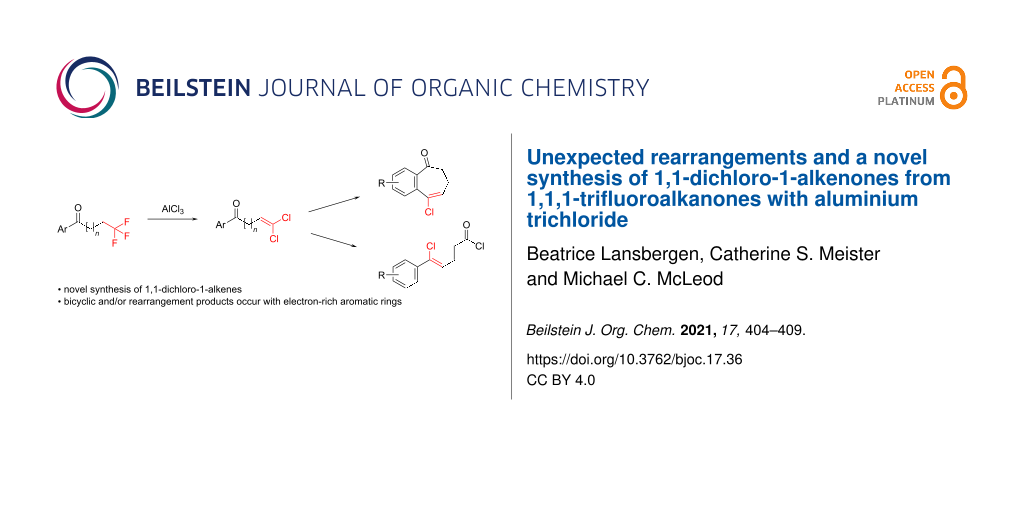
Abstract
A novel reactivity of 1,1,1-trifluoroalkanones is reported, where the reaction with AlCl3 results in the formation of 1,1-dichloro-1-alkenones. The reaction scope was found to be broad, with various chain lengths and aryl substituents tolerated. For substrates containing an electron-rich aromatic ring, further reactions take place, resulting in bicyclic and/or rearrangement products.

Graphical Abstract
Introduction
1,1-Dichloro-1-alkenes are valuable synthetic intermediates and have been employed in Pd-mediated cross couplings of one or both chlorine atoms [1-7], carbonylation reactions [8], and C–H bond alkynylations of heteroarenes [9]. The 1,1-dichloro-1-alkenyl moiety is found in a number of pyrethroid insecticides including permethrin and marine natural products such as caracolamide A [10] (Figure 1).
![[1860-5397-17-36-1]](/bjoc/content/figures/1860-5397-17-36-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of biologically active 1,1-dichloro-1-alkenes.
Figure 1: Examples of biologically active 1,1-dichloro-1-alkenes.
1,1-Dichloro-1-alkenes 2 are commonly prepared from the corresponding aldehydes 1 in one step with PPh3 and CHCl3 [11], CCl4 [12], or with the phosphonate reagent LiCCl2-P(O)(OEt)2 [13,14] (Figure 2a). Alternatively, the aldehydes are converted to trichloromethyl carbinols 3 using various methodologies [15-17], followed by acetylation and elimination to provide the desired dichloroalkenes [18-20]. The preparation of 1,1-dichloro-1-alkenes from hydrazones [21] and from 2,2,2-trichloroethyl carbonates [22] has also been reported. Internal difluoroalkanes have been used to generate chloroalkene products using AlEt2Cl [23,24]. In this article we describe the AlCl3-mediated formation of 1,1-dichloro-1-alkenones 6 from 1,1,1-trifluoroalkanones 5, which in turn are accessed by the Grignard addition of 1,1,1-trifluoroalkylmagnesium halides to nitriles (Figure 2b). It is worth noting that the 1,1,1-trifluoroalkyl halides (n = 1, 2, 3) are commercially available, whereas the corresponding 1,1-dichloroalkenyl halides (or 1,1,1-trichloroalkyl halides) are not. This methodology therefore represents a potentially useful access to the 1,1-dichloro-1-alkenone moiety.
