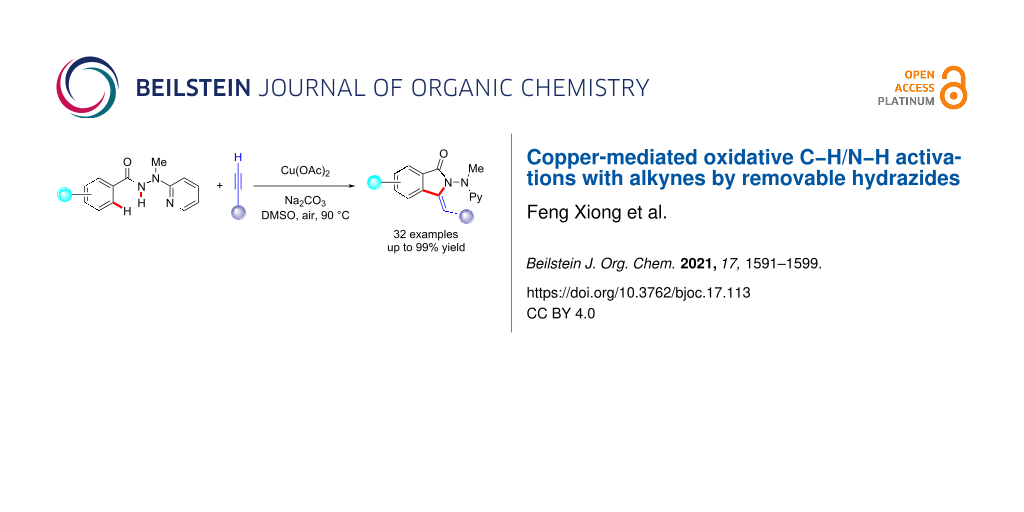
Abstract
The efficient copper-mediated oxidative C–H alkynylation of benzhydrazides was accomplished with terminal alkynes. Thus, a heteroaromatic removable N-2-pyridylhydrazide allowed for domino C–H/N–H functionalization. The approach featured remarkable functional group compatibility and ample substrate scope. Thereby, highly functionalized aromatic and heteroaromatic isoindolin-1-ones were accessed with high efficacy with rate-limiting C–H cleavage.

Graphical Abstract
Introduction
Inexpensive copper-promoted oxidative C−H activations [1-11] have been recognized as competent tools for the efficient assembly and late-stage functionalization of organic molecules due to the natural abundance and versatile reactivity. Early examples of copper-promoted C−H activation of 2-arylpyridines were disclosed by Yu et al. [12] and Chatami et al. [13] independently. Inspired by these studies, various copper-induced C−H functionalizations, such as arylations, alkynylations, cyanations, aminations, nitrations, oxygenations, thiolations, halogenations, and phosphorylations, among others, were accomplished [14-19].
The 3-methyleneisoindolin-1-one moiety represents a key structure motif in natural products [20-23] or important pharmacophores [24]. In this context, You [25], Huang [26], Liu [27], Li [28], and co-workers elegantly disclosed copper-mediated/catalyzed cascade C−H alkynylation and annulation with terminal alkynes to afford 3-methyleneisoindolinone derivatives, through the assistance of 8-aminoquinoline [29] or 2-aminophenyl-1H-pyrazole [30] auxiliaries (Figure 1a). Besides, the cobalt(II)- [31] or nickel(II)-catalyzed [32,33], pyridine oxide (PyO)-directed tandem alkynylation/annulation was realized by Niu and Song et al., which also provided the 3-methyleneisoindolin-1-one scaffolds (Figure 1b). Notably, a sustainable cupraelectro-catalyzed alkyne annulation was very recently achieved by Ackermann et al., which gave rapid access to synthetically meaningful isoindolones (Figure 1c) [34]. In spite of these indisputable advances, the successful removal of the directing groups to deliver the free-NH 3-methyleneisoindolin-1-one has thus far unfortunately proven elusive [35].
![[1860-5397-17-113-1]](/bjoc/content/figures/1860-5397-17-113-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Assembly of 3-methyleneisoindolin-1-one via 3d transition metal-mediated/catalyzed oxidative C−H/N−H activation.
Figure 1: Assembly of 3-methyleneisoindolin-1-one via 3d transition metal-mediated/catalyzed oxidative C−H/N−...
