Scheme 5).
![[1860-5397-17-105-i5]](/bjoc/content/inline/1860-5397-17-105-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Encouraged by the above mentioned results we decided to apply the developed methodology for the preparation of pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline derivatives A (Scheme 1). The previously developed Pd-catalyzed protocol for the preparation of these compounds could be used only for arylation of pyrrole N-benzyl protected starting materials C [17]. The deprotection of the product could be performed only with AlCl3 under harsh conditions and, therefore, cannot be used for the preparation of compounds with acid-sensitive substituents. In addition, the debenzylation of 1-benzyl-3-arylpyrido[2,1-a]pyrrolo[3,2-c]isoquinolines A was found to accompany by isomerization to 2-aryl derivatives, and therefore 2-unsubstituted bases A could not be prepared by a Pd-catalyzed procedure [17].
Optimal reaction conditions, found for the preparation of compounds 3 afforded reasonable results for the synthesis of 3-aryl-substituted heteroaromatics 17 (Table 3). The reaction tolerates various substituents (H, Me, MeO, Ph, 4-MeOC6H4) at the pyridine moiety of 16, with the exception of electron-withdrawing groups (CN, Bz). The reaction of 1H-pyrido[2,1-a]pyrrolo[3,2-c]isoquinolin-4-ium bromides 17a,c–h with KOH at room temperature gave quantitatively bases 18a–h.
Table 3: Synthesis of compounds 16–18.
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-105-i10.svg?max-width=637&scale=1.0)
|
|||||
| entry | Ar, azirine | R1 | R2 = R3 | Yield of 16, % | Yield of 17, % |
| 1 | Ph, 4c | H | H | a, 68 | a, 77 |
| 2 | Ph, 4c | Me | H | b, 64 | b, 46 |
| 3 | Ph, 4c | MeO | H | c, 48 | c, 60 |
| 4 | Ph, 4c | Ph | H | d, 53 | d, 69 |
| 5 | Ph, 4c | 4-MeOС6H4 | H | e, 95 | e, 46 |
| 6 | Ph, 4c | H | Me | f, 35 | f, 64 |
| 7 | Ph, 4c | R1 + R2= (CH=CH)2; R3 = H | g, 44 | g, 49 | |
| 8 | 4-MeOC6H4, 4d | H | H | h, 50 | h, 67 |
| 9 | Ph, 4c | CN | H | i, 18a | –b |
| 10 | Ph, 4c | Bz | H | j, 45a | –b |
aNiBr2·3H2O was used as a base instead of Et3N (see Experimental section). bComplex mixture of unidentified products.
The structure of 17a was confirmed by XRD-analysis (Figure 3).
![[1860-5397-17-105-3]](/bjoc/content/figures/1860-5397-17-105-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure of compound 17a, displacement parameters are drawn at 50% probability level.
Figure 3: Molecular structure of compound 17a, displacement parameters are drawn at 50% probability level.
It was found that both salt 17a and base 18a can be isomerized into isomer 19 under heating in the presence of AlCl3 via migration of the 3-Ph-group into the position 2 (Scheme 6).
![[1860-5397-17-105-i6]](/bjoc/content/inline/1860-5397-17-105-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Isomerization of compounds 17a and 18a.
Scheme 6: Isomerization of compounds 17a and 18a.
Compounds 18a, 19 can be quantitatively N-alkylated by MeI at room temperature and by BnBr at 40 °C (Scheme 7).
![[1860-5397-17-105-i7]](/bjoc/content/inline/1860-5397-17-105-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: N-Alkylation of compounds 18a and 19.
Scheme 7: N-Alkylation of compounds 18a and 19.
A study of photophysical and electrochemical properties of compounds 3 is in progress and will be published elsewhere.