![[1860-5397-17-36-2]](/bjoc/content/figures/1860-5397-17-36-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) Common methods for the preparation of 1,1-dichloro-1-alkenes from aldehydes. b) This work: a two-step synthesis of 1,1-dichloro-1-alkenones from nitriles via 1,1,1-trifluoroalkanones.
Figure 2: a) Common methods for the preparation of 1,1-dichloro-1-alkenes from aldehydes. b) This work: a two...
Findings
Under initial conditions, AlCl3 was shown to invoke the conversion of 1,1,1-trifluoroalkanone 5a [25] to 1,1-dichloro-1-alkenone 6a in 23% yield, without cleavage of the methyl ether [26] (Scheme 1). A similar AlCl3-promoted conversion of vinylic trifluoromethyl groups to 1,1-dichloroalkenes has previously been reported [27,28]. Additionally, 1,1-dichloroalkenes have also been prepared by the elimination of HCl from trichloroalkanes [29]. To the best of our knowledge, the direct conversion of an aliphatic CF3 group to a dichlorovinyl moiety has not been previously reported.
![[1860-5397-17-36-i1]](/bjoc/content/inline/1860-5397-17-36-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The initial attempt for the conversion of 1,1,1-trifluoroalkanone 5a to 1,1-dichloroalkenone 6a.
Scheme 1: The initial attempt for the conversion of 1,1,1-trifluoroalkanone 5a to 1,1-dichloroalkenone 6a.
Encouraged by these initial findings, we performed a screen of reaction conditions using the 3-methoxy derivative 5b (Table 1). During this optimisation process, two things became quickly apparent: (1) the dichloroalkenones 6 are generally volatile and should be handled carefully (water bath <30 °C) and (2) under certain conditions, the trichloroalkanone 8b could also be isolated from the reactions. While the conversion of an aromatic trifluoromethyl to a trichloromethyl group with AlCl3 has been previously reported [30-32], investigations into the analogous transformation of aliphatic CF3 groups has been limited to adamantly-trifluoromethyl moieties [33,34].
Table 1: Optimisation of the reaction conditions for the conversion of 1,1,1-trifluoroalkanone 5b to 1,1-dichloroalkenone 6b.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-36-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | equiv AlCl3 | solvent | temp (°C) | time (h) | ratioa 5b:6b:8b |
| 1 | 5 | CH2Cl2 | 0 | 1.5 | 0.07:1:0.15 |
| 2 | 5 | CH2Cl2 | 0 | 1 | 0.45:1:0.35 |
| 3 | 3 | CH2Cl2 | 0–rt | 18 | 9.7:1:0.7 |
| 4 | 1 | CH2Cl2 | 0–rt | 18 | n.r.b |
| 5 | 5 | PhMe | 0–rt | 18 | complex mixture |
| 6 | 5 | THF | 0–rt | 18 | n.r.b |
| 7 | 5 | DMF | 0–rt | 18 | n.r.b |
| 8 | 5 | MeCN | 0–rt | 18 | n.r.b |
| 9 | 5 | DCE | 0 | 2 | 0.29:1:0.19 |
| 10 | 5 | CH2Cl2 | rt | 2 | 0.05:1:0.05 (78%)c |
| 11 | 5 | CH2Cl2 | 40 | 1 | complex mixture |
aDetermined by 1H NMR analysis; bn.r. = no reaction; cisolated yield of 6b.
An NMR analysis of the crude product mixture obtained using our original reaction conditions (5 equiv AlCl3, CH2Cl2, 0 °C) showed that, while dichloroalkenone 6b was the major product, a small amount of starting material 5b was still present along with significant quantities of the trichloroalkanone 8b (Table 1, entry 1). With a shorter reaction time of 1 h, greater quantities of the trifluoroalkanone starting material 5b and trichloroalkanone 8b were observed (Table 1, entry 2), suggesting that compound 8b is an intermediate structure in the formation of dichloroalkenone 6b. Attempts to reduce the number of equivalents of AlCl3 in the reaction were unsuccessful: only unreacted starting material was observed when 1 equivalent was employed, and 3 equivalents only gave low conversion (Table 1, entries 3 and 4). Replacing dichloromethane with 1,2-dichloroethane as solvent had a negligible effect on the reaction, however, all other solvents that were investigated failed to produce any desired dichloroalkenone product, with only unreacted 5b being observed in most cases (Table 1, entries 5–9). By increasing the temperature to 40 °C, a full conversion of the starting material was observed, along with a decreased amount of the trichloroalkanone 8b, but at the expense of the formation of a number of other unidentified byproducts (Table 1, entry 11). At room temperature, the right balance of starting material conversion and minimisation of byproduct formation was reached, so that dichloroalkenone 6b could be isolated in 78% yield (Table 1, entry 10).