2-(1-Methylhydrazinyl)pyridine (MHP) [36] was identified as a powerful removable bidentate directing group, which found widespread application in various cobalt-catalyzed C−H activations [37-40]. Thus, our group also accomplished a set of electrochemical cobalt-catalyzed C−H activations with the MHP auxiliary [41-44]. In continuation of studies on sustainable 3d transition metal-catalyzed C−H activation [41-49], we have now discovered a robust copper-promoted oxidative C−H/N−H functionalization with terminal alkynes (Figure 1d). Notable advantages of our protocol include: 1) removable MHP auxiliary used for copper-mediated oxidative C–H activations, 2) excellent functional group tolerance and compatibility with valuable heterocycles, and 3) mechanistic studies toward copper-mediated oxidative C−H alkynylations.
Results and Discussion
We initiated our investigation by utilizing benzhydrazide 1a and ethynylbenzene (2a) as the standard substrates (Table 1). After preliminary solvent optimization, we discovered that the desired ortho-selective C−H activation occurred efficiently by the treatment of hydrazide 1a with terminal alkyne 2a and a stoichiometric amount of Cu(OAc)2 in DMSO (Table 1, entries 1–3). Reaction optimization revealed that the most appropriate temperature was 90 °C (Table 1, entries 3–6). An evaluation of bases showed that Na2CO3 was optimal (Table 1, entries 7–11). The best result was obtained when Cu(OAc)2 (1.3 equiv) was utilized in DMSO (6.0 mL, Table 1, entries 12–14). A similar result was obtained when Cu(OAc)2⋅H2O was used instead of Cu(OAc)2 (Table 1, entry 15). Only a trace amount of product 3aa was observed in the absence of either Cu(OAc)2 or Na2CO3 (Table 1, entries 16 and 17). When the reaction was performed under a nitrogen atmosphere, the efficacy was significantly decreased (Table 1, entry 18).
Table 1: Optimization of the copper-mediated C−H/N−H functionalization with terminal alkyne 2a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-113-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | base | T (°C) | Z/E | yield (%) |
| 1 | DMF | Na2CO3 | 90 | — | trace |
| 2 | NMP | Na2CO3 | 90 | — | trace |
| 3 | DMSO | Na2CO3 | 90 | 12:1 | 67 |
| 4 | DMSO | Na2CO3 | 110 | 8:1 | 57 |
| 5 | DMSO | Na2CO3 | 80 | 15:1 | 41 |
| 6 | DMSO | Na2CO3 | 60 | — | 27 |
| 7 | DMSO | NaOAc | 90 | — | 25 |
| 8 | DMSO | NaOPiv | 90 | — | 30 |
| 9 | DMSO | K2CO3 | 90 | 18:1 | 58 |
| 10 | DMSO | Cs2CO3 | 90 | 20:1 | 44 |
| 11 | DMSO | DBU | 90 | — | 13 |
| 12b | DMSO | Na2CO3 | 90 | 12:1 | 42 |
| 13c | DMSO | Na2CO3 | 90 | 9:1 | 83 |
| 14c,d | DMSO | Na2CO3 | 90 | 13:1 | 89 |
| 15d,e | DMSO | Na2CO3 | 90 | 12:1 | 86 |
| 16 | DMSO | — | 90 | — | trace |
| 17f | DMSO | Na2CO3 | 90 | — | trace |
| 18g | DMSO | Na2CO3 | 90 | — | 37 |
aReaction conditions: 1a (0.30 mmol), 2a (0.90 mmol), Cu(OAc)2 (1.1 equiv), base (2.0 equiv), solvent (3.0 mL), 15 h, under air. bCu(OAc)2 (0.8 equiv). cCu(OAc)2 (1.3 equiv). dDMSO (6.0 mL). eCu(OAc)2⋅H2O (1.3 equiv). fWithout Cu(OAc)2. gUnder N2.
We next examined the versatility of the copper-promoted ethynylbenzene (2a) annulation with various benzhydrazides 1 under the optimized reaction conditions (Scheme 1). To our delight, hydrazides 1 with electron-donating or electron-withdrawing substituents were efficiently converted in the C–H/N–H activation annulation process. Notably, a wide range of valuable electrophilic functional groups, such as halogen, methylthio, cyano, amino, and ester groups, were well compatible, which should prove instrumental for the further diversification of the thus obtained 3-methyleneisoindolin-1-ones 3da–ka. For substrates bearing two potential reactive sites, the annulation selectively took place at the less congested ortho-C−H bond (see 3la and 3ma). Moreover, the challenging isonicotinic acid hydrazide 1n was also amenable to this protocol and delivered the desired product 3na with high regioselectivity.
![[1860-5397-17-113-i1]](/bjoc/content/inline/1860-5397-17-113-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Copper-mediated oxidative C−H/N−H functionalization of hydrazides 1 with ethynylbenzene (2a).
Scheme 1: Copper-mediated oxidative C−H/N−H functionalization of hydrazides 1 with ethynylbenzene (2a).
