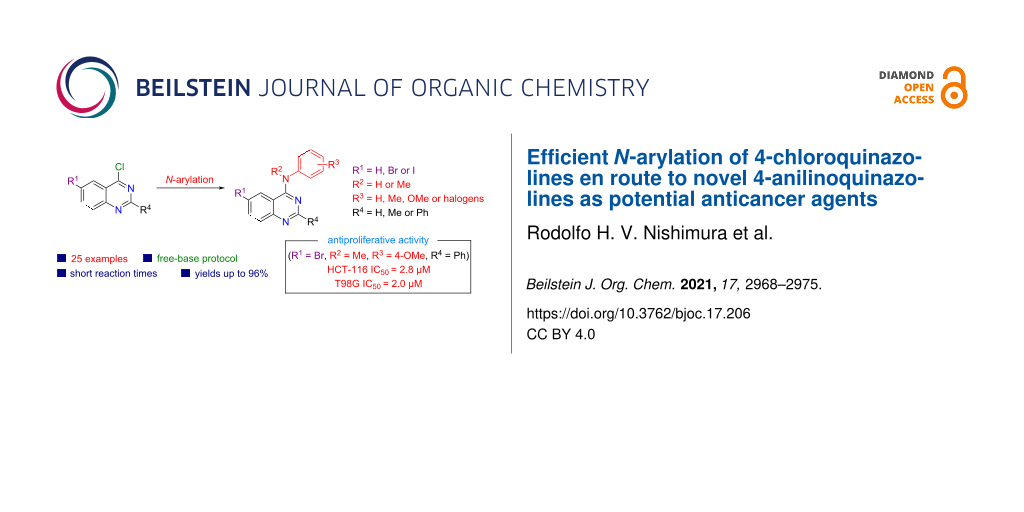
Abstract
Microwave-mediated N-arylation of 4-chloroquinazolines in THF/H2O rapidly and efficiently afforded a library of novel 6-halo-2-phenyl-substituted 4-anilinoquinazolines. The methodology was compatible with numerous ortho-, meta-, and para-substituted N-methylanilines as well as substituted anilines and furnished the corresponding 4-anilinoquinazolines in good yields. Preliminary screening of the synthesized compounds against tumor cells (HCT-116 and T98G) showed promising antiproliferative properties.

Graphical Abstract
Introduction
N-Heterocyclic compounds are commonly present in pharmaceuticals, bioactive natural products, agrochemicals, and synthetic drugs [1,2]. Quinazoline, a benzo-fused N-heterocyclic framework (benzo-1,3-diazine) with relevant biological activities, is recognized as a privileged scaffold in medicinal chemistry [3,4]. Among important quinazoline derivatives, 4-anilinoquinazolines have been widely investigated as antitumor agents because they can inhibit some receptor tyrosine kinases (RTKs) expressed by malignant tumors, including platelet-derived growth factor receptor beta (PDGFR-β), vessel epidermal growth factor receptor (VEGFR-2), and epidermal growth factor receptor (EGFR) [5,6]. In addition, these compounds may act as vascular disrupting agents and tubulin polymerization inhibitors, contributing to apoptosis [7]. Figure 1 highlights the structures of three EGFR inhibitors approved by the United States Food and Drug Administration (FDA) and one known tubulin inhibitor: erlotinib (1), gefitinib (2), lapatinib (3), and MPC-6827 – verubulin (4) [5,7].
![[1860-5397-17-206-1]](/bjoc/content/figures/1860-5397-17-206-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Some antitumor agents containing the 4-anilinoquinazoline moiety.
Figure 1: Some antitumor agents containing the 4-anilinoquinazoline moiety.
Given that 4-anilinoquinazolines are potential antitumor agents, several synthetic methodologies have been developed to prepare these compounds. Amination using primary [6,8-13] or secondary [14-16] amines and 4-chloroquinazolines is among the most employed procedures.
Electron-rich amines (e.g., primary aliphatic amines or hydroxy-substituted anilines) [6,9] readily react with 4-chloroquinazolines to give 4-aminoquinazolines in moderate to good yields under milder reaction conditions (Scheme 1a). On the other hand, long reaction times and low yields may be observed when electron-poor amines are applied in these reactions [17,18]. These limitations can be overcome by using microwave irradiation [12,13,18-20], which promotes fast and efficient anilination reactions when a wide range of anilines bearing both electron-donating and electron-withdrawing groups are employed as nucleophiles (Scheme 1b) [12,13]. Moreover, 4-anilinoquinazolines can be prepared from N-methylanilines under basic [14] or acidic [15,16] (Scheme 1c) conditions.
![[1860-5397-17-206-i1]](/bjoc/content/inline/1860-5397-17-206-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Examples of N-arylation reactions using 4-chloroquinazolines as substrates.
Scheme 1: Examples of N-arylation reactions using 4-chloroquinazolines as substrates.
Due to our interest in the functionalization of aromatic and heterocyclic compounds of medicinal relevance [21-26], we recently reported the preparation of an iodo-substituted analog of the anticancer agent verubulin. Thus, after regioselective 4-chloroquinazoline metalation by an in situ trapping metalation strategy, reaction quenching with iodine allowed us to isolate 4-chloro-8-iodoquinazoline in 83% yield. Surprisingly, further reaction of 4-chloro-8-iodoquinazoline with 4-methoxy-N-methylaniline was not efficient under various reaction conditions and afforded the desired product in low yields even in basic medium (AcONa). To address this issue, we investigated a microwave-mediated base-free amination strategy involving a mixture of THF and water (1:1) as solvent system, which furnished the desired verubulin analog in 87% yield (Scheme 2) [27].