With an optimised set of conditions established for this reaction, we turned our attention to investigating the substrate scope (Scheme 2). The required trifluoroalkanone starting materials were prepared in one step from the corresponding benzonitrile by reaction with the required trifluoroalkylmagnesium iodide (see Supporting Information File 1).
![[1860-5397-17-36-i2]](/bjoc/content/inline/1860-5397-17-36-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope for the reaction of 1,1,1 trifluoroalkanones with AlCl3. aPurification (normal or reversed-phase chromatography) required. bComplex mixture. c5 Days.
Scheme 2: Substrate scope for the reaction of 1,1,1 trifluoroalkanones with AlCl3. aPurification (normal or r...
The substrate scope of the reaction proved quite broad; all phenyl-containing substrates 5a–g could be transformed into the desired products 6a–g. Using these optimised conditions, the isolated yield of our original dichloroalkene 6a (Scheme 1) could be increased from 23% to 78%. Naphthalene 6h and 2-pyridine 6i were also successfully obtained, however, the reactions with the 3- and 4-pyridines 5j and 5k resulted in complex mixtures from which no desired product, nor starting material, could be observed. The scope of this chemistry also extends to substrates with a shorter trifluoroalkanone chain, allowing the preparation of 1,1-dichlorobutenone 6l and 1,1-dichloropropenone 6m, although the latter compound required a considerably longer reaction time (5 days). For some of the above reactions, the crude product obtained after work-up was sufficiently clean (>90% purity as determined by 1H NMR) that no further purification was necessary. The other products needed to be purified by either normal and/or reversed-phase chromatography, resulting in significantly lower yields, in part due to the volatile nature of the products.
The longer reaction time of trifluoropentanone derivative 5m compared to the longer chain substrates (e.g., 5c and 5l) could be explained mechanistically by the formation of a terminal carbocation upon the extraction of a fluorine atom by the aluminium species (Scheme 3). This carbocation could be stabilised to varying degrees by the ketone moiety, depending on the alkyl chain length. For longer chains where n = 1 or 2, this stabilisation would occur via a favoured 5- or 6-ring intermediate, respectively, while for shorter chains (n = 0), an unfavoured 4-membered ring would be required.
![[1860-5397-17-36-i3]](/bjoc/content/inline/1860-5397-17-36-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the formation of dichloroalkene 6 from trifluoroalkanone 5.
Scheme 3: Proposed mechanism for the formation of dichloroalkene 6 from trifluoroalkanone 5.
When the 3-methoxy derivative 5b was treated with 7 (instead of 5) equivalents of AlCl3, a further two products were obtained, whose structures were elucidated as the 8,9-dihydrobenzo[7]annulen-9-ones 9 and 10 (Scheme 4). Presumably, these bicyclic compounds arise via an intramolecular Friedel–Crafts alkylation of 6b, promoted by the ortho/para-directing nature of the methoxy substituent, and a subsequent elimination of HCl. No such transformation was observed with the other trifluoroalkanone substrates, including the 2- and 4-methoxy substrates 5a and 5f, presumably as only the meta-methoxy group is able to sufficiently activate the two positions adjacent to the ketone.
![[1860-5397-17-36-i4]](/bjoc/content/inline/1860-5397-17-36-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Formation of bicyclic ketones 9 and 10 from 1,1,1-trifluoroalkanone 5b using 7 equivalents of AlCl3.
Scheme 4: Formation of bicyclic ketones 9 and 10 from 1,1,1-trifluoroalkanone 5b using 7 equivalents of AlCl3....