We further investigated the viable scope of differently substituted terminal alkynes 2 as the general coupling partners for this transformation. As shown in Scheme 2, a variety of valuable electrophilic substitutes were well tolerated. Moreover, substrates with a highly reactive unprotected amino group also delivered the corresponding product 3cn with good yield. The robustness of this protocol was further highlighted by the excellent reactivity of heterocyclic acetylenes (see 2p–r). However, a complex mixture was observed when an aliphatic terminal alkyne was used, and no annulation product was detected for internal alkynes.
![[1860-5397-17-113-i2]](/bjoc/content/inline/1860-5397-17-113-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Copper-mediated oxidative C−H/N−H functionalization of 1 with alkynes 2.
Scheme 2: Copper-mediated oxidative C−H/N−H functionalization of 1 with alkynes 2.
Our copper-promoted C−H annulation protocol was not restricted to terminal alkynes. Under identical reaction conditions, commercially available alkynylcarboxylic acid 4 also proved to be a viable substrate. Thus, the corresponding isoindolone 3aa was assembled via a tandem decarboxylative C−H/C−C sequence (Scheme 3a). The practical relevance of our approach was reflected by the cleavage of the N-2-pyridylhydrazide group, yielding S-3aa (Scheme 3b).
![[1860-5397-17-113-i3]](/bjoc/content/inline/1860-5397-17-113-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Decaboxylative C−H/N−H activation and cleavage of the directing group.
Scheme 3: Decaboxylative C−H/N−H activation and cleavage of the directing group.
Inspired by the remarkable robustness of the copper-promoted C−H activations with alkynes, we became interested to explore the working mode by a set of experiments. To this end, electron-poor arenes inherently reacted preferentially in intermolecular competition experiments (Scheme 4a). This observation could be explained in terms of a concerted metalation deprotonation (CMD) mechanism [50]. Interestingly, electron-rich alkyne 2f displayed a higher reactivity in the copper-promoted C−H activations as compared to the electron-poor analog 2h (Scheme 4b). A significant H/D scrambling was not detected in the ortho-position of the reisolated benzhydrazide 1c and product 3ca when the reaction was conducted with the isotopically labeled D2O as cosolvent (Scheme 4c). This observation indicated that the C−H cleavage is irreversible. In accordance with this finding, a kinetic isotope effect (KIE) of kH/kD ≈ 6.1 was observed by parallel experiments, again suggesting that the C‒H activation is kinetically relevant (Scheme 4d).
![[1860-5397-17-113-i4]](/bjoc/content/inline/1860-5397-17-113-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Summary of key mechanistic findings.
Scheme 4: Summary of key mechanistic findings.
Based on our mechanistic findings and previous studies, we propose a tentative plausible reaction pathway in Scheme 5. The transformation commences with substrate coordination and subsequent carboxylate-assisted C−H cleavage to deliver copper(II) intermediate A. Next, the copper(III) carboxylate species B is generated. Thereafter, a facile base-assisted ligand exchange is followed by reductive elimination to afford the alkynylated benzamide D. Finally, the desired isoindolone 3 is formed via an intramolecular hydroamination in the presence of base.
![[1860-5397-17-113-i5]](/bjoc/content/inline/1860-5397-17-113-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have reported on the chelation-assisted oxidative copper-promoted cascade C−H alkynylation and intramolecular annulation. The removable N-2-pyridylhydrazide was utilized to facilitate copper(II)-promoted C−H activations. Thus, the robust copper-mediated C−H activation featured remarkable compatibility of synthetically meaningful functional groups, giving facile access to valuable 3-methyleneisoindolin-1-one scaffolds.
Experimental
General information
Yields refer to isolated compounds, estimated to be >95% pure as determined by 1H NMR spectroscopy. Chromatographic separation was carried out on silica gel 60H (200–300 mesh) manufactured by Qingdao Haiyang Chemical Group Co. (China). High-resolution mass spectrometry (HRMS) was measured on a Thermo-DFS mass spectrometer. NMR spectra were recorded on a JEOL 600 NMR device (1H: 600 MHz; 13C: 150 MHz; 19F: 565 MHz) in CDCl3. If not otherwise specified, the chemical shift (δ) is given in ppm.
Materials
Reactions were carried out under an argon atmosphere using predried glassware, if not noted otherwise. Benzhydrazides 1 were synthesized according to a previously described method [36,44]. Other chemicals were obtained from commercial sources and were used without further purification.