![[1860-5397-17-206-i2]](/bjoc/content/inline/1860-5397-17-206-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Inspired by this preliminary result, we decided to investigate this methodology to synthesize other bioactive derivatives, such as 6-halo-2-phenyl-substituted 4-anilinoquinazolines – the presence of an aryl group at C2 and bromo at C6 as substituents in quinazoline ring has been related to increased antiproliferative action of this class of compounds [28]. Therefore, we wish to report the preparation of a library of novel quinazoline-based antitumor candidates through reaction of 4-chloro-6-halo-2-phenylquinazolines with substituted anilines or N-methylanilines bearing electron-withdrawing or -donating groups at the ortho-, meta-, or para-positions.
Results and Discussion
We started by synthesizing 6-bromo-4-chloro-2-phenylquinazoline (8a) and 4-chloro-6-iodo-2-phenylquinazoline (8b) as substrates for the N-arylation study. While anthranilamide (5) bromination with N-bromosuccinimide in acetonitrile at room temperature [29] furnished 2-amino-5-bromobenzamide (6a) in 78% yield, iodination of 5 with iodine in the presence of hydrogen peroxide in water [30] at 50 °C provided 2-amino-5-iodobenzamide (6b) in 89% yield. After that, the cyclocondensation [29] of the halogenated anthranilamides 6a,b with benzaldehyde followed by dehydrogenation promoted by iodine gave the corresponding quinazolin-4(3H)-ones 7a,b, which we used in the next step without purification. Finally, chlorination [31] of quinazolin-4(3H)-ones 7a,b by employing a combination of Cl3CCN/PPh3 afforded 6-bromo-4-chloro-2-phenylquinazoline (8a) and 4-chloro-6-iodo-2-phenylquinazoline (8b) in 78 and 46% yields (over two steps), respectively (Scheme 3).
![[1860-5397-17-206-i3]](/bjoc/content/inline/1860-5397-17-206-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 4-chloro-6-halo-2-phenylquinazolines 8a and 8b. Conditions: a) NBS, CH3CN, 30 min, 25 °C; b) H2O, H2O2, I2, 24 h, 50 °C.
Scheme 3: Synthesis of 4-chloro-6-halo-2-phenylquinazolines 8a and 8b. Conditions: a) NBS, CH3CN, 30 min, 25 ...
With the 4-chloroquinazolines 8a and 8b in hands, we investigated how they reacted with some N-methylanilines (9a–k) by using our established microwave-mediated protocol (Table 1). The reaction of 8a or 8b with N-methylanilines bearing a methoxy group at the para- (9a) or meta- (9b) position furnished the desired 4-anilinoquinazolines 10a–d within 10 min; yields ranged from 63 to 90% (Table 1, entries 1–4). As expected, substituents at the ortho-position of the anilines affected the N-arylation reactions due to the steric hindrance exerted by these groups. In fact, reactions with 2-methoxy-substituted N-methylaniline 9c were slower, but we obtained high conversions within 20 min under microwave heating, which allowed us to isolate the 4-anilinoquinazoline derivatives 10e and 10f in 87 and 84% yields, respectively (Table 1, entries 5 and 6). In contrast, no N-arylated products emerged when we used the ortho-methyl-substituted N-methylaniline 9d as nucleophile, even when we conducted the reactions at 120 °C for 1 h (Table 1, entries 7 and 8). Subsequently, we employed the meta-methyl-substituted derivative 9e as nucleophile under the established conditions, to obtain the desired anilinated derivatives 10i and 10j in 80 and 84% yields, respectively (Table 1, entries 9 and 10).
Table 1: 4-Anilinoquinazoline derivatives 10 obtained from N-arylation reactions involving 4-chloroquinazolines 8 and N-methylanilines 9.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-206-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | X | R | Product | Yield (%)a |
| 1 | I | 4-OMe (9a) | 10a | 86b |
| 2 | Br | 4-OMe (9a) | 10b | 63b |
| 3 | I | 3-OMe (9b) | 10c | 90b |
| 4 | Br | 3-OMe (9b) | 10d | 84b |
| 5 | I | 2-OMe (9c) | 10e | 87c |
| 6 | Br | 2-OMe (9c) | 10f | 84c |
| 7 | I | 2-Me (9d) | 10g | no reactiond,e |
| 8 | Br | 2-Me (9d) | 10h | no reactiond,e |
| 9 | I | 3-Me (9e) | 10i | 80b |
| 10 | Br | 3-Me (9e) | 10j | 84b |
| 11 | I | 3-Br (9f) | 10k | 72b |
| 12 | Br | 3-Br (9f) | 10l | 73b |
| 13 | I | 4-F (9g) | 10m | 84f |
| 14 | Br | 4-F (9g) | 10n | 75f |
| 15 | I | 3-F (9h) | 10o | 72c |
| 16 | Br | 3-F (9h) | 10p | 70b |
| 17 | I | 2-F (9i) | 10q | no reactiond,e |
| 18 | Br | 2-F (9i) | 10r | no reactiond,e |
| 19 | I | 4-CN (9j) | 10s | no reactiond,e |
| 20 | Br | 4-CN (9j) | 10t | no reactiond,e |
| 21 | Br | 4-NO2 (9k) | 10u | no reactiong |
aIsolated yield; breaction time = 10 min; creaction time = 20 min; dreaction time = 1 h; etemperature = 120 °C; freaction time = 40 min; greaction time = 2 h.