When the 4-methoxy derivative 5n was treated with AlCl3 using our standard conditions, none of the expected 1,1-dichloroalkenone 6n was obtained. Instead, the acid chloride 13 was isolated in an excellent yield (Scheme 5). The structure of this product was elucidated using 2D-NMR experiments, with an HMBC cross-peak between the aryl ring and the alkene moiety proving most illuminating (see Supporting Information File 1). The geometry of the double bond was determined from a NOE interaction between the alkene proton and the ring hydrogen atoms. This molecule could not however be detected by mass spectrometry and so it was converted to carboxylic acids 14 and 15 by the treatment with silica gel and NaOH, respectively, in order to confirm its identity. A possible mechanism for the formation of 13 from 5n could involve the formation of a terminal carbocation from trichloroalkanone 8n, followed by a favoured 6-endo-trig cyclisation driven by the 4-methoxy group to form the 6,6-spirocycle 11. The addition of water and subsequent ring-opening would then form the acid 12, which upon elimination of water would then provide the observed acid chloride 13.
![[1860-5397-17-36-i5]](/bjoc/content/inline/1860-5397-17-36-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Conversion of 1,1,1-trifluoroalkanone 5n to acid chloride 13 with AlCl3, and possible mechanism.
Scheme 5: Conversion of 1,1,1-trifluoroalkanone 5n to acid chloride 13 with AlCl3, and possible mechanism.
We wished to also determine whether the described reactivity would be observed with substrates not containing a ketone linker. Replacing the ketone moiety with an oxygen or sulphur atom proved unsuccessful, with only complex mixtures being obtained the upon treatment with AlCl3 (see Supporting Information File 1). This observation suggests that the role of the ketone moiety is not only to stabilise the putative carbocation as discussed earlier, but also to deactivate the aromatic ring to Friedel–Craft-type alkylations.
However, when the trifluoropentylbenzene 16 was prepared and subjected to the standard AlCl3 conditions (Scheme 6), a single product was isolated from the reaction that, after careful elucidation (see Supporting Information File 1), was identified as the bicycle 17. This compound could potentially arise via an initial conversion to the dichloroalkyl cation 18, followed by a ring closure and elimination of HCl to form the 6,6-spirocycle 19. A 1,2-rearrangement would then produce the observed product 17. Presumably the analogous dichloroalkenone 6d does not undergo this cyclisation due to the deactivation of the ring by the ketone towards nucleophilic attack.
![[1860-5397-17-36-i6]](/bjoc/content/inline/1860-5397-17-36-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Conversion of 1,1,1-trifluoroalkane 16 to bicycle 17 upon treatment with AlCl3, and possible mechanism.
Scheme 6: Conversion of 1,1,1-trifluoroalkane 16 to bicycle 17 upon treatment with AlCl3, and possible mechan...
Conclusion
In conclusion, we have shown that 1,1,1-trifluoroalkanones can be converted into 1,1-dichloro-1-alkenes when treated with AlCl3, allowing for an alternate and rapid route to this sought-after moiety. With 3- and 4-methoxy-substituted substrates, the initially formed dichloroalkene products are sufficiently electron rich to allow further reactions, giving rise to [6,7]bicyclic ketones 9, and 10 and the linear acid chloride 13, respectively. We have also found that when the ketone is not present, the trifluoroalkyl substrate is converted into bicyclic vinyl chloride 17 when treated with AlCl3. We hope that this short communication will inspire other researchers to utilise this functional group conversion in future syntheses of 1,1-dichloro-1-alkenes and to further investigate the unexpected reactivity of these compounds.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and NMR spectra for all compounds referenced. | ||