General procedure for the copper-promoted oxidative C−H/N−H activation with alkynes
To a 25 mL Schlenk tube were added benzhydrazide 1 (0.30 mmol, 1.00 equiv), the alkyne (0.90 mmol, 3.0 equiv), Cu(OAc)2 (71 mg, 0.39 mmol, 1.30 equiv), and Na2CO3 (64 mg, 0.60 mmol, 2.00 equiv) under an air atmosphere. The mixture was stirred at 90 °C for 15 h. At ambient temperature, H2O (15 mL) and Et3N (0.5 mL) were added, and a suspension was formed immediately. After filtrated through a Celite® pad, the reaction mixture was extracted with EtOAc (3 × 20 mL). The combined organic phase was washed with brine (20 mL) and dried over Na2SO4. Then, Et3N (0.5 mL) and silica gel (0.8 g) were added, and the combined solvent was removed under reduced pressure. The residue solid sample was purified by column chromatography on silica gel (petroleum/EtOAc 5:1 to 2:1, with 1% Et3N), yielding the desired product 3.
(Z)-3-Benzylidene-2-(methyl[pyridin-2-yl]amino)isoindolin-1-one (3aa)
The general procedure was followed using hydrazide 1a (68.2 mg, 0.30 mmol) and alkyne 2a (91.9 mg, 0.90 mmol). Purification by column chromatography on silica gel (petroleum/EtOAc 20:1, with 1% Et3N) yielded 3aa (87.4 mg, 89%, Z/E = 13:1) as a light yellow solid. mp 67–68 °C; 1H NMR (CDCl3, 600 MHz) δ 8.13 (ddd, J = 5.0; 1.9; 0.9 Hz, 1H), 7.90 (dd, J = 7.6; 1.0 Hz, 1H), 7.85–7.82 (m, 1H), 7.70 (d, J = 1.2 Hz, 1H), 7.56 (dd, J = 7.6; 0.9 Hz, 1H), 7.44 (ddd, J = 8.8; 7.1; 1.9 Hz, 1H), 7.17–7.05 (m, 5H), 6.85 (d, J = 0.9 Hz, 1H), 6.67 (ddd, J = 7.2; 5.0; 0.9 Hz, 1H), 6.44–6.41 (m, 1H), 3.01 (s, 3H); 13C{1H} NMR (CDCl3, 150 MHz) δ 165.7 (Cq), 157.6 (Cq), 147.7 (CH), 137.4 (CH), 136.2 (Cq), 133.2 (Cq), 132.8 (CH), 132.1 (Cq), 129.3 (CH), 128.7 (CH), 127.3 (CH), 127.3 (CH), 126.5 (Cq), 123.8 (CH), 119.8 (CH), 114.3 (CH), 107.8 (CH), 106.4 (CH), 36.7 (CH3); HRESIMS (m∕z): [M + H]+ calcd for C21H18N3O, 328.1444; found, 328.1439.
Supporting Information
| Supporting Information File 1: Characterization data for 3 and copies of 1H, 13C, and 19F NMR spectra. | ||
| Format: PDF | Size: 6.1 MB | Download |
References
-
Gandeepan, P.; Finger, L. H.; Meyer, T. H.; Ackermann, L. Chem. Soc. Rev. 2020, 49, 4254–4272. doi:10.1039/d0cs00149j
Return to citation in text: [1] -
Ellman, J. A.; Ackermann, L.; Shi, B.-F. J. Org. Chem. 2019, 84, 12701–12704. doi:10.1021/acs.joc.9b02663
Return to citation in text: [1] -
Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A. K.; Lei, A. Chem. Rev. 2017, 117, 9016–9085. doi:10.1021/acs.chemrev.6b00620
Return to citation in text: [1] -
Wei, Y.; Hu, P.; Zhang, M.; Su, W. Chem. Rev. 2017, 117, 8864–8907. doi:10.1021/acs.chemrev.6b00516
Return to citation in text: [1] -
Rao, W.-H.; Shi, B.-F. Org. Chem. Front. 2016, 3, 1028–1047. doi:10.1039/c6qo00156d
Return to citation in text: [1] -
Zheng, Q.-Z.; Jiao, N. Chem. Soc. Rev. 2016, 45, 4590–4627. doi:10.1039/c6cs00107f
Return to citation in text: [1] -
Wencel-Delord, J.; Glorius, F. Nat. Chem. 2013, 5, 369–375. doi:10.1038/nchem.1607
Return to citation in text: [1] -
Rouquet, G.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11726–11743. doi:10.1002/anie.201301451
Return to citation in text: [1] -
Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788–802. doi:10.1021/ar200185g