Conclusion
A wide range of derivatives with new pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline skeleton was synthesized by free-radical intramolecular cyclization of o-bromophenyl-substituted pyrrolylpyridinium salts using the TTMSS/AIBN system. The cyclization provides generally good yields of pyrido[2,1-a]pyrrolo[3,4-c]isoquinoline hydrobromides having no additional radical-sensitive substituents. The developed protocol does not require column chromatography to purify target compounds and can be performed on a gram scale. The free bases can be obtained from the synthesized hydrobromides in quantitative yield by basification at rt. The use 2/4-(ortho-bromo/iodophenyl)-substituted pyrrolylpyridinium bromides allows control over the chemoselectivity of the intramolecular arylation to give the monocyclization product starting from bromo/iodo-substituents or biscyclization product starting from dibromo-derivatives to give the new 24π-electron hexacyclic phenalenoid doped with two nitrogens. The protocol is also applicable for the construction of the pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline backbone from N-unprotected pyrrolylpyridinium salts and, unlike the Pd-catalyzed version cyclization, avoids the deprotection step that is accompanied by isomerization.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, X-ray diffraction experiment, and copies of NMR spectra of new compounds. | ||
| Format: PDF | Size: 48.7 MB | Download |
Funding
We gratefully acknowledge the financial support of the Russian Science Foundation project 16-13-10036. This research was carried out using resources of the Centre for Magnetic Resonance, the Research Centre for X-ray Diffraction Studies, the Centre for Chemical Analysis and Materials, and the Computer Centre of the Science Park of Saint Petersburg State University.
References
-
Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076
Return to citation in text: [1] [2] -
Lakshminarayana, A. N.; Chi, C. N-Containing Polycyclic Heteroarenes. In Polycyclic Arenes and Heteroarenes: Synthesis, Properties, and Applications; Miao, Q., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp 225–249. doi:10.1002/9783527689545.ch9
Return to citation in text: [1] -
Hou, Y.; Jiang, G.; Gong, J.; Sha, R.; Wang, J. Chem. Res. Chin. Univ. 2021, 37, 73–82. doi:10.1007/s40242-021-0396-z
Return to citation in text: [1] [2] -
Zhen, X.; Qu, R.; Chen, W.; Wu, W.; Jiang, X. Biomater. Sci. 2021, 9, 285–300. doi:10.1039/d0bm00819b
Return to citation in text: [1] [2] -
Wang, W.-W.; Wang, Y.; Wu, W.-N.; Zhao, X.-L.; Xu, Z.-Q.; Xu, Z.-H.; Li, X.-X.; Fan, Y.-C. Spectrochim. Acta, Part A 2020, 226, 117592. doi:10.1016/j.saa.2019.117592
Return to citation in text: [1] -
Ren, F.; Shi, J.; Tong, B.; Cai, Z.; Dong, Y. Mater. Chem. Front. 2019, 3, 2072–2076. doi:10.1039/c9qm00372j
Return to citation in text: [1] -