| Format: PDF | Size: 4.3 MB | Download |
References
-
Minato, A.; Suzuki, K.; Tamao, K. J. Am. Chem. Soc. 1987, 109, 1257–1258. doi:10.1021/ja00238a052
Return to citation in text: [1] -
Minato, A. J. Org. Chem. 1991, 56, 4052–4056. doi:10.1021/jo00012a048
Return to citation in text: [1] -
Shi, J.-c.; Negishi, E.-i. J. Organomet. Chem. 2003, 687, 518–524. doi:10.1016/j.jorganchem.2003.09.008
Return to citation in text: [1] -
Shi, J.-c.; Zeng, X.; Negishi, E.-i. Org. Lett. 2003, 5, 1825–1828. doi:10.1021/ol030017x
Return to citation in text: [1] -
Tan, Z.; Negishi, E.-i. Angew. Chem., Int. Ed. 2006, 45, 762–765. doi:10.1002/anie.200503519
Return to citation in text: [1] -
Andrei, D.; Wnuk, S. F. J. Org. Chem. 2006, 71, 405–408. doi:10.1021/jo051980e
Return to citation in text: [1] -
Liron, F.; Fosse, C.; Pernolet, A.; Roulland, E. J. Org. Chem. 2007, 72, 2220–2223. doi:10.1021/jo061908w
Return to citation in text: [1] -
Arthuis, M.; Lecup, A.; Roulland, E. Chem. Commun. 2010, 46, 7810–7812. doi:10.1039/c0cc02517h
Return to citation in text: [1] -
Ackermann, L.; Kornhaass, C.; Zhu, Y. Org. Lett. 2012, 14, 1824–1826. doi:10.1021/ol300514d
Return to citation in text: [1] -
Naman, C. B.; Almaliti, J.; Armstrong, L.; Caro-Díaz, E. J.; Pierce, M. L.; Glukhov, E.; Fenner, A.; Spadafora, C.; Debonsi, H. M.; Dorrestein, P. C.; Murray, T. F.; Gerwick, W. H. J. Nat. Prod. 2017, 80, 2328–2334. doi:10.1021/acs.jnatprod.7b00367
Return to citation in text: [1] -
Speziale, A. J.; Ratts, K. W. J. Am. Chem. Soc. 1962, 84, 854–859. doi:10.1021/ja00864a035
Return to citation in text: [1] -
Rabinowitz, R.; Marcus, R. J. Am. Chem. Soc. 1962, 84, 1312–1313. doi:10.1021/ja00866a056
Return to citation in text: [1] -
Seyferth, D.; Marmor, R. S. J. Organomet. Chem. 1973, 59, 237–245. doi:10.1016/s0022-328x(00)95040-x
Return to citation in text: [1] -
Villieras, J.; Perriot, P.; Normant, J. F. Synthesis 1975, 458–461. doi:10.1055/s-1975-23805
Return to citation in text: [1] -
Tanaka, H.; Yamashita, S.; Yamanoue, M.; Torii, S. J. Org. Chem. 1989, 54, 444–450. doi:10.1021/jo00263a035
Return to citation in text: [1] -
Corey, E. J.; Link, J. O.; Shao, Y. Tetrahedron Lett. 1992, 33, 3435–3438. doi:10.1016/s0040-4039(00)92656-x
Return to citation in text: [1] -
Aggarwal, V. K.; Mereu, A. J. Org. Chem. 2000, 65, 7211–7212. doi:10.1021/jo000584n
Return to citation in text: [1] -
Kryshtal, G. V.; Bogdanov, V. S.; Yanovskaya, L. A.; Volkov, Yu. P.; Trusova, E. I. Tetrahedron Lett. 1982, 23, 3607–3610. doi:10.1016/s0040-4039(00)87684-4
Return to citation in text: [1] -
Zhdankina, G. M.; Kryshtal', G. V.; Serebreyakov, É. P.; Yanovaskaya, L. A. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1988, 37, 2589–2591. doi:10.1007/bf00952654
Return to citation in text: [1] -
Wang, Z.; Campagna, S.; Xu, G.; Pierce, M. E.; Fortunak, J. M.; Confalone, P. N. Tetrahedron Lett. 2000, 41, 4007–4009. doi:10.1016/s0040-4039(00)00547-5
Return to citation in text: [1] -
Shastin, A. V.; Korotchenko, V. N.; Nenajdenko, V. G.; Balenkova, E. S. Tetrahedron 2000, 56, 6557–6563. doi:10.1016/s0040-4020(00)00606-2
Return to citation in text: [1] -
Ram, R. N.; Tittal, R. K. Tetrahedron Lett. 2014, 55, 4342–4345. doi:10.1016/j.tetlet.2014.05.097
Return to citation in text: [1] -
Wang, J.; Ogawa, Y.; Shibata, N. iScience 2019, 17, 132–143. doi:10.1016/j.isci.2019.06.018
Return to citation in text: [1] -
Wang, J.; Ogawa, Y.; Shibata, N. Sci. Rep. 2019, 9, 19113. doi:10.1038/s41598-019-55206-7
Return to citation in text: [1] -
Deprez, N. R.; Reddy, R. P.; Sharpe, P. L.; Stevenson, T. M. Pyrimidinyloxy benzene derivatives as herbicides. WO Pat. Appl. WO2015/108779 A1, July 23, 2015.
