

Abstract
Pyrrolidones are common heterocyclic fragments in various biologically active compounds. Here, a two-step radical-based approach to γ-lactams bearing three to four stereocenters starting from epoxides, N-allylic silylacetamides and TEMPO is reported. The sequence starts with a new tandem nucleophilic substitution/Brook rearrangement/single electron transfer-induced radical oxygenation furnishing orthogonally protected α,γ-dioxygenated N-allylamides with wide scope, mostly good yields, and partly good diastereo- and enantioselectivity for defined combinations of chiral epoxides and chiral amides. This represents a very rare example of an oxidative geminal C–C/C–O difunctionalization next to carbonyl groups. The resulting dioxygenated allylic amides are subsequently subjected to persistent radical effect-based 5-exo-trig radical cyclization reactions providing functionalized pyrrolidones in high yields as diastereomeric mixtures. They converge to 3,4-trans-γ-lactams by base-mediated equilibration, which can be easily further diversified. Stereochemical models for both reaction types were developed.
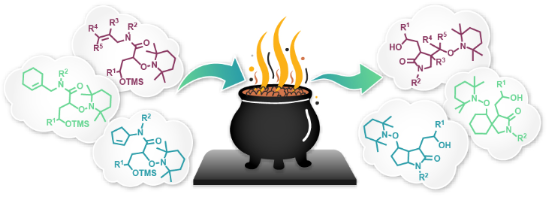
Graphical Abstract
Introduction
Nitrogen-containing heterocycles are widely distributed in biologically active compounds [1-4]. Saturated nitrogen heterocycles such as pyrrolidines [5-9], piperidines, pyrrolizidines or indolizidines [10-16] are central fragments in various natural products, which are often synthesized from lactams by reduction. This makes them important building blocks in the total syntheses of alkaloids and their non-natural analogs [17-21]. However, the γ-lactam substructure itself is a central fragment of numerous bioactive alkaloids, such as pyrrocidine B (I) [22], fusarin C (II) [23], fusarisetin A (III) [24], pseurotin A (IV) [25], (−)-salinosporamide A (V) [26], parvistemoline (VI) [27], glochidine (VII) [28], and other alkaloids [29-34] (Figure 1). Moreover, functionalized synthetic γ-lactams are important lead compounds, e.g., derivatives with antibacterial activity were discovered, what gains importance with respect to the increasing bacterial resistance toward traditional β-lactam antibiotics [35-39].
![[1860-5397-17-58-1]](/bjoc/content/figures/1860-5397-17-58-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected alkaloids containing the pyrrolidone motif.
Figure 1: Selected alkaloids containing the pyrrolidone motif.
Various synthetic pathways can be applied for the construction of the γ-lactam scaffold [40-44]. The pyrrolidinone fragment is often synthesized by transition metal- [45-50] or Lewis acid-catalyzed cyclization reactions [51-54]. The Diels–Alder reaction can also be used for the preparation of functionalized γ-lactams in a single step [55]. Radical 5-exo or 5-endo cyclizations of substituted N-allyl or N-vinyl α-halo amides VIII [56-61] or X [62-66] using atom transfer and other chain reactions, as well as non-chain methods [67-73] have been used to approach diverse γ-lactam-containing skeletons of the general structure IX or XI, respectively (Scheme 1A).
![[1860-5397-17-58-i1]](/bjoc/content/inline/1860-5397-17-58-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A) Classical γ-lactam synthesis by atom transfer radical cyclizations; B) previously developed tandem epoxide opening/Brook rearrangement/radical oxygenation and radical carbocyclizations; C) proposed approach to functionalized γ-lactams 10 by Brook rearrangement/radical oxygenation and subsequent 5-exo cyclization reactions.
Scheme 1: A) Classical γ-lactam synthesis by atom transfer radical cyclizations; B) previously developed tand...
Recently, we became interested in merging sigmatropic rearrangements with radical oxygenation reactions since profound changes in the connectivity patterns during both reaction modes will potentially significantly simplify the access to complex target molecules [74,75]. The principle is illustrated for a merger of nucleophilic opening of allylepoxides 1 with silylacetamides 2/Brook rearrangement [76-78] and oxygenation with TEMPO (3) leading to γ-(silyloxy)-α-(aminoxy)amides 5, which can be subsequently subjected to thermal radical cyclization reactions according to the persistent radical effect [79,80] forming functionalized cyclopentanes 6 (Scheme 1B).
Based on this sequence various other reaction pathways can be envisaged. Among them we hypothesized that the nucleophilic ring opening of simple epoxides 7 by N-allylic 2-silylacetamides would provide an intermediate alkoxide from which the Brook rearrangement and subsequent oxygenation could proceed (Scheme 1C). This represents in the event a geminal C–C/C–O difunctionalization of amide 8 and results in the α,γ-dioxygenated N-allylic amides 9. Thermal radical cyclizations to lactams of type 10 based on the persistent radical effect (PRE) are unknown and may provide a simple access to 3,4-disubstituted γ-lactams.
We report here that tandem nucleophilic epoxide ring-opening/Brook rearrangement/radical oxygenation reactions are indeed very effective for the synthesis of diverse N-allylic α-(aminoxy)amides 9 from various epoxides 7 and a range of N-allylic α-silylamides 8. α-(Aminoxy)amides 9 serve well for the synthesis of polysubstituted γ-lactams 10 with moderate diastereoselectivities. The stereochemistry of the initial cyclization products can be however simplified by further useful reaction steps.
Results
Tandem nucleophilic substitution/Brook rearrangement/radical α-oxygenation reactions
The N-allylic α-(trimethylsilyl)acetamides 8a–m were efficiently prepared by a two-step sequence. First, N-allyl acetamides 11a–m were synthesized by N-acetylation of the corresponding acyclic or cyclic allylic amines in very good yields (see Supporting Information File 1 for details). Their subsequent α-deprotonation by LDA followed by treatment with chlorotrimethylsilane at −78 °C [81] resulted in clean C-silylation of the corresponding enolate providing silylacetamides 8a–m in good to very good yields (Table 1, entries 1–13).