We also evaluated how the halogen substituents present in the anilines affected the N-arylation reactions. When we used 3-bromo-N-methylaniline (9f), we isolated derivatives 10k and 10l in 72 and 73% yields, respectively (Table 1, entries 11 and 12). Similarly, the reactions using 4-fluoro- or 3-fluoro-N-methylaniline (9g and 9h, respectively) allowed us to isolate the expected derivatives 10m–p in yields ranging from 70 to 84% (Table 1, entries 13–16). The electron-withdrawing effect of the fluoro substituent appeared to affect the reactivity of amine 9g in those reactions drastically, so a longer reaction time (40 min) was necessary to obtain the desired products in good yield. Moreover, we did not observe any products when we used the 2-fluoro-substituted aniline 9i even at higher temperature and for longer reaction time (Table 1, entries 17 and 18). Furthermore, no aminated products were observed when 8a and 8b were reacted with 4-cyano- or 4-nitro-N-methylaniline (Table 1, entries 19–21).
Given our interest in preparing 4-anilinoquinazolines 10g, 10h, 10q, and 10r, we decided to investigate a two-step strategy starting by N-arylation using o-methyl- and o-fluoro-substituted primary anilines, followed by N-methylation [15]. The microwave-mediated reaction of 4-chloro-6-halo-2-phenylquinazolines 8a or 8b with o-toluidine (14a) in THF/H2O could be accomplished within 2 h and afforded the corresponding quinazoline derivatives 15a and 15b in 74% and 78% isolated yields, respectively. In addition, when we used 2-fluoroaniline (14b), we obtained derivatives 15c and 15d in 60 and 56% yields, respectively, after heating for 40 min (Scheme 4). Then, the derivatives 15a–d were efficiently N-methylated with iodomethane in the presence of sodium hydride, to afford the corresponding methylated 4-anilinoquinazolines 10g, 10h, 10q, and 10r in yields ranging from 76 to 89% (Scheme 4). Moreover, to demonstrate the applicability of meta- and para-substituted primary anilines employing our N-arylation procedure, we evaluated reactions using 3-chloro-, 4-fluoro- and 4-methoxyaniline (14c–e, respectively) with 6-bromo-4-chloro-2-phenylquinazoline (8a), which provided the respective products 15e–g in yields ranging from 92 to 96% (Scheme 4).
![[1860-5397-17-206-i4]](/bjoc/content/inline/1860-5397-17-206-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: N-Arylation reactions using ortho-, meta-, and para-substituted primary anilines of type 14 followed by methylation of ortho-derivatives 15a–d, to obtain the desired 4-anilinoquinazolines of type 10.
Scheme 4: N-Arylation reactions using ortho-, meta-, and para-substituted primary anilines of type 14 followe...
Finally, to illustrate the importance and scope of the methodology, we used the commercially available 4-chloroquinazoline (16) and 4-chloro-2-methylquinazoline (17), which was prepared by chlorination of the corresponding quinazolinone (see Supporting Information File 1), in the N-arylation reaction. N-Arylation of 16 with N-methylaniline (9l) afforded N-methyl-N-phenylquinazolin-4-amine (18) in 81% yield (Scheme 5). Subsequently, the reaction of 17 with N-methylanilines 9a, 9g, and 9l provided the desired 4-anilinoquinazolines 4, 19, and 20 in yields ranging from 90 to 95% (Scheme 5), among which we can highlight the anticancer agent verubulin (4).
![[1860-5397-17-206-i5]](/bjoc/content/inline/1860-5397-17-206-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: N-Arylation reactions using 4-chloroquinazoline (16) and 4-chloro-2-methylquinazoline (17) to achieve the desired 4-anilinoquinazolines (4,18–20).
Scheme 5: N-Arylation reactions using 4-chloroquinazoline (16) and 4-chloro-2-methylquinazoline (17) to achie...
The methodology reported herein allowed us to prepare numerous novel 4-anilinoquinazolines derivatives in good yield within short reaction times. To the best of our knowledge, only few examples of the use of ortho-substituted anilines for the anilination of 4-chloroquinazolines have been reported [6,18]. Thus, the established methodology proved an important alternative to synthesize these compounds from ortho-substituted anilines bearing both electron-withdrawing and -donating groups. Furthermore, compared to a previously reported protocol that uses AcONa as base and a THF/H2O 3:1 mixture as solvent [11,32], the protocol we developed here is base-free and requires a reduced amount of organic solvent, thereby being a more environmentally friendly methodology.
To evaluate the antiproliferative properties of the 4-anilinoquinazolines we obtained in this study, we screened the synthesized compounds against a set of tumor cell lines including HCT-116 (human colorectal carcinoma), MCF-7 (human breast adenocarcinoma), and T98G (human glioblastoma). Initially, we evaluated all compounds at 50 µM, and we considered active the compounds that inhibited cell proliferation by over 75% for each cell line. Compounds 10b, 10c, 10g, 10k, 10l, 15a, 15b, and 15d were active against T98G cells, and compounds 10b, 15a, and 15b were also active against HCT-116 cells. However, none of the tested compounds was able to inhibit MCF-7 cell proliferation (Figure S1 in Supporting Information File 1).