Return to citation in text: [1] -
Kawamura, Y.; Takatsuki, H.; Torii, F.; Horie, T. Bull. Chem. Soc. Jpn. 1994, 67, 511–515. doi:10.1246/bcsj.67.511
Return to citation in text: [1] -
Van Der Puy, M. J. Org. Chem. 1997, 62, 6466–6468. doi:10.1021/jo970655r
Return to citation in text: [1] -
Liu, Z.; Tu, X.-S.; Guo, L.-T.; Wang, X.-C. Chem. Sci. 2020, 11, 11548–11553. doi:10.1039/d0sc03883k
Return to citation in text: [1] -
Fontana, G.; Frenna, V.; Lamartina, L.; Natoli, M. C.; Noto, R. J. Phys. Org. Chem. 2002, 15, 108–114. doi:10.1002/poc.459
Return to citation in text: [1] -
Henne, A. L.; Newman, M. S. J. Am. Chem. Soc. 1938, 60, 1697–1698. doi:10.1021/ja01274a046
Return to citation in text: [1] -
Riera, J.; Castañer, J.; Carilla, J.; Robert, A. Tetrahedron Lett. 1989, 30, 3825–3828. doi:10.1016/s0040-4039(01)80668-7
Return to citation in text: [1] -
Goh, K. K. K.; Sinha, A.; Fraser, C.; Young, R. D. RSC Adv. 2016, 6, 42708–42712. doi:10.1039/c6ra09429e
Return to citation in text: [1] -
Sorochinskii, A. E.; Aleksandrov, A. M.; Kukhar, V. P. Zh. Org. Khim. 1979, 15, 1555–1556.
Return to citation in text: [1] -
Duddeck, H.; Spitzer, M.; Bolte, G. Liebigs Ann. Chem. 1985, 545–554. doi:10.1002/jlac.198519850315
Return to citation in text: [1]
| 30. | Henne, A. L.; Newman, M. S. J. Am. Chem. Soc. 1938, 60, 1697–1698. doi:10.1021/ja01274a046 |
| 31. | Riera, J.; Castañer, J.; Carilla, J.; Robert, A. Tetrahedron Lett. 1989, 30, 3825–3828. doi:10.1016/s0040-4039(01)80668-7 |
| 32. | Goh, K. K. K.; Sinha, A.; Fraser, C.; Young, R. D. RSC Adv. 2016, 6, 42708–42712. doi:10.1039/c6ra09429e |
| 33. | Sorochinskii, A. E.; Aleksandrov, A. M.; Kukhar, V. P. Zh. Org. Khim. 1979, 15, 1555–1556. |
| 34. | Duddeck, H.; Spitzer, M.; Bolte, G. Liebigs Ann. Chem. 1985, 545–554. doi:10.1002/jlac.198519850315 |
| 1. | Minato, A.; Suzuki, K.; Tamao, K. J. Am. Chem. Soc. 1987, 109, 1257–1258. doi:10.1021/ja00238a052 |
| 2. | Minato, A. J. Org. Chem. 1991, 56, 4052–4056. doi:10.1021/jo00012a048 |
| 3. | Shi, J.-c.; Negishi, E.-i. J. Organomet. Chem. 2003, 687, 518–524. doi:10.1016/j.jorganchem.2003.09.008 |
| 4. | Shi, J.-c.; Zeng, X.; Negishi, E.-i. Org. Lett. 2003, 5, 1825–1828. doi:10.1021/ol030017x |
| 5. | Tan, Z.; Negishi, E.-i. Angew. Chem., Int. Ed. 2006, 45, 762–765. doi:10.1002/anie.200503519 |
| 6. | Andrei, D.; Wnuk, S. F. J. Org. Chem. 2006, 71, 405–408. doi:10.1021/jo051980e |
| 7. | Liron, F.; Fosse, C.; Pernolet, A.; Roulland, E. J. Org. Chem. 2007, 72, 2220–2223. doi:10.1021/jo061908w |
| 11. | Speziale, A. J.; Ratts, K. W. J. Am. Chem. Soc. 1962, 84, 854–859. doi:10.1021/ja00864a035 |
| 27. | Van Der Puy, M. J. Org. Chem. 1997, 62, 6466–6468. doi:10.1021/jo970655r |