Table 1: Preparation of tertiary N-allylic α-(trimethylsilyl)amides 8a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-58-i12.svg?max-width=637&scale=1.0)
|
|||||||
| entry | 11 | R1 | R2 | R3 | R4 | R5 | 8, % |
| 1 | a | allyl | H | H | H | H | a, 76 |
| 2 | b | CH3 | H | H | H | H | b, 70 |
| 3 | c | Bn | CH3 | H | H | H | c, 75 |
| 4 | d | Bn | H | CH3 | CH3 | H | d, 72 |
| 5 | e | (S)-PhCHCH3 | H | H | H | H | e, 89 |
| 6 | f | (S)-β-NapCHCH3b | H | H | H | H | f, 95 |
| 7 | g | (S)-PhCHCH3 | H | CH3 | CH3 | H | g, 88 |
| 8 | h | (S)-PhCHCH3 | CH3 | H | H | H | h, 93 |
| 9 | i | (S)-PhCHCH3 | -(CH2)3- | H | H | i, 84 | |
| 10 | j | Bn | -(CH2)3- | H | H | j, 82 | |
| 11 | k | Bn | -(CH2)4- | H | H | k, 85 | |
| 12 | l | Bn | H | H | -(CH2)2- | l, 91 | |
| 13 | m | Bn | H | H | (R)-(CH2)3- | m, 76 | |
aGeneral conditions: 11 (1 equiv), iPr2NH (1.1 equiv), n-BuLi (1.1 equiv), TMSCl (1.05 equiv), −78 °C, 1 h; bNap = naphthyl.
For the synthesis of the targeted orthogonally protected α-(aminoxy)-γ-(silyloxy)amides 9 α-silylacetamides 8a–k were deprotonated by s-BuLi and treated with commercially available racemic epoxides 7a–d,f (Table 2, entries 1–7 and 13–15) or with enantiomerically pure epoxides (S)-7b, (R)-7b, or (S)-7e (Table 2, entries 8–12) at 0 °C. The epoxide opening/Brook rearrangement steps were typically complete after an hour, except for cyclohexene oxide 7f for which the nucleophilic opening and Brook rearrangement steps took 24 h (Table 2, entry 13). Ferrocenium hexafluorophosphate (4) and TEMPO (3) were subsequently added to trigger the single electron oxidation of the formed amide enolates and radical oxygenation affording α-(aminoxy)amides 9a–n in good 51–77% isolated yields. Cyclic units in the allylic N-substituent (Table 2, entries 14 and 15) and the epoxide (Table 2, entry 13) are tolerated. Most dioxygenated amides 9b–h,m,n were isolated as inseparable 1–1.2:1 anti/syn mixtures of unassigned diastereomers (Table 2, entries 2–8, 14 and 15), thus the silyloxy group in γ-position did not influence the face selectivity of radical coupling with TEMPO (3). However, since for all dioxygenated amides 9 the stereocenter at the alkoxyamine unit will be destroyed in the subsequent radical reaction, the low diastereoselectivity at that center is not of concern. In contrast, in the reactions of silylamides 8e–g bearing enantiomerically pure 1-arylethyl substituents with enantiomerically pure (S)-propylene oxide ((S)-7b) the dioxygenated amides (2R,4S)-9i–k were predominately formed with 3:1 anti/syn diastereoselectivity in the radical coupling with TEMPO irrespective of the aryl substituent (Table 2, entries 9, 11, and 12), whereas the sequence of amide 8e with (R)-propylene oxide ((R)-7b) provided the product (2S,4R)-9i in a much better 8:1 anti/syn coupling diastereoselectivity (Table 2, entry 10).
Table 2: Tandem nucleophilic ring opening/Brook rearrangement/radical oxygenationa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-58-i13.svg?max-width=637&scale=1.0)
|
|||||||||||
| entry | 8 | 7b | R1 | R2 | R3 | R4 | R5 | R6 | R7 | 9, % | anti/syn |
| 1 | a | a | allyl | H | H | H | H | CH3 | CH3 | a, 61 | – |
| 2 | a | b | allyl | H | H | H | H | CH3 | H | b, 77 | 1.1:1 |
| 3 | a | c | allyl | H | H | H | H | C4H9 | H | c, 71 | 1:1 |
| 4 | a | d | allyl | H | H | H | H | Ph | H | d, 56 | 1.1:1 |
| 5 | b | b | CH3 | H | H | H | H | CH3 | H | e, 68 | 1.1:1 |
| 6 | c | b | Bn | CH3 | H | H | H | CH3 | H | f, 51 | 1:1 |
| 7 | d | b | Bn | H | CH3 | CH3 | H | CH3 | H | g, 53 | 1:1 |
| 8 | a | (S)-e | allyl | H | H | H | H | Ac | H | h, 65 | 1.1:1 |
| 9 | e | (S)-b | Bc | H | H | H | H | CH3 | H | i, 64 | 3:1 |
| 10 | e | (R)-b | Bc | H | H | H | H | CH3 | H | i, 63 | 8:1 |
| 11 | f | (S)-b | Cc | H | H | H | H | CH3 | H | j, 62 | 3:1 |
| 12 | g | (S)-b | Bc | H | CH3 | CH3 | H | CH3 | H | k, 61 | 3:1 |
| 13d | a | f | allyl | H | H | H | -(CH2)4- | H | l, 63 | 7:1 | |
| 14 | j | b | Bn | -(CH2)3- | H | H | CH3 | H | m, 61 | 1.2:1 | |
| 15 | k | b | Bn | -(CH2)4- | H | H | CH3 | H | n, 62 | 1.1:1 | |
aGeneral conditions: s-BuLi (1.1 equiv), 8 (1 equiv), LiCl (6 equiv), 0 °C, 30 min, 7 (1.05 equiv), 0 °C, then 3 (1.05 equiv), 4 (1.2 equiv), −78 °C; bepoxides 7 are racemic unless indicated otherwise; cA = (S)-CH2OBn, B = (S)-PhCHCH3, C = (S)-β-NapCHCH3; dnucleophilic opening of 7e complete after 24 h at room temperature.
The configuration of the major anti-diastereomer of alkoxyamine 9j was determined by X-ray crystallography after desilylation and hydrochloride formation (see Supporting Information File 1 for details). The (R)-configuration at C2 as well as the (S)-configuration at both, C4 and the N-arylethyl group were established (Figure 2).
![[1860-5397-17-58-2]](/bjoc/content/figures/1860-5397-17-58-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of the major (2R,4S)-alkoxyamine hydrochloride derived from 9j. Displacement ellipsoids at 30% probability level and a disordered CHCl3 solvent molecule is not shown for clarity.
Figure 2: X-ray crystal structure of the major (2R,4S)-alkoxyamine hydrochloride derived from 9j. Displacemen...
A good diastereoselectivity was also observed for the ring-opening/Brook rearrangement/oxygenation sequence with cyclohexene oxide 7f, which furnished the dioxygenated amide 9l with a 7:1 anti/syn diastereoselectivity for the radical coupling (Table 2, entry 13). When the reaction was quenched after completion of the Brook rearrangement, N-allyl-N-propyl-2-(2-((trimethylsilyl)oxy)cyclohexyl)acetamide was obtained as a single diastereomer because of the stereospecific epoxide ring-opening in 80% yield (not shown, see Supporting Information File 1 for details).