Next, we determined the IC50 of the active compounds against both HCT-116 and T98G cells; we employed doxorubicin as positive control. We also investigated the potency of erlotinib hydrochloride, gefitinib, and verubulin against T98G cells, which were the most sensitive to the novel 4-anilinoquinazolines (Figure S2 in Supporting Information File 1). Compared to the reference drug, doxorubicin, derivative 10b showed promising IC50 values (2.8 and 2.0 µM against HCT-116 and T98G cells, respectively). Additionally, the C2 phenyl ring in compound 10b may be subjected to future replacements for optimization as less bulky groups at this position seem to be of great interest for gain in activity (e.g., 2-methyl group in verubulin). The EGFR inhibitor drugs bearing the 4-anilinoquinazoline moiety did not show potent cytotoxic activity against T98G cells (21.3 µM for erlotinib, and 37.8 µM for gefitinib). However, the tubulin polymerization inhibitor (verubulin), which contains 4-anilinoquinazoline in its chemical structure, was strongly cytotoxic to T98G cells (0.2 nM). In fact, the promising activity of verubulin in glioma is known, which has prompted phase 2 clinical trials in patients with recurrent glioblastoma, but the studies have been interrupted due to the observed adverse events [33]. Nonetheless, further studies aiming at optimizing the activity of compound 10b and better understanding its mechanism of action and selectivity are underway in our research group.
Table 2: Evaluation of HCT-116 and T98G cell growth inhibition upon exposure to 4-anilinoquinazolines and compounds with the same scaffolds (erlotinib, gefitinib, and verubulin), and to positive control doxorubicin (MTT assay after treatment for 72 h).
| HCT-116 cell line | ||
| Compound | IC50 (µM)a | CI 95% (µM)b |
| 10b | 2.8 | 2.0–4.0 |
| 15a | 26.2 | 20.7–32.9 |
| 15b | 33.4 | 26.7–41.5 |
| doxorubicin | 0.1 | 0.1–0.1 |
| T98G cell line | ||
| Compound | IC50 (µM)a | CI 95% (µM)b |
| 10b | 2.0 | 1.1–3.8 |
| 10c | 7.7 | 6.2–ndc |
| 10g | 33.6 | 26.1–ndc |
| 10k | 38.4 | 30.9–ndc |
| 10l | >50 | ndc |
| 15a | 28.6 | 21.6–ndc |
| 15b | >50 | 42.5–67.9 |
| 15d | 3.1 | 2.2–4.2 |
| erlotinib | 21.3 | 16.1–28.1 |
| gefitinib | 37.8 | ndc |
| verubulin | 0.2 (nM) | 0.1–0.3 (nM) |
| doxorubicin | 0.6 | 0.4–0.9 |
aIC50: half-maximal inhibitory concentration; bCI 95%: confidence interval; cnd: not determined.
Conclusion
We have synthesized a library of novel 6-halo-2-phenyl-substituted 4-anilinoquinazolines through N-arylation of 4-chloro-substituted substrates with different anilines. The developed base-free protocol was compatible with several anilines bearing substituents at the ortho-, meta-, or para-positions, giving the corresponding N-arylated derivatives in yields of up to 96%. Moreover, it allowed fast and efficient reactions that required reduced amounts of organic solvents, so it is more sustainable than the protocols reported in the literature. Preliminary screening against selected tumor cells lines (HCT-116, MCF-7, and T98G) demonstrated the medicinal relevance of the 4-anilinoquinazolines synthesized here: many compounds (10b, 10c, 10g, 10k, 10l, 15a, 15b, and 15d) proved active against at least one of the tested tumor cell lines. Compound 10b afforded the most promising IC50 values (2.8 and 2.0 µM against HCT-116 and T98G cells, respectively). The scope of the developed methodology and its applicability toward the synthesis of other biologically active molecules are currently being investigated in our laboratories.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C spectra and additional information of antiproliferative assay. | ||
| Format: PDF | Size: 5.9 MB | Download |
Acknowledgements
The authors acknowledge the assistance of Mrs. Izabel C. C. Turatti, Mr. José C. Tomaz and Mr. Vinicius Palaretti in the GC–MS, HRMS and NMR analysis, respectively.
Funding
The authors gratefully thank financial support for this work by the Brazilian foundations: Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (Grant numbers: 2015/01466-9, 2015/21364-6, 2015/17177-6, 2017/18235-5, 2020/08987-2 and 2018/14150-8); Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq (Grant number: 140137/2018-1 and 140146/2020-2); and Coordenação de Aperfeiçoamento Pessoal de Nível Superior - CAPES.