Return to citation in text: [1] -
Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960–9009. doi:10.1002/anie.201201666
Return to citation in text: [1] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n
Return to citation in text: [1] -
Chen, X.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 6790–6791. doi:10.1021/ja061715q
Return to citation in text: [1] -
Uemura, T.; Imoto, S.; Chatani, N. Chem. Lett. 2006, 35, 842–843. doi:10.1246/cl.2006.842
Return to citation in text: [1] -
Kim, H.; Heo, J.; Kim, J.; Baik, M.-H.; Chang, S. J. Am. Chem. Soc. 2018, 140, 14350–14356. doi:10.1021/jacs.8b08826
Return to citation in text: [1] -
Takamatsu, K.; Hirano, K.; Miura, M. Angew. Chem., Int. Ed. 2017, 56, 5353–5357. doi:10.1002/anie.201701918
Return to citation in text: [1] -
Wang, S.; Guo, R.; Wang, G.; Chen, S.-Y.; Yu, X.-Q. Chem. Commun. 2014, 50, 12718–12721. doi:10.1039/c4cc06246a
Return to citation in text: [1] -
Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2013, 52, 4457–4461. doi:10.1002/anie.201300587
Return to citation in text: [1] -
Kim, J.; Kim, H.; Chang, S. Org. Lett. 2012, 14, 3924–3927. doi:10.1021/ol301674m
Return to citation in text: [1] -
Zhang, L.; Liu, Z.; Li, H.; Fang, G.; Barry, B.-D.; Belay, T. A.; Bi, X.; Liu, Q. Org. Lett. 2011, 13, 6536–6539. doi:10.1021/ol2028288
Return to citation in text: [1] -
Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Org. Biomol. Chem. 2007, 5, 1466–1471. doi:10.1039/b701661a
Return to citation in text: [1] -
Rys, V.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 2003, 59, 6615–6619. doi:10.1016/s0040-4020(03)01067-6
Return to citation in text: [1] -
Chia, Y.-C.; Chang, F.-R.; Teng, C.-M.; Wu, Y.-C. J. Nat. Prod. 2000, 63, 1160–1163. doi:10.1021/np000063v
Return to citation in text: [1] -
Blaskó, G.; Gula, D. J.; Shamma, M. J. Nat. Prod. 1982, 45, 105–122. doi:10.1021/np50020a001
Return to citation in text: [1] -
Botero Cid, H. M.; Tränkle, C.; Baumann, K.; Pick, R.; Mies-Klomfass, E.; Kostenis, E.; Mohr, K.; Holzgrabe, U. J. Med. Chem. 2000, 43, 2155–2164. doi:10.1021/jm991136e
Return to citation in text: [1] -
Dong, J.; Wang, F.; You, J. Org. Lett. 2014, 16, 2884–2887. doi:10.1021/ol501023n
Return to citation in text: [1] -
Zhang, Y.; Wang, Q.; Yu, H.; Huang, Y. Org. Biomol. Chem. 2014, 12, 8844–8850. doi:10.1039/c4ob01312c
Return to citation in text: [1] -
Zhu, W.; Wang, B.; Zhou, S.; Liu, H. Beilstein J. Org. Chem. 2015, 11, 1624–1631. doi:10.3762/bjoc.11.177
Return to citation in text: [1] -
Lee, W.-C. C.; Wang, W.; Li, J. J. J. Org. Chem. 2018, 83, 2382–2388. doi:10.1021/acs.joc.7b02893
Return to citation in text: [1] -
Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154–13155. doi:10.1021/ja054549f
Return to citation in text: [1] -
Lee, W.-C. C.; Shen, Y.; Gutierrez, D. A.; Li, J. J. Org. Lett. 2016, 18, 2660–2663. doi:10.1021/acs.orglett.6b01105
Return to citation in text: [1] -
Zhang, L.-B.; Hao, X.-Q.; Liu, Z.-J.; Zheng, X.-X.; Zhang, S.-K.; Niu, J.-L.; Song, M.-P. Angew. Chem., Int. Ed. 2015, 54, 10012–10015. doi:10.1002/anie.201504962
Return to citation in text: [1] -