N-Cyclopent-2-enyl and N-cyclohex-2-enylamides 8l,m provided the oxygenated products 9o,p in 68% and 63% yields, respectively, as 2:2:1:1 mixture of diastereomers (Scheme 2). Thus, the chiral cyclic amide substituent on the nitrogen atom influences the selectivity of the radical coupling with TEMPO to some extent, though less than the 1-arylethyl groups.
![[1860-5397-17-58-i2]](/bjoc/content/inline/1860-5397-17-58-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of the α-(aminoxy)amides 9o,p.
Scheme 2: Formation of the α-(aminoxy)amides 9o,p.
Somewhat surprisingly, amides 8h,i did not react with propylene oxide 7b neither at 0 °C nor at room temperature. In order to confirm enolate formation from 8h,i with s-BuLi, a deuterium quenching experiment with D2O was performed. Analysis by 1H NMR spectroscopy revealed 87 and 91% deuterium incorporation, respectively, indicating that a deprotonation occurred, but the epoxide opening was hampered by the combination of a sterically more demanding branched amide substituent and the R2 substituent at the internal carbon of the allylic unit.
Transformation of α-(aminoxy)amides 9 to lactams 12 by persistent radical effect-based cyclization reactions
The acyclic α-(aminoxy)amides 9a–k are suitable precursors for thermal radical cyclization reactions based on the persistent radical effect. Heating them to 150 °C in tert-butanol provided diverse 1,3,4-trisubstituted pyrrolidones (Table 3). For stability reasons the initially obtained silyl-protected lactams 10a–k were deprotected without isolation by TBAF in THF affording hydroxy lactams 12a–k in 66–93% yields. The thermal cyclization of α-(aminoxy)amide 9a provided two diastereomers of lactam 12a in a 2:1 trans/cis ratio (Table 3, entry 1). The N-allylic amides 9b–f provided lactams 12b–f as mixtures of four inseparable diastereomers in which those with trans orientation of the substituents at C3 and C4 of the formed 2-pyrrolidone ring predominated with moderate selectivity (Table 3, entries 2–7). The diastereomeric ratio is more or less independent of the nature of the amide substituent R1 or the substituent R4. Amide 9f with a methyl group at the internal position of the alkene unit cyclized exclusively in the 5-exo-trig mode and provided the pyrrolidone 12f with a quaternary center at C4 in moderate yield and similar diastereoselectivity as for 12a–d (Table 3, entry 7); a product of potentially competing 6-endo cyclization was not detected. Amides 9g,k with trisubstituted alkene units furnished 4-isopropenylpyrrolidones 12g,k in very good yields as mixtures of inseparable diastereomers (Table 3, entries 7 and 11); no alkoxyamine-containing products were isolated. Lactam 12h with a defined hydroxy group configuration in the side chain at C3 of the lactam as well as pyrrolidones 12i–k bearing configurationally defined 1-arylethyl groups at the amide nitrogen and the hydroxy group in the C3 side chain were obtained as inseparable mixtures of four diastereomers (Table 3, entries 8–11). The two possible trans-diastereomers predominated with moderate selectivity. These results indicate negligible asymmetric inductions from the chiral centers, both at the exocyclic hydroxy substituent in the C3 side chain (Table 3, entry 8) and/or of the 1-arylethyl group during the radical cyclization under the reaction conditions (Table 3, entries 9–11). The size of the arylethyl substituent at the nitrogen atom also plays essentially no role for the diastereoselectivity of the cyclization (Table 3, entry 10 vs entry 9).
Table 3: Hydroxyalkyl-γ-lactams 12 by PRE-based radical 5-exo cyclizationsa.
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-58-i14.svg?max-width=637&scale=1.0)
|
||||||||
| entry | 9 | R1 | R2 | R3 | R4 | R5 | 12, % | dr |
| 1 | a | allyl | H | H | CH3 | CH3 | a, 91 | 2:1 |
| 2 | b | allyl | H | H | CH3 | H | b, 87 | 2.5:2.5:1:1 |
| 3 | c | allyl | H | H | C4H9 | H | c, 86 | 2:2:1:1 |
| 4 | d | allyl | H | H | Ph | H | d, 86 | 3:3:1:1 |
| 5 | e | CH3 | H | H | CH3 | H | e, 82 | 2:2:1:1 |
| 6 | f | Bn | CH3 | H | CH3 | H | f, 66 | 3:3:1:1 |
| 7 | g | Bn | H | CH3 | CH3 | H | g, 93 | 2:2:1:1 |
| 8 | h | allyl | H | H | (S)-CH2OBn | H | h, 72 | 4:4:1:1b |
| 9 | i | (S)-PhCHCH3 | H | H | (S)-CH3 | H | i, 82 | 2:2:1:1 |
| 10 | j | (S)-β-NapCHCH3 | H | H | (S)-CH3 | H | j, 77 | 2:2:1:1 |
| 11 | k | (S)-PhCHCH3 | H | CH3 | (S)-CH3 | H | k, 92 | 2:2:1:1 |
aGeneral conditions: 1) 9 (1 equiv), t-BuOH, 150 °C, 1 h; 2) TBAF (1.2 equiv), THF, 0 °C, 30 min; bthe enantiomeric product is shown for clarity and simplicity.
The minor cis-diastereomer of N-(1-β-naphthylethyl)pyrrolidone 12j crystallized after oxidation to ketone 13j and its configuration was unequivocally established by X-ray crystallography (Figure 3, vide infra). Similarly, the minor cis-diastereomer of hydroxy lactam 12k crystallized and its configuration was confirmed. The configuration of the other lactams was assigned by analogy, by base-mediated equilibration and oxidation experiments (vide infra).
![[1860-5397-17-58-3]](/bjoc/content/figures/1860-5397-17-58-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: X-ray crystal structure of the minor cis-diastereomers of the keto lactam 13j (left) and the hydroxy lactam 12k (right). Displacement ellipsoids are drawn at the 30% probability level.
Figure 3: X-ray crystal structure of the minor cis-diastereomers of the keto lactam 13j (left) and the hydrox...