Leventis, A.; Royakkers, J.; Rapidis, A. G.; Goodeal, N.; Corpinot, M. K.; Frost, J. M.; Bučar, D.-K.; Blunt, M. O.; Cacialli, F.; Bronstein, H. J. Am. Chem. Soc. 2018, 140, 1622–1626. doi:10.1021/jacs.7b13447
Return to citation in text: [1] -
Li, Y.; Luo, X.; Guo, Q.; Nie, Y.; Wang, T.; Zhang, C.; Huang, Z.; Wang, X.; Liu, Y.; Chen, Y.; Zheng, J.; Yang, S.; Fan, Y.; Xiang, R. J. Med. Chem. 2018, 61, 3166–3192. doi:10.1021/acs.jmedchem.8b00209
Return to citation in text: [1] -
Santra, M.; Jun, Y. W.; Bae, J.; Sarkar, S.; Choi, W.; Gryko, D. T.; Ahn, K. H. Asian J. Org. Chem. 2017, 6, 278–281. doi:10.1002/ajoc.201600613
Return to citation in text: [1] -
Ge, Y.; Liu, A.; Dong, J.; Duan, G.; Cao, X.; Li, F. Sens. Actuators, B 2017, 247, 46–52. doi:10.1016/j.snb.2017.02.157
Return to citation in text: [1] -
Yu, C.; Wu, Q.; Wang, J.; Wei, Y.; Hao, E.; Jiao, L. J. Org. Chem. 2016, 81, 3761–3770. doi:10.1021/acs.joc.6b00414
Return to citation in text: [1] -
Sareen, D.; Lee, J. H.; Hwang, H.; Yoo, S.; Lee, C.-H. Chem. Commun. 2016, 52, 5852–5855. doi:10.1039/c6cc01679k
Return to citation in text: [1] -
Koshel, E. I.; Tomashenko, O. A.; Khlebnikov, A. F.; Gaginskaya, E. R.; Saifitdinova, A. F.; Tunik, S. P. Chromosome Res. 2016, 24 (Suppl. 1), 29–30. doi:10.1007/s10577-016-9532-x
Return to citation in text: [1] [2] -
Khlebnikov, A. F.; Golovkina, M. V.; Novikov, M. S.; Yufit, D. S. Org. Lett. 2012, 14, 3768–3771. doi:10.1021/ol3016594
Return to citation in text: [1] [2] -
Galenko, E. E.; Tomashenko, O. A.; Khlebnikov, A. F.; Novikov, M. S. Org. Biomol. Chem. 2015, 13, 9825–9833. doi:10.1039/c5ob01537e
Return to citation in text: [1] -
Funt, L. D.; Tomashenko, O. A.; Khlebnikov, A. F.; Novikov, M. S.; Ivanov, A. Y. J. Org. Chem. 2016, 81, 11210–11221. doi:10.1021/acs.joc.6b02200
Return to citation in text: [1] -
Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Tomashenko, O. A.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2017, 82, 616–623. doi:10.1021/acs.joc.6b02627
Return to citation in text: [1] [2] -
Funt, L. D.; Tomashenko, O. A.; Mosiagin, I. P.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2017, 82, 7583–7594. doi:10.1021/acs.joc.7b01341
Return to citation in text: [1] [2] -
Shakirova, J. R.; Tomashenko, O. A.; Grachova, E. V.; Starova, G. L.; Sizov, V. V.; Khlebnikov, A. F.; Tunik, S. P. Eur. J. Inorg. Chem. 2017, 4180–4186. doi:10.1002/ejic.201700731
Return to citation in text: [1] -
Shakirova, J. R.; Tomashenko, O. A.; Galenko, E. E.; Khlebnikov, A. F.; Hirva, P.; Starova, G. L.; Su, S.-H.; Chou, P.-T.; Tunik, S. P. Inorg. Chem. 2018, 57, 6853–6864. doi:10.1021/acs.inorgchem.8b00390
Return to citation in text: [1] -
Shakirova, J. R.; Shevchenko, N. N.; Baigildin, V. A.; Chelushkin, P. S.; Khlebnikov, A. F.; Tomashenko, O. A.; Solomatina, A. I.; Starova, G. L.; Tunik, S. P. ACS Appl. Polym. Mater. 2020, 2, 537–547. doi:10.1021/acsapm.9b00952