Zheng, X.-X.; Du, C.; Zhao, X.-M.; Zhu, X.; Suo, J.-F.; Hao, X.-Q.; Niu, J.-L.; Song, M.-P. J. Org. Chem. 2016, 81, 4002–4011. doi:10.1021/acs.joc.6b00129
Return to citation in text: [1] -
Hao, X.-Q.; Du, C.; Zhu, X.; Li, P.-X.; Zhang, J.-H.; Niu, J.-L.; Song, M.-P. Org. Lett. 2016, 18, 3610–3613. doi:10.1021/acs.orglett.6b01632
Return to citation in text: [1] -
Tian, C.; Dhawa, U.; Scheremetjew, A.; Ackermann, L. ACS Catal. 2019, 9, 7690–7696. doi:10.1021/acscatal.9b02348
Return to citation in text: [1] -
Fitzgerald, L. S.; O'Duill, M. L. Chem. – Eur. J. 2021, 27, 8411–8436. doi:10.1002/chem.202100093
Return to citation in text: [1] -
Zhai, S.; Qiu, S.; Chen, X.; Wu, J.; Zhao, H.; Tao, C.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. Chem. Commun. 2018, 54, 98–101. doi:10.1039/c7cc08533h
Return to citation in text: [1] [2] -
Zhao, H.; Wang, T.; Qing, Z.; Zhai, H. Chem. Commun. 2020, 56, 5524–5527. doi:10.1039/d0cc01582b
Return to citation in text: [1] -
Zhao, H.; Shao, X.; Wang, T.; Zhai, S.; Qiu, S.; Tao, C.; Wang, H.; Zhai, H. Chem. Commun. 2018, 54, 4927–4930. doi:10.1039/c8cc01774c
Return to citation in text: [1] -
Zhai, S.; Qiu, S.; Chen, X.; Tao, C.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. ACS Catal. 2018, 8, 6645–6649. doi:10.1021/acscatal.8b01720
Return to citation in text: [1] -
Qiu, S.; Zhai, S.; Wang, H.; Tao, C.; Zhao, H.; Zhai, H. Adv. Synth. Catal. 2018, 360, 3271–3276. doi:10.1002/adsc.201800388
Return to citation in text: [1] -
Mei, R.; Fang, X.; He, L.; Sun, J.; Zou, L.; Ma, W.; Ackermann, L. Chem. Commun. 2020, 56, 1393–1396. doi:10.1039/c9cc09076b
Return to citation in text: [1] [2] -
Sau, S. C.; Mei, R.; Struwe, J.; Ackermann, L. ChemSusChem 2019, 12, 3023–3027. doi:10.1002/cssc.201900378
Return to citation in text: [1] [2] -
Mei, R.; Ma, W.; Zhang, Y.; Guo, X.; Ackermann, L. Org. Lett. 2019, 21, 6534–6538. doi:10.1021/acs.orglett.9b02463
Return to citation in text: [1] [2] -
Mei, R.; Sauermann, N.; Oliveira, J. C. A.; Ackermann, L. J. Am. Chem. Soc. 2018, 140, 7913–7921. doi:10.1021/jacs.8b03521
Return to citation in text: [1] [2] [3] -
Mei, R.; Dhawa, U.; Samanta, R. C.; Ma, W.; Wencel‐Delord, J.; Ackermann, L. ChemSusChem 2020, 13, 3306–3356. doi:10.1002/cssc.202000024
Return to citation in text: [1] -
Sauermann, N.; Mei, R.; Ackermann, L. Angew. Chem., Int. Ed. 2018, 57, 5090–5094. doi:10.1002/anie.201802206
Return to citation in text: [1] -
Mei, R.; Wang, H.; Warratz, S.; Macgregor, S. A.; Ackermann, L. Chem. – Eur. J. 2016, 22, 6759–6763. doi:10.1002/chem.201601101
Return to citation in text: [1] -
Mei, R.; Loup, J.; Ackermann, L. ACS Catal. 2016, 6, 793–797. doi:10.1021/acscatal.5b02661
Return to citation in text: [1] -
Mei, R.; Ackermann, L. Adv. Synth. Catal. 2016, 358, 2443–2448. doi:10.1002/adsc.201600384
Return to citation in text: [1] -
Ackermann, L. Chem. Rev. 2011, 111, 1315–1345. doi:10.1021/cr100412j
Return to citation in text: [1]
| 41. | Mei, R.; Fang, X.; He, L.; Sun, J.; Zou, L.; Ma, W.; Ackermann, L. Chem. Commun. 2020, 56, 1393–1396. doi:10.1039/c9cc09076b |
| 42. | Sau, S. C.; Mei, R.; Struwe, J.; Ackermann, L. ChemSusChem 2019, 12, 3023–3027. doi:10.1002/cssc.201900378 |
| 43. | Mei, R.; Ma, W.; Zhang, Y.; Guo, X.; Ackermann, L. Org. Lett. 2019, 21, 6534–6538. doi:10.1021/acs.orglett.9b02463 |