The α-(aminoxy)-γ-(silyloxy)amides 9l–p with cyclic units are also suitable precursors for radical cyclization reactions (Scheme 3). 3-(2-Hydroxycyclohexyl)-2-pyrrolidone 12l was obtained by the thermal cyclization of α-(aminoxy)cyclohexylacetamide 9l as a mixture of two major trans isomers 12lA and 12lB, which were accompanied by traces of a C3–C4 cis-diastereomer (not shown). The relative configuration of pyrrolidone 12l was assigned by NOE experiments of 3-(2-oxocyclohexyl)lactams 13l prepared from 12l by Dess–Martin oxidation (vide infra). The thermal cyclization of compounds 9m,n with N-cycloalkenylmethyl substituents provided spirolactams 12m,n in good yields, but with overall low diastereoselectivity. In the cyclization of 9m lactams 12mA and 12mB with trans orientation at C4 and the newly introduced aminoxy group at C5 were the major diastereomers, which results in an overall 4.5:1 trans/cis cyclization diastereoselectivity. The radical coupling with TEMPO (3) proceeded with moderate 2:1 diastereoselectivity for both pairs of the cyclized diastereomers. In the case of azaspiro[4,5]decanone 12n with a spirocyclohexyl substituent the cyclization diastereoselectivity was with 2.5:1 lower than that for 12m. However, the coupling diastereoselectivity with TEMPO (3) amounted to 4:1 for the trans diastereomers 12nA,B and exclusive for the cis isomer 12nC. The relative configuration of diastereomer 12nA was unequivocally established by X-ray crystallography of the hydrochloride adduct of the keto lactam 13nA (Scheme 3, insert). The relative configuration of the compounds 12m,n was determined by analogy, by ROESY investigations for the keto lactams 13m,n, and by isomerization experiments for 12n (vide infra). Amides 9o,p with cycloalkenyl substituents on the nitrogen were transformed to fused lactams 12o,p with good diastereoselectivity. Azabicyclo[3.3.0]octanone 12o prepared from amide 9o with a racemic cyclopent-2-enyl group on nitrogen was obtained as an inseparable mixture of four diastereomers 12oA and 12oB. In major 12oA the hydroxypropyl and tetramethylpiperidinyloxy groups reside on the convex face of the bicyclic system. Amide 9p with an enantiomerically enriched cyclohex-2-enyl substituent cyclized with exclusive diastereoselectivity and only two diastereomers 12pA differing in the orientation of the hydroxy group were obtained in high yield. The radical coupling with TEMPO also occurred exclusively at the convex face of the bicyclic system. The configurations of the fused lactams were assigned by chemical derivatization and NOE experiments (vide infra).
![[1860-5397-17-58-i3]](/bjoc/content/inline/1860-5397-17-58-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Thermal radical cyclization reactions of amides 9l–p bearing cyclic units. Conditions: a) t-BuOH, 150 °C, 1 h; b) TBAF, THF, 0 °C. All = allyl.
Scheme 3: Thermal radical cyclization reactions of amides 9l–p bearing cyclic units. Conditions: a) t-BuOH, 1...
Functionalization reactions of lactams 12
Base-mediated isomerization reactions
Lactams 12 are mixtures of two, four, six or eight diastereomers (vide supra) and the analysis and further application of such diastereomeric mixtures is problematic. To improve the diastereomeric ratio and to establish the relative configuration, lactam mixtures 12 were subjected to epimerization at C3 (Table 4). Indeed, 50 mol % KOt-Bu in t-BuOH under thermodynamic equilibration conditions at room temperature or at 50 °C for 24 h induced conversion of the minor cis isomers to the corresponding trans diastereomers in good yield (Table 4, entries 1–4, and 8). Resubjecting several trans-enriched compounds to the reaction conditions did not improve the results, indicating that the reactions are at thermodynamic equilibrium after 24 h. Surprisingly, for N-methyl-substituted lactam 12e the diastereomeric enrichment remained rather moderate despite variations of temperature and time (Table 4, entry 5), whereas the diastereomeric ratio of lactam 12f with a quaternary center at C4 did not change at all, neither at room temperature nor at 50 °C (Table 4, entry 6). Lactam 12g with a benzyl group as well as pyrrolidones 12i–k bearing branched 1-arylethyl groups on the nitrogen required warming to 50 °C to induce epimerization at C3 (Table 4, entries 9–11). Thus, the nitrogen substituent exerted a significant influence on the facility and position of the trans/cis equilibrium of the lactams 12. The influence of an isopropenyl or tetramethylpiperidinyloxymethyl group at C4 has in contrast only little influence on the thermodynamic equilibrium (Table 4, entries 7 and 11 vs entries 1 and 9).
Table 4: Isomerization of lactams 12 using KOt-Bu in t-BuOHa.
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-58-i15.svg?max-width=637&scale=1.0)
|
|||||||
| entry | 12 | R1 | R2 | R3 | R4 | trans-12 | dr |
| 1 | a | allyl | H | CH3 | CH3 | a | 17:1 |
| 2 | b | allyl | H | CH3 | H | b | 12:12:1:1 |
| 3 | c | allyl | H | C4H9 | H | c | 16:16:1:1 |
| 4 | d | allyl | H | Ph | H | d | 13:13:1:1 |
| 5 | e | CH3 | H | CH3 | H | e | 4:4:1:1 |
| 6b | f | Bn | CH3 | CH3 | H | f | 3:3:1:1 |
| 7b | g | Bn | H | CH3 | H | g | 7:7:1:0 |
| 8b | h | allyl | H | (S)-CH2OBn | H | h | 17:17:1:1 |
| 9b | i | (S)-PhCHCH3 | H | (S)-CH3 | H | i | 1:1:0:0 |
| 10b | j | (S)-β-NapCHCH3 | H | (S)-CH3 | H | j | 4:4:1:1 |
| 11b | k | (S)-PhCHCH3 | H | (S)-CH3 | H | k | 1:1:0:0 |
aGeneral conditions: 12 (1 equiv), KOt-Bu (0.5 equiv), t-BuOH, room temperature, 24 h; breaction at 50 °C.
Attempts to influence the diastereomeric ratio of the cyclization products by irreversible stoichiometric deprotonation of the lactams 12d,f,i by LDA at −78 °C and subsequent protonation by methanol did not lead to substantial changes of the initial diastereomeric ratios. To confirm enolate formation, lactam 12f was deprotonated by LDA at −78 °C and quenched with D2O, resulting in lactam 12f with 86% deuterium incorporation, but no change in the diastereomeric ratio.
Spirolactams 12m,n were also subjected to epimerization (Scheme 4). The change of the diastereomeric ratio A–D was not significant for 12m, whereas during equilibration of 12n the content of the cis isomers 12nC in the mixture decreased.
![[1860-5397-17-58-i4]](/bjoc/content/inline/1860-5397-17-58-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Epimerization of spirolactams 12m,n.
Scheme 4: Epimerization of spirolactams 12m,n.