Return to citation in text: [1] -
Liao, J.; Yang, X.; Ouyang, L.; Lai, Y.; Huang, J.; Luo, R. Org. Chem. Front. 2021, 8, 1345–1363. doi:10.1039/d0qo01453b
Return to citation in text: [1] [2] -
Landais, Y., Ed. Free-Radical Synthesis and Functionalization of Heterocycles; Springer Int. Publ. AG: Switzerland, 2018. doi:10.1007/978-3-319-89521-5
Return to citation in text: [1] [2] -
Majumdar, K. C.; Basu, P. K.; Chattopadhyay, S. K. Tetrahedron 2007, 63, 793–826. doi:10.1016/j.tet.2006.09.049
Return to citation in text: [1] [2] -
Bowman, W. R.; Cloonan, M. O.; Krintel, S. L. J. Chem. Soc., Perkin Trans. 1 2001, 2885–2902. doi:10.1039/a909340k
Return to citation in text: [1] [2] -
Allin, S. M.; Bowman, W. R.; Elsegood, M. R. J.; McKee, V.; Karim, R.; Rahman, S. S. Tetrahedron 2005, 61, 2689–2696. doi:10.1016/j.tet.2005.01.064
Return to citation in text: [1] -
Kotani, M.; Yamago, S.; Satoh, A.; Tokuyama, H.; Fukuyama, T. Synlett 2005, 1893–1896. doi:10.1055/s-2005-871931
Return to citation in text: [1] -
Yamakawa, T.; Ideue, E.; Shimokawa, J.; Fukuyama, T. Angew. Chem., Int. Ed. 2010, 49, 9262–9265. doi:10.1002/anie.201004963
Return to citation in text: [1] -
Petrovskii, P. P.; Tomashenko, O. A.; Novikov, M. S.; Khlebnikov, A. F.; Stoeckli-Evans, H. Chem. Heterocycl. Compd. 2017, 53, 909–912. doi:10.1007/s10593-017-2144-3
Return to citation in text: [1] [2] -
Chatgilialoglu, C.; Lalevée, J. Molecules 2012, 17, 527–555. doi:10.3390/molecules17010527
Return to citation in text: [1] -
Castillo, R. R.; Burgos, C.; Vaquero, J. J.; Alvarez-Builla, J. Eur. J. Org. Chem. 2011, 619–628. doi:10.1002/ejoc.201001071
Return to citation in text: [1]
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
| 1. | Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076 |
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
| 1. | Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076 |
| 2. | Lakshminarayana, A. N.; Chi, C. N-Containing Polycyclic Heteroarenes. In Polycyclic Arenes and Heteroarenes: Synthesis, Properties, and Applications; Miao, Q., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp 225–249. doi:10.1002/9783527689545.ch9 |
| 3. | Hou, Y.; Jiang, G.; Gong, J.; Sha, R.; Wang, J. Chem. Res. Chin. Univ. 2021, 37, 73–82. doi:10.1007/s40242-021-0396-z |
| 4. | Zhen, X.; Qu, R.; Chen, W.; Wu, W.; Jiang, X. Biomater. Sci. 2021, 9, 285–300. doi:10.1039/d0bm00819b |
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
| 31. | Chatgilialoglu, C.; Lalevée, J. Molecules 2012, 17, 527–555. doi:10.3390/molecules17010527 |
| 32. | Castillo, R. R.; Burgos, C.; Vaquero, J. J.; Alvarez-Builla, J. Eur. J. Org. Chem. 2011, 619–628. doi:10.1002/ejoc.201001071 |
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
| 18. | Tomashenko, O. A.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2017, 82, 616–623. doi:10.1021/acs.joc.6b02627 |