References
-
Liu, S.; Zhao, Z.; Wang, Y. Chem. – Eur. J. 2019, 25, 2423–2441. doi:10.1002/chem.201803960
Return to citation in text: [1] -
Khan, I.; Ibrar, A.; Ahmed, W.; Saeed, A. Eur. J. Med. Chem. 2015, 90, 124–169. doi:10.1016/j.ejmech.2014.10.084
Return to citation in text: [1] -
Gupta, T.; Rohilla, A.; Pathak, A.; Akhtar, M. J.; Haider, M. R.; Yar, M. S. Synth. Commun. 2018, 48, 1099–1127. doi:10.1080/00397911.2018.1431282
Return to citation in text: [1] -
Ajani, O. O.; Audu, O. Y.; Aderohunmu, D. V.; Owolabi, F. E.; Olomieja, A. O. Am. J. Drug Discovery Dev. 2017, 7, 1–24. doi:10.3923/ajdd.2017.1.24
Return to citation in text: [1] -
Bansal, R.; Malhotra, A. Eur. J. Med. Chem. 2021, 211, 113016. doi:10.1016/j.ejmech.2020.113016
Return to citation in text: [1] [2] -
Xu, P.; Chu, J.; Li, Y.; Wang, Y.; He, Y.; Qi, C.; Chang, J. Bioorg. Med. Chem. 2019, 27, 114938. doi:10.1016/j.bmc.2019.06.001
Return to citation in text: [1] [2] [3] [4] -
Banerjee, S.; Arnst, K. E.; Wang, Y.; Kumar, G.; Deng, S.; Yang, L.; Li, G.-b.; Yang, J.; White, S. W.; Li, W.; Miller, D. D. J. Med. Chem. 2018, 61, 1704–1718. doi:10.1021/acs.jmedchem.7b01858
Return to citation in text: [1] [2] -
Wei, H.; Duan, Y.; Gou, W.; Cui, J.; Ning, H.; Li, D.; Qin, Y.; Liu, Q.; Li, Y. Eur. J. Med. Chem. 2019, 181, 111552. doi:10.1016/j.ejmech.2019.07.055
Return to citation in text: [1] -
Fan, Y.-H.; Ding, H.-W.; Liu, D.-D.; Song, H.-R.; Xu, Y.-N.; Wang, J. Bioorg. Med. Chem. 2018, 26, 1675–1685. doi:10.1016/j.bmc.2018.02.015
Return to citation in text: [1] [2] -
Mphahlele, M. J.; Maluleka, M. M.; Aro, A.; McGaw, L. J.; Choong, Y. S. J. Enzyme Inhib. Med. Chem. 2018, 33, 1516–1528. doi:10.1080/14756366.2018.1510919
Return to citation in text: [1] -
Van Horn, K. S.; Burda, W. N.; Fleeman, R.; Shaw, L. N.; Manetsch, R. J. Med. Chem. 2014, 57, 3075–3093. doi:10.1021/jm500039e
Return to citation in text: [1] [2] -
Krapf, M. K.; Gallus, J.; Spindler, A.; Wiese, M. Eur. J. Med. Chem. 2019, 161, 506–525. doi:10.1016/j.ejmech.2018.10.026
Return to citation in text: [1] [2] [3] -
Krapf, M. K.; Gallus, J.; Wiese, M. Eur. J. Med. Chem. 2017, 139, 587–611. doi:10.1016/j.ejmech.2017.08.020
Return to citation in text: [1] [2] [3] -
Anderson, M. B.; Willardsen, J. A.; Weiner, W. S.; Yungai, A.; Halter, R. J.; Klimova, Y.; Suzuki, K.; Reeder, M. Compounds and Therapeutical Use Thereof. U.S. Pat. Appl. US 2010/0069383 A1, March 18, 2010.
Return to citation in text: [1] [2] -
Sirisoma, N.; Kasibhatla, S.; Pervin, A.; Zhang, H.; Jiang, S.; Willardsen, J. A.; Anderson, M. B.; Baichwal, V.; Mather, G. G.; Jessing, K.; Hussain, R.; Hoang, K.; Pleiman, C. M.; Tseng, B.; Drewe, J.; Cai, S. X. J. Med. Chem. 2008, 51, 4771–4779. doi:10.1021/jm8003653
Return to citation in text: [1] [2] [3] -
Choudhary, S.; Doshi, A.; Luckett-Chastain, L.; Ihnat, M.; Hamel, E.; Mooberry, S. L.; Gangjee, A. Bioorg. Med. Chem. 2021, 35, 116061. doi:10.1016/j.bmc.2021.116061
Return to citation in text: [1] [2] -
Liu, G.; Yang, S.; Song, B.; Xue, W.; Hu, D.; Jin, L.; Lu, P. Molecules 2006, 11, 272–278. doi:10.3390/11040272
Return to citation in text: [1] -
Gellis, A.; Kieffer, C.; Primas, N.; Lanzada, G.; Giorgi, M.; Verhaeghe, P.; Vanelle, P. Tetrahedron 2014, 70, 8257–8266. doi:10.1016/j.tet.2014.09.024
Return to citation in text: [1] [2] [3] -
Murie, V. E.; Marques, L. M. M.; Souza, G. E. P.; Oliveira, A. R. M.; Lopes, N. P.; Clososki, G. C. J. Braz. Chem. Soc. 2016, 27, 1121–1128. doi:10.5935/0103-5053.20160005
Return to citation in text: [1] -
Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. doi:10.1002/anie.200400655