Oxidation of the side chain hydroxy group at C3
Hydroxy lactams 12 can be transformed to keto pyrrolidones 13 by a Dess–Martin oxidation (Table 5). For the isomerized ketones trans-12b–d this leads essentially to single keto lactams 13b–d (Table 5, entries 1, 3, and 4). The non-equilibrated ketones can also be used as exemplified for 12b providing an unchanged 2.5:1 trans/cis diastereomeric mixture (Table 5, entry 2, cf. Table 3, entry 2). The trans and cis orientation of the substituents at C3 and C4 of the pyrrolidone ring of 13b was confirmed by NOE experiments (see Supporting Information File 1 for details). The trans/cis mixture of 12f was similarly oxidized providing the pyrrolidone diastereomers 13f in good yield and unchanged ratio (Table 5, entry 5). The diastereomerically enriched lactams trans-12i,j with 1-arylethyl substituents also provided the unchanged diastereomeric mixtures 13i,j (Table 5, entries 6 and 7), thus confirming that they have opposite trans arrangement at C3 and C4 based on the fixed (S)-configuration of the 1-arylethyl substituents. One of the minor cis diastereomers of 13j crystallized and its configuration was unequivocally established by X-ray crystallography (Figure 3, vide supra).
Table 5: Oxidation of hydroxy lactams trans-12 to keto lactams 13 by the Dess–Martin periodinanea.
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-58-i16.svg?max-width=637&scale=1.0)
|
||||||
| entry | trans-12 | R1 | R2 | R3 | 13 | dr |
| 1 | b | allyl | H | CH3 | b 88 | 1:0 |
| 2b | b | allyl | H | CH3 | b 86 | 2.5:1 |
| 3 | c | allyl | H | C4H9 | c 87 | 1:0 |
| 4 | d | allyl | H | Ph | d 80 | 1:0 |
| 5 | f | Bn | CH3 | CH3 | f 90 | 3:1 |
| 6 | i | (S)-PhCHCH3 | H | (S)-CH3 | i 77 | 1:1:0:0 |
| 7 | j | (S)-β-NapCHCH3 | H | (S)-CH3 | j 81 | 4:4:1:1 |
aGeneral conditions: trans-12 (1 equiv), DMP (1.3 equiv), t-BuOH (10 mol %), CH2Cl2, room temperature, 1 h; bnon-isomerized 12b was used.
Lactams 12l–o with cyclic subunits were also subjected to a Dess–Martin oxidation to confirm their relative configuration (Scheme 5). The diastereomeric mixture of lactam 12lA,B gave two trans-diastereomers of 13lA,B in an unchanged 1.2:1 ratio. Their opposite relative configuration at C3 and C4 was established by NOE experiments (see Supporting Information File 1). The rather complex diastereomeric mixture of spirolactams equil-12m,nA–D simplified on oxidation to partly separable mixtures of four and three diastereomers of keto lactams 13m,n, respectively. During purification of lactam 13m a major fraction was isolated in a 1.8:1 ratio, whereas the minor consisted of a 3:1 ratio, reflecting the initial 13mA,B:13mC,D ratio. The mixture of keto lactams 13nA–C was similarly separated to a 5:1 mixture of the lactams 13nA,B and the minor 13nC, respectively. This lends support to the C3–C4 trans arrangement for 13m,nA and 13m,nB as well as to the respective cis orientation in 13m,nC and 13mD. The oxidation of the hydroxy group in the bicyclic annulated racemic compound 12o reduced the number of diastereomers as expected to two, 13oA and 13oB, in a 4:1 ratio. The relative configurations of the major and the minor diastereomers were determined by NOE experiments (see Supporting Information File 1).
![[1860-5397-17-58-i5]](/bjoc/content/inline/1860-5397-17-58-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The Dess–Martin oxidation of lactams 12l–o. Conditions: a) DMP (1.3 equiv), t-BuOH (10 mol %), CH2Cl2, room temperature, 1 h.
Scheme 5: The Dess–Martin oxidation of lactams 12l–o. Conditions: a) DMP (1.3 equiv), t-BuOH (10 mol %), CH2Cl...
Further functionalization of lactams 12
Derivatizations of lactams 12 lead to valuable compound classes for further elaboration. The alkoxyamine functionality of lactam trans-12b was reductively cleaved by excess zinc in the presence of acetic acid at elevated temperature (Scheme 6); the dihydroxy lactam 14 was obtained in 82% yield. In the presence of LiAlH4 pyrrolidone trans-12b was reduced to pyrrolidine trans-15 in 95% yield, whereas no reaction was observed with DIBAL-H, NaBH4 or LiHBEt3 as the reducing agents; the starting material was typically recovered in 95% yield. However, the reduction of trans-12b by Red-Al in the presence of KOt-Bu was effective for the reduction of the lactam function to the hemiaminal followed by a nucleophilic exchange providing bicyclic hemiaminal 16 as 1:1 mixture of diastereomers in 83% yield. The stereochemical assignment of 16 is based on the relative configuration of the starting material. The N-allyl group in trans-12b can be also easily deprotected by a rhodium-catalyzed isomerization to the corresponding N-propenyl lactam followed by osmium tetroxide-catalyzed oxidative cleavage, providing lactam trans-17 in 62% yield. Additionally, the diastereomeric mixture of lactam 12o was subjected to oxidative N–O bond cleavage by mCPBA furnishing bicyclo[3.3.0]octandiones 18. The isolation of a mixture of four diastereomers as in the starting material indicates that radical coupling by TEMPO proceeds with exclusive diastereoselectivity (vide infra).
![[1860-5397-17-58-i6]](/bjoc/content/inline/1860-5397-17-58-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Selected transformations of the lactams trans-12b and 12o.
Scheme 6: Selected transformations of the lactams trans-12b and 12o.