| 44. | Mei, R.; Sauermann, N.; Oliveira, J. C. A.; Ackermann, L. J. Am. Chem. Soc. 2018, 140, 7913–7921. doi:10.1021/jacs.8b03521 |
| 36. | Zhai, S.; Qiu, S.; Chen, X.; Wu, J.; Zhao, H.; Tao, C.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. Chem. Commun. 2018, 54, 98–101. doi:10.1039/c7cc08533h |
| 37. | Zhao, H.; Wang, T.; Qing, Z.; Zhai, H. Chem. Commun. 2020, 56, 5524–5527. doi:10.1039/d0cc01582b |
| 38. | Zhao, H.; Shao, X.; Wang, T.; Zhai, S.; Qiu, S.; Tao, C.; Wang, H.; Zhai, H. Chem. Commun. 2018, 54, 4927–4930. doi:10.1039/c8cc01774c |
| 39. | Zhai, S.; Qiu, S.; Chen, X.; Tao, C.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. ACS Catal. 2018, 8, 6645–6649. doi:10.1021/acscatal.8b01720 |
| 40. | Qiu, S.; Zhai, S.; Wang, H.; Tao, C.; Zhao, H.; Zhai, H. Adv. Synth. Catal. 2018, 360, 3271–3276. doi:10.1002/adsc.201800388 |
| 1. | Gandeepan, P.; Finger, L. H.; Meyer, T. H.; Ackermann, L. Chem. Soc. Rev. 2020, 49, 4254–4272. doi:10.1039/d0cs00149j |
| 2. | Ellman, J. A.; Ackermann, L.; Shi, B.-F. J. Org. Chem. 2019, 84, 12701–12704. doi:10.1021/acs.joc.9b02663 |
| 3. | Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A. K.; Lei, A. Chem. Rev. 2017, 117, 9016–9085. doi:10.1021/acs.chemrev.6b00620 |
| 4. | Wei, Y.; Hu, P.; Zhang, M.; Su, W. Chem. Rev. 2017, 117, 8864–8907. doi:10.1021/acs.chemrev.6b00516 |
| 5. | Rao, W.-H.; Shi, B.-F. Org. Chem. Front. 2016, 3, 1028–1047. doi:10.1039/c6qo00156d |
| 6. | Zheng, Q.-Z.; Jiao, N. Chem. Soc. Rev. 2016, 45, 4590–4627. doi:10.1039/c6cs00107f |
| 7. | Wencel-Delord, J.; Glorius, F. Nat. Chem. 2013, 5, 369–375. doi:10.1038/nchem.1607 |
| 8. | Rouquet, G.; Chatani, N. Angew. Chem., Int. Ed. 2013, 52, 11726–11743. doi:10.1002/anie.201301451 |
| 9. | Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788–802. doi:10.1021/ar200185g |
| 10. | Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960–9009. doi:10.1002/anie.201201666 |
| 11. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n |
| 20. | Lamblin, M.; Couture, A.; Deniau, E.; Grandclaudon, P. Org. Biomol. Chem. 2007, 5, 1466–1471. doi:10.1039/b701661a |
| 21. | Rys, V.; Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 2003, 59, 6615–6619. doi:10.1016/s0040-4020(03)01067-6 |
| 22. | Chia, Y.-C.; Chang, F.-R.; Teng, C.-M.; Wu, Y.-C. J. Nat. Prod. 2000, 63, 1160–1163. doi:10.1021/np000063v |
| 23. | Blaskó, G.; Gula, D. J.; Shamma, M. J. Nat. Prod. 1982, 45, 105–122. doi:10.1021/np50020a001 |
| 34. | Tian, C.; Dhawa, U.; Scheremetjew, A.; Ackermann, L. ACS Catal. 2019, 9, 7690–7696. doi:10.1021/acscatal.9b02348 |
| 14. | Kim, H.; Heo, J.; Kim, J.; Baik, M.-H.; Chang, S. J. Am. Chem. Soc. 2018, 140, 14350–14356. doi:10.1021/jacs.8b08826 |
| 15. | Takamatsu, K.; Hirano, K.; Miura, M. Angew. Chem., Int. Ed. 2017, 56, 5353–5357. doi:10.1002/anie.201701918 |
| 16. | Wang, S.; Guo, R.; Wang, G.; Chen, S.-Y.; Yu, X.-Q. Chem. Commun. 2014, 50, 12718–12721. doi:10.1039/c4cc06246a |
| 17. | Nishino, M.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2013, 52, 4457–4461. doi:10.1002/anie.201300587 |
| 18. | Kim, J.; Kim, H.; Chang, S. Org. Lett. 2012, 14, 3924–3927. doi:10.1021/ol301674m |