| 19. | Funt, L. D.; Tomashenko, O. A.; Mosiagin, I. P.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2017, 82, 7583–7594. doi:10.1021/acs.joc.7b01341 |
| 14. | Khlebnikov, A. F.; Golovkina, M. V.; Novikov, M. S.; Yufit, D. S. Org. Lett. 2012, 14, 3768–3771. doi:10.1021/ol3016594 |
| 14. | Khlebnikov, A. F.; Golovkina, M. V.; Novikov, M. S.; Yufit, D. S. Org. Lett. 2012, 14, 3768–3771. doi:10.1021/ol3016594 |
| 15. | Galenko, E. E.; Tomashenko, O. A.; Khlebnikov, A. F.; Novikov, M. S. Org. Biomol. Chem. 2015, 13, 9825–9833. doi:10.1039/c5ob01537e |
| 16. | Funt, L. D.; Tomashenko, O. A.; Khlebnikov, A. F.; Novikov, M. S.; Ivanov, A. Y. J. Org. Chem. 2016, 81, 11210–11221. doi:10.1021/acs.joc.6b02200 |
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
| 18. | Tomashenko, O. A.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2017, 82, 616–623. doi:10.1021/acs.joc.6b02627 |
| 19. | Funt, L. D.; Tomashenko, O. A.; Mosiagin, I. P.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2017, 82, 7583–7594. doi:10.1021/acs.joc.7b01341 |
| 23. | Liao, J.; Yang, X.; Ouyang, L.; Lai, Y.; Huang, J.; Luo, R. Org. Chem. Front. 2021, 8, 1345–1363. doi:10.1039/d0qo01453b |
| 24. | Landais, Y., Ed. Free-Radical Synthesis and Functionalization of Heterocycles; Springer Int. Publ. AG: Switzerland, 2018. doi:10.1007/978-3-319-89521-5 |
| 25. | Majumdar, K. C.; Basu, P. K.; Chattopadhyay, S. K. Tetrahedron 2007, 63, 793–826. doi:10.1016/j.tet.2006.09.049 |
| 26. | Bowman, W. R.; Cloonan, M. O.; Krintel, S. L. J. Chem. Soc., Perkin Trans. 1 2001, 2885–2902. doi:10.1039/a909340k |
| 27. | Allin, S. M.; Bowman, W. R.; Elsegood, M. R. J.; McKee, V.; Karim, R.; Rahman, S. S. Tetrahedron 2005, 61, 2689–2696. doi:10.1016/j.tet.2005.01.064 |
| 28. | Kotani, M.; Yamago, S.; Satoh, A.; Tokuyama, H.; Fukuyama, T. Synlett 2005, 1893–1896. doi:10.1055/s-2005-871931 |
| 29. | Yamakawa, T.; Ideue, E.; Shimokawa, J.; Fukuyama, T. Angew. Chem., Int. Ed. 2010, 49, 9262–9265. doi:10.1002/anie.201004963 |
| 30. | Petrovskii, P. P.; Tomashenko, O. A.; Novikov, M. S.; Khlebnikov, A. F.; Stoeckli-Evans, H. Chem. Heterocycl. Compd. 2017, 53, 909–912. doi:10.1007/s10593-017-2144-3 |
| 3. | Hou, Y.; Jiang, G.; Gong, J.; Sha, R.; Wang, J. Chem. Res. Chin. Univ. 2021, 37, 73–82. doi:10.1007/s40242-021-0396-z |
| 4. | Zhen, X.; Qu, R.; Chen, W.; Wu, W.; Jiang, X. Biomater. Sci. 2021, 9, 285–300. doi:10.1039/d0bm00819b |
| 5. | Wang, W.-W.; Wang, Y.; Wu, W.-N.; Zhao, X.-L.; Xu, Z.-Q.; Xu, Z.-H.; Li, X.-X.; Fan, Y.-C. Spectrochim. Acta, Part A 2020, 226, 117592. doi:10.1016/j.saa.2019.117592 |
| 6. | Ren, F.; Shi, J.; Tong, B.; Cai, Z.; Dong, Y. Mater. Chem. Front. 2019, 3, 2072–2076. doi:10.1039/c9qm00372j |
| 7. | Leventis, A.; Royakkers, J.; Rapidis, A. G.; Goodeal, N.; Corpinot, M. K.; Frost, J. M.; Bučar, D.-K.; Blunt, M. O.; Cacialli, F.; Bronstein, H. J. Am. Chem. Soc. 2018, 140, 1622–1626. doi:10.1021/jacs.7b13447 |