Discussion
The sequence nucleophilic epoxide opening/Brook rearrangement/single electron transfer-induced radical oxygenation proceeds efficiently with silylacetamides 8a–g,j,k giving α,γ-dioxygenated amides 9a–p in good yields (cf. Table 2). This transformation represents a rare geminal C–C/C–O functionalization of the starting silylacetamides 8. However, neither the silyloxy group in γ-position nor the size of the N-substituent influence the diastereoselectivity of radical coupling with TEMPO (3). In contrast, the chiral N-(1-phenylethyl)- and N-(1-naphthylethyl)-substituted amides 9i–k were obtained from (S)-propylene oxide (S)-7b with moderate 3:1 anti/syn-diastereoselectivity and from (R)-propylene oxide (R)-7b with good 8:1 anti/syn-diastereoselectivity. This makes the following oxygenation course most likely (Scheme 7). Silylacetamides 8e–g exist as approximately 4:1 rotameric mixtures of (Z/E)-isomers as determined by 1H NMR spectroscopy at room temperature and ROESY investigations (see Supporting Information File 1). This corresponds to the previously reported data for N-benzylacetamides [82]. It can be assumed that the amide enolates after the Brook rearrangement and the α-amide radicals (4S,S)-19i–k and (4R,S)-19i, respectively, resulting after SET oxidation have a similarly preferred (Z)-orientation, since the environment around the amide does not change significantly during these elementary steps. A zig-zag conformation of the main chain places the bulky silyloxy and the methyl group of the 1-arylethyl unit in (4S,S)-19i–k, but the silyloxy group and the sterically more demanding phenyl ring of the 1-arylethyl group in (4R,S)-19i at the β-face shielding it in both radicals for the approach of TEMPO (3), but significantly more effectively in (4R,S)-19i (Scheme 7). The α-face is in contrast much less crowded and allows smooth radical coupling providing a good 8:1 anti/syn-diastereoselectivity for (2S,4R)-9i, but only 3:1 for (2R,4S)-9i–k. Thus, the configurations of both, the epoxide 7 and the silylacetamide 8 are important in the nucleophilic ring opening/Brook rearrangement/radical oxygenation sequence for obtaining optimal anti-diastereoselectivity. Opposite absolute configurations in both components, 7 and 8, are displaying a synergistic effect for optimal stereocontrol in the radical oxygenation step with TEMPO (3).
![[1860-5397-17-58-i7]](/bjoc/content/inline/1860-5397-17-58-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Diastereoselectivity for the formation of α-(aminoxy)amides 9i–k.
Scheme 7: Diastereoselectivity for the formation of α-(aminoxy)amides 9i–k.
A good diastereoselectivity of the oxygenation was also observed for the formation of 9l (cf. Table 2). Assuming a preferred conformation in radical 20, in which the interactions of the carbonyl group and the cyclohexane ring are minimized, the β-face at the radical center is significantly blocked by the silyloxy group hindering the approach of TEMPO (3), whereas the α-face is free for radical coupling resulting in the formation of (2R*,4S*)-9l as the major product (Scheme 8).
![[1860-5397-17-58-i8]](/bjoc/content/inline/1860-5397-17-58-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Rationalization of the diastereoselectivity for the formation of the α-(aminoxy)amide 9l.
Scheme 8: Rationalization of the diastereoselectivity for the formation of the α-(aminoxy)amide 9l.
The outcome of the thermal radical cyclizations of the dioxygenated amides 9 is dependent on the structure of the alkene unit, but the general trend is similar (Scheme 9). Amides with a terminal alkene unit furnished dioxygenated pyrrolidones 12a–f,h–j as products, amides 9g,k with trisubstituted alkene units exclusively lead to isopropenylpyrrolidones 12g,k. A similar reactivity was observed before in thermal radical cyclizations leading to cyclopentane derivatives [74,75]. All γ-silyloxy amides 9a–k cyclize in the 5-exo mode via envelope transition states 21a–k in which both the amide resonance and the resonance of the radical with the carbonyl group are disturbed [83,84]. Reactions via transition states trans-21a–k are energetically favored over the corresponding sterically more hindered cis-oriented transition states cis-21a–k, however, the diastereoselectivity remains moderate under the thermal conditions. This is in line with the previously reported radical cyclization reactions to pyrrolidones [38,85-88]. There is apparently no energy difference between the pairs of trans-21 or cis-21, thus the γ-silyloxy group exerts no influence. The cyclized radicals 22 couple subsequently with TEMPO providing lactams 12a–k after deprotection of the TMS groups. The cyclized tertiary alkoxyamines (R3 = Me) are known to be thermally labile [89-92] and consequently 4-isopropenylpyrrolidones 12g,k are isolated as the exclusive products. The cyclization of the 2-silyloxycyclohexyl-substituted amide 9l proceeds similarly providing almost equal amounts of two trans diastereomers 12l as single isomers because of the constrained conformation of radical 20, which allows a cyclization essentially only from the α-face (not shown, cf. Scheme 8).
![[1860-5397-17-58-i9]](/bjoc/content/inline/1860-5397-17-58-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Rationalization of the thermal radical cyclization diastereoselectivity of alkoxyamines 9a–k. (S)-Configuration at silyl ether function displayed as for 9i–k and 12i–k.
Scheme 9: Rationalization of the thermal radical cyclization diastereoselectivity of alkoxyamines 9a–k. (S)-C...
Amides 9m,n with cyclopent-1-en-1-ylmethyl or cyclohex-1-en-1-ylmethyl substituents on nitrogen similarly cyclize via envelope transition states trans-21m,n and cis-21m,n with preferred trans orientation of the olefin unit and the silyloxy-bearing side chain (Scheme 10). Thus, similarly as for 12a–k, two cyclized diastereomeric radicals trans-22m,n and two radicals cis-22m,n result, which differ in their orientation with respect to the racemic silyloxy group (cf. Scheme 9). The situation in the azaspiro[4,4] and azaspiro[4,5] radicals 22m,n is, however, more complex since they are prochiral, and thus eight diastereomers result. The coupling of 22m,n with TEMPO (3) occurs predominately from the more accessible β-face of the cyclopentyl or cyclohexyl radicals, since the α-face is partially blocked by the oxygenated alkyl chain at C4. However, the diastereoselectivity is also dependent on the ring size of the spirocyclic radical and proved to be better for the cyclohexyl radicals 22n, where coupling with TEMPO proceeded with a 4:1 12nA/12nB selectivity and exclusive diastereoselectivity for 12nC (cf. Scheme 3).
![[1860-5397-17-58-i10]](/bjoc/content/inline/1860-5397-17-58-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: The stereochemical course for the formation of products 12m,n by thermal radical cyclization of alkoxyamines 9m,n.
Scheme 10: The stereochemical course for the formation of products 12m,n by thermal radical cyclization of alk...
Fused lactams 12o,p were obtained with good to excellent diastereoselectivity from N-cyclopent-2-enyl or N-cyclohex-2-enyl amides 9o,p (cf. Scheme 3). The 5-exo cyclization step of radicals 21o,p proceeds with good to exclusive trans diastereoselectivity forming the azabicyclo[3.3.0]octyl or azabicyclo[4.3.0]nonyl radicals 22o,p (Scheme 11). The subsequent coupling of 22o,p with TEMPO (3) occurs exclusively from the accessible convex face of the bicyclic radicals providing the lactams 12o,p.
![[1860-5397-17-58-i11]](/bjoc/content/inline/1860-5397-17-58-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
The diastereomeric mixtures resulting from the thermal radical cyclization converge easily to the corresponding trans isomers by base-mediated isomerization reactions, if C4 is not disubstituted.