Return to citation in text: [1] -
Murie, V. E.; Nishimura, R. H. V.; Rolim, L. A.; Vessecchi, R.; Lopes, N. P.; Clososki, G. C. J. Org. Chem. 2018, 83, 871–880. doi:10.1021/acs.joc.7b02855
Return to citation in text: [1] -
dos Santos, T.; Orenha, H. P.; Murie, V. E.; Vessecchi, R.; Clososki, G. C. Org. Lett. 2021, 23, 7396–7400. doi:10.1021/acs.orglett.1c02572
Return to citation in text: [1] -
Bozzini, L. A.; Batista, J. H. C.; de Mello, M. B. M.; Vessecchi, R.; Clososki, G. C. Tetrahedron Lett. 2017, 58, 4186–4190. doi:10.1016/j.tetlet.2017.09.051
Return to citation in text: [1] -
Bozzini, L. A.; dos Santos, T.; Murie, V. E.; de Mello, M. B. M.; Vessecchi, R.; Clososki, G. C. J. Org. Chem. 2021, 86, 1204–1215. doi:10.1021/acs.joc.0c02369
Return to citation in text: [1] -
Murie, V. E.; Nicolino, P. V.; dos Santos, T.; Gambacorta, G.; Nishimura, R. H. V.; Perovani, I. S.; Furtado, L. C.; Costa-Lotufo, L. V.; Moraes de Oliveira, A.; Vessecchi, R.; Baxendale, I. R.; Clososki, G. C. J. Org. Chem. 2021, 86, 13402–13419. doi:10.1021/acs.joc.1c01521
Return to citation in text: [1] -
Bertallo, C. R. d. S.; Arroio, T. R.; Toledo, M. F. Z. J.; Sadler, S. A.; Vessecchi, R.; Steel, P. G.; Clososki, G. C. Eur. J. Org. Chem. 2019, 5205–5213. doi:10.1002/ejoc.201900608
Return to citation in text: [1] -
Nishimura, R. H. V.; Murie, V. E.; Vessecchi, R.; Clososki, G. C. ChemistrySelect 2020, 5, 11106–11111. doi:10.1002/slct.202002589
Return to citation in text: [1] -
Paumo, H.; Makhafola, T.; Mphahlele, M. Molecules 2016, 21, 1366. doi:10.3390/molecules21101366
Return to citation in text: [1] -
Paumo, H. K.; Mphahlele, M. J.; Rhyman, L.; Ramasami, P. Tetrahedron 2016, 72, 123–133. doi:10.1016/j.tet.2015.11.014
Return to citation in text: [1] [2] -
Gallo, R. D. C.; Ferreira, I. M.; Casagrande, G. A.; Pizzuti, L.; Oliveira-Silva, D.; Raminelli, C. Tetrahedron Lett. 2012, 53, 5372–5375. doi:10.1016/j.tetlet.2012.07.102
Return to citation in text: [1] -
Kijrungphaiboon, W.; Chantarasriwong, O.; Chavasiri, W. Tetrahedron Lett. 2012, 53, 674–677. doi:10.1016/j.tetlet.2011.11.123
Return to citation in text: [1] -
Cao, D.; Wang, X.; Lei, L.; Ma, L.; Yang, Z.; Wang, F.; Chen, L. Chem. Biol. Drug Des. 2020, 96, 1084–1094. doi:10.1111/cbdd.12706
Return to citation in text: [1] -
Chamberlain, M. C.; Grimm, S.; Phuphanich, S.; Recht, L.; Zhu, J. Z.; Kim, L.; Rosenfeld, S.; Fadul, C. E.; Brain Tumor Investigational Consortium (BTIC). J. Neuro-Oncol. 2014, 118, 335–343. doi:10.1007/s11060-014-1437-y
Return to citation in text: [1]
| 29. | Paumo, H. K.; Mphahlele, M. J.; Rhyman, L.; Ramasami, P. Tetrahedron 2016, 72, 123–133. doi:10.1016/j.tet.2015.11.014 |
| 29. | Paumo, H. K.; Mphahlele, M. J.; Rhyman, L.; Ramasami, P. Tetrahedron 2016, 72, 123–133. doi:10.1016/j.tet.2015.11.014 |
| 30. | Gallo, R. D. C.; Ferreira, I. M.; Casagrande, G. A.; Pizzuti, L.; Oliveira-Silva, D.; Raminelli, C. Tetrahedron Lett. 2012, 53, 5372–5375. doi:10.1016/j.tetlet.2012.07.102 |
| 1. | Liu, S.; Zhao, Z.; Wang, Y. Chem. – Eur. J. 2019, 25, 2423–2441. doi:10.1002/chem.201803960 |
| 2. | Khan, I.; Ibrar, A.; Ahmed, W.; Saeed, A. Eur. J. Med. Chem. 2015, 90, 124–169. doi:10.1016/j.ejmech.2014.10.084 |
| 5. | Bansal, R.; Malhotra, A. Eur. J. Med. Chem. 2021, 211, 113016. doi:10.1016/j.ejmech.2020.113016 |
| 7. | Banerjee, S.; Arnst, K. E.; Wang, Y.; Kumar, G.; Deng, S.; Yang, L.; Li, G.-b.; Yang, J.; White, S. W.; Li, W.; Miller, D. D. J. Med. Chem. 2018, 61, 1704–1718. doi:10.1021/acs.jmedchem.7b01858 |
| 27. | Nishimura, R. H. V.; Murie, V. E.; Vessecchi, R.; Clososki, G. C. ChemistrySelect 2020, 5, 11106–11111. doi:10.1002/slct.202002589 |