| 19. | Zhang, L.; Liu, Z.; Li, H.; Fang, G.; Barry, B.-D.; Belay, T. A.; Bi, X.; Liu, Q. Org. Lett. 2011, 13, 6536–6539. doi:10.1021/ol2028288 |
| 35. | Fitzgerald, L. S.; O'Duill, M. L. Chem. – Eur. J. 2021, 27, 8411–8436. doi:10.1002/chem.202100093 |
| 13. | Uemura, T.; Imoto, S.; Chatani, N. Chem. Lett. 2006, 35, 842–843. doi:10.1246/cl.2006.842 |
| 31. | Zhang, L.-B.; Hao, X.-Q.; Liu, Z.-J.; Zheng, X.-X.; Zhang, S.-K.; Niu, J.-L.; Song, M.-P. Angew. Chem., Int. Ed. 2015, 54, 10012–10015. doi:10.1002/anie.201504962 |
| 12. | Chen, X.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 6790–6791. doi:10.1021/ja061715q |
| 32. | Zheng, X.-X.; Du, C.; Zhao, X.-M.; Zhu, X.; Suo, J.-F.; Hao, X.-Q.; Niu, J.-L.; Song, M.-P. J. Org. Chem. 2016, 81, 4002–4011. doi:10.1021/acs.joc.6b00129 |
| 33. | Hao, X.-Q.; Du, C.; Zhu, X.; Li, P.-X.; Zhang, J.-H.; Niu, J.-L.; Song, M.-P. Org. Lett. 2016, 18, 3610–3613. doi:10.1021/acs.orglett.6b01632 |
| 27. | Zhu, W.; Wang, B.; Zhou, S.; Liu, H. Beilstein J. Org. Chem. 2015, 11, 1624–1631. doi:10.3762/bjoc.11.177 |
| 29. | Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154–13155. doi:10.1021/ja054549f |
| 36. | Zhai, S.; Qiu, S.; Chen, X.; Wu, J.; Zhao, H.; Tao, C.; Li, Y.; Cheng, B.; Wang, H.; Zhai, H. Chem. Commun. 2018, 54, 98–101. doi:10.1039/c7cc08533h |
| 44. | Mei, R.; Sauermann, N.; Oliveira, J. C. A.; Ackermann, L. J. Am. Chem. Soc. 2018, 140, 7913–7921. doi:10.1021/jacs.8b03521 |
| 26. | Zhang, Y.; Wang, Q.; Yu, H.; Huang, Y. Org. Biomol. Chem. 2014, 12, 8844–8850. doi:10.1039/c4ob01312c |
| 30. | Lee, W.-C. C.; Shen, Y.; Gutierrez, D. A.; Li, J. J. Org. Lett. 2016, 18, 2660–2663. doi:10.1021/acs.orglett.6b01105 |
| 25. | Dong, J.; Wang, F.; You, J. Org. Lett. 2014, 16, 2884–2887. doi:10.1021/ol501023n |
| 41. | Mei, R.; Fang, X.; He, L.; Sun, J.; Zou, L.; Ma, W.; Ackermann, L. Chem. Commun. 2020, 56, 1393–1396. doi:10.1039/c9cc09076b |
| 42. | Sau, S. C.; Mei, R.; Struwe, J.; Ackermann, L. ChemSusChem 2019, 12, 3023–3027. doi:10.1002/cssc.201900378 |
| 43. | Mei, R.; Ma, W.; Zhang, Y.; Guo, X.; Ackermann, L. Org. Lett. 2019, 21, 6534–6538. doi:10.1021/acs.orglett.9b02463 |
| 44. | Mei, R.; Sauermann, N.; Oliveira, J. C. A.; Ackermann, L. J. Am. Chem. Soc. 2018, 140, 7913–7921. doi:10.1021/jacs.8b03521 |
| 45. | Mei, R.; Dhawa, U.; Samanta, R. C.; Ma, W.; Wencel‐Delord, J.; Ackermann, L. ChemSusChem 2020, 13, 3306–3356. doi:10.1002/cssc.202000024 |
| 46. | Sauermann, N.; Mei, R.; Ackermann, L. Angew. Chem., Int. Ed. 2018, 57, 5090–5094. doi:10.1002/anie.201802206 |
| 47. | Mei, R.; Wang, H.; Warratz, S.; Macgregor, S. A.; Ackermann, L. Chem. – Eur. J. 2016, 22, 6759–6763. doi:10.1002/chem.201601101 |
| 48. | Mei, R.; Loup, J.; Ackermann, L. ACS Catal. 2016, 6, 793–797. doi:10.1021/acscatal.5b02661 |
| 49. | Mei, R.; Ackermann, L. Adv. Synth. Catal. 2016, 358, 2443–2448. doi:10.1002/adsc.201600384 |
| 24. | Botero Cid, H. M.; Tränkle, C.; Baumann, K.; Pick, R.; Mies-Klomfass, E.; Kostenis, E.; Mohr, K.; Holzgrabe, U. J. Med. Chem. 2000, 43, 2155–2164. doi:10.1021/jm991136e |
| 28. | Lee, W.-C. C.; Wang, W.; Li, J. J. J. Org. Chem. 2018, 83, 2382–2388. doi:10.1021/acs.joc.7b02893 |
© 2021 Xiong et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)