Conclusion
We developed a two-step methodology for the synthesis of diverse γ-lactam scaffolds. A tandem reaction combining nucleophilic epoxide opening by N-allylic silylacetamides, Brook rearrangement, and radical oxygenation serves for the preparation of N-allylic α-(aminoxy)-γ-(silyloxy)amides 9, which represents an oxidative C–C/C–O difunctionalization at the α-position of the amides. With correct configuration combination of chiral epoxides 7 and chiral amides 8, 2,4-dioxygenated amides 9 can be obtained with good anti-diastereoselectivity and enantioselectivity. Dioxygenated amides 9 are convenient precursors for radical 5-exo cyclization reactions based on the persistent radical effect. The N,3,4-trisubstituted lactams 12 were obtained in good yields, but with moderate trans/cis diastereoselectivity. The use of N-cycloalkenyl amides enables access to fused and spirolactams. The diastereomeric mixtures resulting from thermal radical cyclization converge to trans-3,4-disubstituted lactams by basic epimerization in 3-position of the lactam under thermodynamic conditions. The pyrrolidones can be easily further diversified by oxidation and reduction reactions. Thus, this methodology is suitable for the synthesis of functionalized γ-lactams, which can be used as building blocks for the synthesis of natural products or biologically active compounds.
Experimental
Tandem nucleophilic epoxide opening/Brook rearrangement/α-oxygenation (general procedure)
In a similar manner as described in [74]: LiCl (252 mg, 6 mmol) was added to a round-bottomed flask containing a stirring bar, which was sealed with a septum, and dried under vacuum by a heat gun. Dry THF (8 mL) and amide 8 (1.0 mmol) were added under argon. The mixture was cooled to 0 °C in an ice/water bath, sec-butyllithium (1.4 M solution in cyclohexane, 0.8 mL, 1.1 mmol) was added dropwise by a syringe, and the mixture was stirred at 0 °C for 15 min. Then, the epoxide 7 (1.05 mmol) was added at once by syringe and the reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was cooled to −78 °C, diluted with dry THF (8 mL), and TEMPO (3, 164 mg, 1.05 mmol) was added as a solid in a single portion. Ferrocenium hexafluorophosphate (4, 397 mg, 1.2 mmol) was added in small portions with vigorous stirring until a dark blue-green color of the reaction mixture persisted for 20 min. The reaction mixture was quenched by saturated NH4Cl solution (5 drops), diluted with diethyl ether (10 mL), and filtered through a pad of silica gel, which was washed with a fresh portion of diethyl ether. The filtrate was evaporated, and the crude inhomogeneous mixture was purified by flash chromatography (gradient, hexanes/EtOAc 20:1 to 5:1) to give pure α-(aminoxy)amides 9.
Thermal radical cyclization of compounds 9 and further deprotection (general procedure)
The α-(aminoxy)amide 9 (0.65 mmol) was heated in t-BuOH (6 mL) in a microwave reactor at 150 °C for 1 h. The reaction mixture was diluted with diethyl ether (5 mL), transferred into a round-bottomed Schlenk flask and evaporated. The crude residue was dissolved in THF (5 mL), the reaction mixture was cooled to 0 °C in an ice/water bath, tetrabutylammonium fluoride (1 M solution in THF, 0.96 mL, 0.96 mmol) was added and the mixture was stirred at this temperature for 30 min. The reaction was quenched by saturated NH4Cl solution, diluted with water (5 mL) and diethyl ether (5 mL), the organic layer separated, and the aqueous layer extracted with diethyl ether (2 × 5 mL). The combined organic extracts were dried over MgSO4, filtered, and evaporated. The crude mixture was purified by column chromatography (gradient, hexanes/EtOAc 5:1 to 1:1) to give pure lactams 12 as diastereomeric mixtures.
Equilibration of lactams 12 (general procedure)
A solution of KOt-Bu (1 M in THF, 0.25 mL, 0.25 mmol) was added to a stirred solution of hydroxy lactam 12 (0.50 mmol) in t-BuOH (3 mL) at room temperature or 50 °C and the reaction mixture was stirred for 24 h. The reaction was quenched by the addition of saturated NH4Cl solution and diluted with water (3 mL) and diethyl ether (5 mL). The organic layer was separated and the aqueous layer was extracted with diethyl ether (3 × 5 mL). The combined organic extracts were dried over MgSO4 and filtered. The filtrate was evaporated and the diastereomeric ratio was determined by 1H NMR spectroscopy. The crude mixture was purified by flash chromatography (gradient, hexanes/EtOAc 10:1 to 1:1) to give lactam trans-12.
Dess–Martin oxidation of hydroxylactams 12 (general procedure)
A solution of hydroxy lactam 12 or trans-12 (0.7 mmol) in dichloromethane (4 mL) was added to a stirred solution of Dess–Martin periodinane (386 mg, 0.9 mmol) and t-BuOH (0.1 mL, 1.1 mmol) in dichloromethane (4 mL) at room temperature. After 30 min, saturated Na2CO3 solution (2 mL) and saturated Na2S2O3 solution (2 mL) were added. After 5 min of vigorous stirring, the mixture was diluted with dichloromethane (10 mL), the organic layer was separated, washed with brine, dried over MgSO4, and the solvent was evaporated. The residue was purified by column chromatography (gradient, hexanes/EtOAc 10:1 to 1:1) to give keto lactam 13.
Supporting Information
| Supporting Information File 1: Experimental details and spectral data. | ||
| Format: PDF | Size: 17.8 MB | Download |
Funding
We thank the Grant Agency of the Czech Republic (16-18513S), the Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic (RVO: 61388963) and the Gilead Sciences & IOCB Research Center for generous financial support. I.C. thanks the Ministry of Education, Youth and Sports of the Czech Republic (MSM0021620857) for financial support.