| 7. | Banerjee, S.; Arnst, K. E.; Wang, Y.; Kumar, G.; Deng, S.; Yang, L.; Li, G.-b.; Yang, J.; White, S. W.; Li, W.; Miller, D. D. J. Med. Chem. 2018, 61, 1704–1718. doi:10.1021/acs.jmedchem.7b01858 |
| 28. | Paumo, H.; Makhafola, T.; Mphahlele, M. Molecules 2016, 21, 1366. doi:10.3390/molecules21101366 |
| 5. | Bansal, R.; Malhotra, A. Eur. J. Med. Chem. 2021, 211, 113016. doi:10.1016/j.ejmech.2020.113016 |
| 6. | Xu, P.; Chu, J.; Li, Y.; Wang, Y.; He, Y.; Qi, C.; Chang, J. Bioorg. Med. Chem. 2019, 27, 114938. doi:10.1016/j.bmc.2019.06.001 |
| 15. | Sirisoma, N.; Kasibhatla, S.; Pervin, A.; Zhang, H.; Jiang, S.; Willardsen, J. A.; Anderson, M. B.; Baichwal, V.; Mather, G. G.; Jessing, K.; Hussain, R.; Hoang, K.; Pleiman, C. M.; Tseng, B.; Drewe, J.; Cai, S. X. J. Med. Chem. 2008, 51, 4771–4779. doi:10.1021/jm8003653 |
| 16. | Choudhary, S.; Doshi, A.; Luckett-Chastain, L.; Ihnat, M.; Hamel, E.; Mooberry, S. L.; Gangjee, A. Bioorg. Med. Chem. 2021, 35, 116061. doi:10.1016/j.bmc.2021.116061 |
| 33. | Chamberlain, M. C.; Grimm, S.; Phuphanich, S.; Recht, L.; Zhu, J. Z.; Kim, L.; Rosenfeld, S.; Fadul, C. E.; Brain Tumor Investigational Consortium (BTIC). J. Neuro-Oncol. 2014, 118, 335–343. doi:10.1007/s11060-014-1437-y |
| 3. | Gupta, T.; Rohilla, A.; Pathak, A.; Akhtar, M. J.; Haider, M. R.; Yar, M. S. Synth. Commun. 2018, 48, 1099–1127. doi:10.1080/00397911.2018.1431282 |
| 4. | Ajani, O. O.; Audu, O. Y.; Aderohunmu, D. V.; Owolabi, F. E.; Olomieja, A. O. Am. J. Drug Discovery Dev. 2017, 7, 1–24. doi:10.3923/ajdd.2017.1.24 |
| 21. | Murie, V. E.; Nishimura, R. H. V.; Rolim, L. A.; Vessecchi, R.; Lopes, N. P.; Clososki, G. C. J. Org. Chem. 2018, 83, 871–880. doi:10.1021/acs.joc.7b02855 |
| 22. | dos Santos, T.; Orenha, H. P.; Murie, V. E.; Vessecchi, R.; Clososki, G. C. Org. Lett. 2021, 23, 7396–7400. doi:10.1021/acs.orglett.1c02572 |
| 23. | Bozzini, L. A.; Batista, J. H. C.; de Mello, M. B. M.; Vessecchi, R.; Clososki, G. C. Tetrahedron Lett. 2017, 58, 4186–4190. doi:10.1016/j.tetlet.2017.09.051 |
| 24. | Bozzini, L. A.; dos Santos, T.; Murie, V. E.; de Mello, M. B. M.; Vessecchi, R.; Clososki, G. C. J. Org. Chem. 2021, 86, 1204–1215. doi:10.1021/acs.joc.0c02369 |
| 25. | Murie, V. E.; Nicolino, P. V.; dos Santos, T.; Gambacorta, G.; Nishimura, R. H. V.; Perovani, I. S.; Furtado, L. C.; Costa-Lotufo, L. V.; Moraes de Oliveira, A.; Vessecchi, R.; Baxendale, I. R.; Clososki, G. C. J. Org. Chem. 2021, 86, 13402–13419. doi:10.1021/acs.joc.1c01521 |
| 26. | Bertallo, C. R. d. S.; Arroio, T. R.; Toledo, M. F. Z. J.; Sadler, S. A.; Vessecchi, R.; Steel, P. G.; Clososki, G. C. Eur. J. Org. Chem. 2019, 5205–5213. doi:10.1002/ejoc.201900608 |
| 17. | Liu, G.; Yang, S.; Song, B.; Xue, W.; Hu, D.; Jin, L.; Lu, P. Molecules 2006, 11, 272–278. doi:10.3390/11040272 |
| 18. | Gellis, A.; Kieffer, C.; Primas, N.; Lanzada, G.; Giorgi, M.; Verhaeghe, P.; Vanelle, P. Tetrahedron 2014, 70, 8257–8266. doi:10.1016/j.tet.2014.09.024 |
| 12. | Krapf, M. K.; Gallus, J.; Spindler, A.; Wiese, M. Eur. J. Med. Chem. 2019, 161, 506–525. doi:10.1016/j.ejmech.2018.10.026 |
| 13. | Krapf, M. K.; Gallus, J.; Wiese, M. Eur. J. Med. Chem. 2017, 139, 587–611. doi:10.1016/j.ejmech.2017.08.020 |
| 6. | Xu, P.; Chu, J.; Li, Y.; Wang, Y.; He, Y.; Qi, C.; Chang, J. Bioorg. Med. Chem. 2019, 27, 114938. doi:10.1016/j.bmc.2019.06.001 |
| 18. | Gellis, A.; Kieffer, C.; Primas, N.; Lanzada, G.; Giorgi, M.; Verhaeghe, P.; Vanelle, P. Tetrahedron 2014, 70, 8257–8266. doi:10.1016/j.tet.2014.09.024 |