| 28. | Liu, Z.; Tu, X.-S.; Guo, L.-T.; Wang, X.-C. Chem. Sci. 2020, 11, 11548–11553. doi:10.1039/d0sc03883k |
| 10. | Naman, C. B.; Almaliti, J.; Armstrong, L.; Caro-Díaz, E. J.; Pierce, M. L.; Glukhov, E.; Fenner, A.; Spadafora, C.; Debonsi, H. M.; Dorrestein, P. C.; Murray, T. F.; Gerwick, W. H. J. Nat. Prod. 2017, 80, 2328–2334. doi:10.1021/acs.jnatprod.7b00367 |
| 29. | Fontana, G.; Frenna, V.; Lamartina, L.; Natoli, M. C.; Noto, R. J. Phys. Org. Chem. 2002, 15, 108–114. doi:10.1002/poc.459 |
| 9. | Ackermann, L.; Kornhaass, C.; Zhu, Y. Org. Lett. 2012, 14, 1824–1826. doi:10.1021/ol300514d |
| 25. | Deprez, N. R.; Reddy, R. P.; Sharpe, P. L.; Stevenson, T. M. Pyrimidinyloxy benzene derivatives as herbicides. WO Pat. Appl. WO2015/108779 A1, July 23, 2015. |
| 8. | Arthuis, M.; Lecup, A.; Roulland, E. Chem. Commun. 2010, 46, 7810–7812. doi:10.1039/c0cc02517h |
| 26. | Kawamura, Y.; Takatsuki, H.; Torii, F.; Horie, T. Bull. Chem. Soc. Jpn. 1994, 67, 511–515. doi:10.1246/bcsj.67.511 |
| 18. | Kryshtal, G. V.; Bogdanov, V. S.; Yanovskaya, L. A.; Volkov, Yu. P.; Trusova, E. I. Tetrahedron Lett. 1982, 23, 3607–3610. doi:10.1016/s0040-4039(00)87684-4 |
| 19. | Zhdankina, G. M.; Kryshtal', G. V.; Serebreyakov, É. P.; Yanovaskaya, L. A. Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.) 1988, 37, 2589–2591. doi:10.1007/bf00952654 |
| 20. | Wang, Z.; Campagna, S.; Xu, G.; Pierce, M. E.; Fortunak, J. M.; Confalone, P. N. Tetrahedron Lett. 2000, 41, 4007–4009. doi:10.1016/s0040-4039(00)00547-5 |
| 22. | Ram, R. N.; Tittal, R. K. Tetrahedron Lett. 2014, 55, 4342–4345. doi:10.1016/j.tetlet.2014.05.097 |
| 15. | Tanaka, H.; Yamashita, S.; Yamanoue, M.; Torii, S. J. Org. Chem. 1989, 54, 444–450. doi:10.1021/jo00263a035 |
| 16. | Corey, E. J.; Link, J. O.; Shao, Y. Tetrahedron Lett. 1992, 33, 3435–3438. doi:10.1016/s0040-4039(00)92656-x |
| 17. | Aggarwal, V. K.; Mereu, A. J. Org. Chem. 2000, 65, 7211–7212. doi:10.1021/jo000584n |
| 23. | Wang, J.; Ogawa, Y.; Shibata, N. iScience 2019, 17, 132–143. doi:10.1016/j.isci.2019.06.018 |
| 24. | Wang, J.; Ogawa, Y.; Shibata, N. Sci. Rep. 2019, 9, 19113. doi:10.1038/s41598-019-55206-7 |
| 13. | Seyferth, D.; Marmor, R. S. J. Organomet. Chem. 1973, 59, 237–245. doi:10.1016/s0022-328x(00)95040-x |
| 14. | Villieras, J.; Perriot, P.; Normant, J. F. Synthesis 1975, 458–461. doi:10.1055/s-1975-23805 |
| 12. | Rabinowitz, R.; Marcus, R. J. Am. Chem. Soc. 1962, 84, 1312–1313. doi:10.1021/ja00866a056 |
| 21. | Shastin, A. V.; Korotchenko, V. N.; Nenajdenko, V. G.; Balenkova, E. S. Tetrahedron 2000, 56, 6557–6563. doi:10.1016/s0040-4020(00)00606-2 |
© 2021 Lansbergen et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)