| 8. | Li, Y.; Luo, X.; Guo, Q.; Nie, Y.; Wang, T.; Zhang, C.; Huang, Z.; Wang, X.; Liu, Y.; Chen, Y.; Zheng, J.; Yang, S.; Fan, Y.; Xiang, R. J. Med. Chem. 2018, 61, 3166–3192. doi:10.1021/acs.jmedchem.8b00209 |
| 9. | Santra, M.; Jun, Y. W.; Bae, J.; Sarkar, S.; Choi, W.; Gryko, D. T.; Ahn, K. H. Asian J. Org. Chem. 2017, 6, 278–281. doi:10.1002/ajoc.201600613 |
| 10. | Ge, Y.; Liu, A.; Dong, J.; Duan, G.; Cao, X.; Li, F. Sens. Actuators, B 2017, 247, 46–52. doi:10.1016/j.snb.2017.02.157 |
| 11. | Yu, C.; Wu, Q.; Wang, J.; Wei, Y.; Hao, E.; Jiao, L. J. Org. Chem. 2016, 81, 3761–3770. doi:10.1021/acs.joc.6b00414 |
| 12. | Sareen, D.; Lee, J. H.; Hwang, H.; Yoo, S.; Lee, C.-H. Chem. Commun. 2016, 52, 5852–5855. doi:10.1039/c6cc01679k |
| 13. | Koshel, E. I.; Tomashenko, O. A.; Khlebnikov, A. F.; Gaginskaya, E. R.; Saifitdinova, A. F.; Tunik, S. P. Chromosome Res. 2016, 24 (Suppl. 1), 29–30. doi:10.1007/s10577-016-9532-x |
| 30. | Petrovskii, P. P.; Tomashenko, O. A.; Novikov, M. S.; Khlebnikov, A. F.; Stoeckli-Evans, H. Chem. Heterocycl. Compd. 2017, 53, 909–912. doi:10.1007/s10593-017-2144-3 |
| 22. | Shakirova, J. R.; Shevchenko, N. N.; Baigildin, V. A.; Chelushkin, P. S.; Khlebnikov, A. F.; Tomashenko, O. A.; Solomatina, A. I.; Starova, G. L.; Tunik, S. P. ACS Appl. Polym. Mater. 2020, 2, 537–547. doi:10.1021/acsapm.9b00952 |
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
| 21. | Shakirova, J. R.; Tomashenko, O. A.; Galenko, E. E.; Khlebnikov, A. F.; Hirva, P.; Starova, G. L.; Su, S.-H.; Chou, P.-T.; Tunik, S. P. Inorg. Chem. 2018, 57, 6853–6864. doi:10.1021/acs.inorgchem.8b00390 |
| 23. | Liao, J.; Yang, X.; Ouyang, L.; Lai, Y.; Huang, J.; Luo, R. Org. Chem. Front. 2021, 8, 1345–1363. doi:10.1039/d0qo01453b |
| 24. | Landais, Y., Ed. Free-Radical Synthesis and Functionalization of Heterocycles; Springer Int. Publ. AG: Switzerland, 2018. doi:10.1007/978-3-319-89521-5 |
| 25. | Majumdar, K. C.; Basu, P. K.; Chattopadhyay, S. K. Tetrahedron 2007, 63, 793–826. doi:10.1016/j.tet.2006.09.049 |
| 26. | Bowman, W. R.; Cloonan, M. O.; Krintel, S. L. J. Chem. Soc., Perkin Trans. 1 2001, 2885–2902. doi:10.1039/a909340k |
| 20. | Shakirova, J. R.; Tomashenko, O. A.; Grachova, E. V.; Starova, G. L.; Sizov, V. V.; Khlebnikov, A. F.; Tunik, S. P. Eur. J. Inorg. Chem. 2017, 4180–4186. doi:10.1002/ejic.201700731 |
| 13. | Koshel, E. I.; Tomashenko, O. A.; Khlebnikov, A. F.; Gaginskaya, E. R.; Saifitdinova, A. F.; Tunik, S. P. Chromosome Res. 2016, 24 (Suppl. 1), 29–30. doi:10.1007/s10577-016-9532-x |
| 17. | Tomashenko, O. A.; Khlebnikov, A. F.; Mosiagin, I. P.; Novikov, M. S.; Grachova, E. V.; Shakirova, J. R.; Tunik, S. P. RSC Adv. 2015, 5, 94551–94561. doi:10.1039/c5ra17755c |
© 2021 Mosiagin et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)