| 6. | Xu, P.; Chu, J.; Li, Y.; Wang, Y.; He, Y.; Qi, C.; Chang, J. Bioorg. Med. Chem. 2019, 27, 114938. doi:10.1016/j.bmc.2019.06.001 |
| 9. | Fan, Y.-H.; Ding, H.-W.; Liu, D.-D.; Song, H.-R.; Xu, Y.-N.; Wang, J. Bioorg. Med. Chem. 2018, 26, 1675–1685. doi:10.1016/j.bmc.2018.02.015 |
| 14. | Anderson, M. B.; Willardsen, J. A.; Weiner, W. S.; Yungai, A.; Halter, R. J.; Klimova, Y.; Suzuki, K.; Reeder, M. Compounds and Therapeutical Use Thereof. U.S. Pat. Appl. US 2010/0069383 A1, March 18, 2010. |
| 11. | Van Horn, K. S.; Burda, W. N.; Fleeman, R.; Shaw, L. N.; Manetsch, R. J. Med. Chem. 2014, 57, 3075–3093. doi:10.1021/jm500039e |
| 32. | Cao, D.; Wang, X.; Lei, L.; Ma, L.; Yang, Z.; Wang, F.; Chen, L. Chem. Biol. Drug Des. 2020, 96, 1084–1094. doi:10.1111/cbdd.12706 |
| 14. | Anderson, M. B.; Willardsen, J. A.; Weiner, W. S.; Yungai, A.; Halter, R. J.; Klimova, Y.; Suzuki, K.; Reeder, M. Compounds and Therapeutical Use Thereof. U.S. Pat. Appl. US 2010/0069383 A1, March 18, 2010. |
| 15. | Sirisoma, N.; Kasibhatla, S.; Pervin, A.; Zhang, H.; Jiang, S.; Willardsen, J. A.; Anderson, M. B.; Baichwal, V.; Mather, G. G.; Jessing, K.; Hussain, R.; Hoang, K.; Pleiman, C. M.; Tseng, B.; Drewe, J.; Cai, S. X. J. Med. Chem. 2008, 51, 4771–4779. doi:10.1021/jm8003653 |
| 16. | Choudhary, S.; Doshi, A.; Luckett-Chastain, L.; Ihnat, M.; Hamel, E.; Mooberry, S. L.; Gangjee, A. Bioorg. Med. Chem. 2021, 35, 116061. doi:10.1016/j.bmc.2021.116061 |
| 31. | Kijrungphaiboon, W.; Chantarasriwong, O.; Chavasiri, W. Tetrahedron Lett. 2012, 53, 674–677. doi:10.1016/j.tetlet.2011.11.123 |
| 6. | Xu, P.; Chu, J.; Li, Y.; Wang, Y.; He, Y.; Qi, C.; Chang, J. Bioorg. Med. Chem. 2019, 27, 114938. doi:10.1016/j.bmc.2019.06.001 |
| 8. | Wei, H.; Duan, Y.; Gou, W.; Cui, J.; Ning, H.; Li, D.; Qin, Y.; Liu, Q.; Li, Y. Eur. J. Med. Chem. 2019, 181, 111552. doi:10.1016/j.ejmech.2019.07.055 |
| 9. | Fan, Y.-H.; Ding, H.-W.; Liu, D.-D.; Song, H.-R.; Xu, Y.-N.; Wang, J. Bioorg. Med. Chem. 2018, 26, 1675–1685. doi:10.1016/j.bmc.2018.02.015 |
| 10. | Mphahlele, M. J.; Maluleka, M. M.; Aro, A.; McGaw, L. J.; Choong, Y. S. J. Enzyme Inhib. Med. Chem. 2018, 33, 1516–1528. doi:10.1080/14756366.2018.1510919 |
| 11. | Van Horn, K. S.; Burda, W. N.; Fleeman, R.; Shaw, L. N.; Manetsch, R. J. Med. Chem. 2014, 57, 3075–3093. doi:10.1021/jm500039e |
| 12. | Krapf, M. K.; Gallus, J.; Spindler, A.; Wiese, M. Eur. J. Med. Chem. 2019, 161, 506–525. doi:10.1016/j.ejmech.2018.10.026 |
| 13. | Krapf, M. K.; Gallus, J.; Wiese, M. Eur. J. Med. Chem. 2017, 139, 587–611. doi:10.1016/j.ejmech.2017.08.020 |
| 12. | Krapf, M. K.; Gallus, J.; Spindler, A.; Wiese, M. Eur. J. Med. Chem. 2019, 161, 506–525. doi:10.1016/j.ejmech.2018.10.026 |
| 13. | Krapf, M. K.; Gallus, J.; Wiese, M. Eur. J. Med. Chem. 2017, 139, 587–611. doi:10.1016/j.ejmech.2017.08.020 |
| 18. | Gellis, A.; Kieffer, C.; Primas, N.; Lanzada, G.; Giorgi, M.; Verhaeghe, P.; Vanelle, P. Tetrahedron 2014, 70, 8257–8266. doi:10.1016/j.tet.2014.09.024 |
| 19. | Murie, V. E.; Marques, L. M. M.; Souza, G. E. P.; Oliveira, A. R. M.; Lopes, N. P.; Clososki, G. C. J. Braz. Chem. Soc. 2016, 27, 1121–1128. doi:10.5935/0103-5053.20160005 |
| 20. | Kappe, C. O. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. doi:10.1002/anie.200400655 |
| 15. | Sirisoma, N.; Kasibhatla, S.; Pervin, A.; Zhang, H.; Jiang, S.; Willardsen, J. A.; Anderson, M. B.; Baichwal, V.; Mather, G. G.; Jessing, K.; Hussain, R.; Hoang, K.; Pleiman, C. M.; Tseng, B.; Drewe, J.; Cai, S. X. J. Med. Chem. 2008, 51, 4771–4779. doi:10.1021/jm8003653 |
© 2021 Nishimura et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.