References
-
Mal, D.; Shome, B.; Dinda, B. K. Pyrrole and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 187–220. doi:10.1002/9783527634880.ch6
Return to citation in text: [1] -
Kiuru, P.; Yli-Kauhaluoma, J. Pyridine and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 267–297. doi:10.1002/9783527634880.ch8
Return to citation in text: [1] -
Martins, P.; Jesus, J.; Santos, S.; Raposo, L. R.; Roma-Rodrigues, C.; Baptista, P. V.; Fernandes, A. R. Molecules 2015, 20, 16852–16891. doi:10.3390/molecules200916852
Return to citation in text: [1] -
Taylor, A. P.; Robinson, R. P.; Fobian, Y. M.; Blakemore, D. C.; Jones, L. H.; Fadeyi, O. Org. Biomol. Chem. 2016, 14, 6611–6637. doi:10.1039/c6ob00936k
Return to citation in text: [1] [2] -
Pyne, S. G.; Davis, A. S.; Gates, N. J.; Hartley, J. P.; Lindsay, K. B.; Machan, T.; Tang, M. Synlett 2004, 2670–2680. doi:10.1055/s-2004-834801
Return to citation in text: [1] [2] -
Milen, M.; Abranyi-Balogh, P.; Keglevich, G. Curr. Org. Synth. 2014, 11, 889–901. doi:10.2174/1570179411666140818210247
Return to citation in text: [1] -
Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193
Return to citation in text: [1] -
Li, J.; Ye, Y.; Zhang, Y. Org. Chem. Front. 2018, 5, 864–892. doi:10.1039/c7qo01077j
Return to citation in text: [1] -
Shibano, M.; Tsukamoto, D.; Masuda, A.; Tanaka, Y.; Kusano, G. Chem. Pharm. Bull. 2001, 49, 1362–1365. doi:10.1248/cpb.49.1362
Return to citation in text: [1] -
Bhat, C.; Tilve, S. G. RSC Adv. 2014, 4, 5405–5452. doi:10.1039/c3ra44193h
Return to citation in text: [1] -
O’Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d
Return to citation in text: [1] -
Pinder, A. R. Nat. Prod. Rep. 1986, 3, 171–180. doi:10.1039/np9860300171
Return to citation in text: [1] -
Moreira, R.; Pereira, D. M.; Valentão, P.; Andrade, P. B. Int. J. Mol. Sci. 2018, 19, 1668. doi:10.3390/ijms19061668
Return to citation in text: [1] -
Robertson, J.; Stevens, K. Nat. Prod. Rep. 2017, 34, 62–89. doi:10.1039/c5np00076a
Return to citation in text: [1] -
Ratmanova, N. K.; Andreev, I. A.; Leontiev, A. V.; Momotova, D.; Novoselov, A. M.; Ivanova, O. A.; Trushkov, I. V. Tetrahedron 2020, 76, 131031. doi:10.1016/j.tet.2020.131031
Return to citation in text: [1] -
Bronner, S. M.; Im, G.-Y. J.; Garg, N. K. Indoles and Indolizidines. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 221–265. doi:10.1002/9783527634880.ch7
Return to citation in text: [1] -
Takao, K.-i.; Aoki, S.-y.; Tadano, K.-i. J. Synth. Org. Chem., Jpn. 2007, 65, 460–469. doi:10.5059/yukigoseikyokaishi.65.460
Return to citation in text: [1] -
Lebedev, A. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2007, 43, 673–684. doi:10.1007/s10593-007-0110-1
Return to citation in text: [1] -
Nagao, Y.; Matsunaga, H.; Kumagai, T.; Inoue, Y.; Miwa, Y.; Taga, T. J. Chem. Soc., Chem. Commun. 1992, 437–439. doi:10.1039/c39920000437
Return to citation in text: [1] -
Ishibashi, H.; Sasaki, M.; Taniguchi, T. Tetrahedron 2008, 64, 7771–7773. doi:10.1016/j.tet.2008.06.005
Return to citation in text: [1] -
Yin, X.; Ma, K.; Dong, Y.; Dai, M. Org. Lett. 2020, 22, 5001–5004. doi:10.1021/acs.orglett.0c01570
Return to citation in text: [1] -
He, H.; Yang, H. Y.; Bigelis, R.; Solum, E. H.; Greenstein, M.; Carter, G. T. Tetrahedron Lett. 2002, 43, 1633–1636. doi:10.1016/s0040-4039(02)00099-0
Return to citation in text: [1] -
Gelderblom, W. C. A.; Marasas, W. F. O.; Steyn, P. S.; Thiel, P. G.; van der Merwe, K. J.; van Rooyen, P. H.; Vleggaar, R.; Wessels, P. L. J. Chem. Soc., Chem. Commun. 1984, 122–124. doi:10.1039/c39840000122
Return to citation in text: [1] -
Jang, J.-H.; Asami, Y.; Jang, J.-P.; Kim, S.-O.; Moon, D. O.; Shin, K.-S.; Hashizume, D.; Muroi, M.; Saito, T.; Oh, H.; Kim, B. Y.; Osada, H.; Ahn, J. S. J. Am. Chem. Soc. 2011, 133, 6865–6867. doi:10.1021/ja1110688
Return to citation in text: [1] -
Bloch, P.; Tamm, C.; Bollinger, P.; Petcher, T. J.; Weber, H. P. Helv. Chim. Acta 1976, 59, 133–137. doi:10.1002/hlca.19760590114
Return to citation in text: [1] -
Shibasaki, M.; Kanai, M.; Fukuda, N. Chem. – Asian J. 2007, 2, 20–38. doi:10.1002/asia.200600310
Return to citation in text: [1] -
Pilli, R. A.; Rosso, G. B.; de Oliveira, M. d. C. F. Nat. Prod. Rep. 2010, 27, 1908–1937. doi:10.1039/c005018k
Return to citation in text: [1] -
Lee, Y. S.; Kim, S. H.; Jung, S. H.; Lee, S. J.; Park, H.-K. Heterocycles 1994, 37, 303–309. doi:10.3987/com-93-s2
Return to citation in text: [1] -
Matsumoto, T.; Nakamura, S.; Nakashima, S.; Ohta, T.; Yano, M.; Tsujihata, J.; Tsukioka, J.; Ogawa, K.; Fukaya, M.; Yoshikawa, M.; Matsuda, H. J. Nat. Med. 2016, 70, 376–383. doi:10.1007/s11418-015-0963-z
Return to citation in text: [1] -
Inoue, T.; Konishi, T.; Kiyosawa, S.; Fujiwara, Y. Chem. Pharm. Bull. 1994, 42, 154–155. doi:10.1248/cpb.42.154
Return to citation in text: [1] -
Ujihara, Y.; Nakayama, K.; Sengoku, T.; Takahashi, M.; Yoda, H. Org. Lett. 2012, 14, 5142–5145. doi:10.1021/ol3024506
Return to citation in text: [1] -
Kim, K.-H.; Lee, I.-K.; Park, K.-M.; Kim, W.-K.; Lee, K.-R. Bull. Korean Chem. Soc. 2008, 29, 1591–1593. doi:10.5012/bkcs.2008.29.8.1591
Return to citation in text: [1] -
Zhang, Y.; Zhao, X.-c.; Xie, Y.-g.; Fan, C.; Huang, Y.-y.; Yan, S.-k.; Zhang, Y.; Jin, H.-z.; Zhang, W.-d. Fitoterapia 2017, 118, 80–86. doi:10.1016/j.fitote.2017.03.001
Return to citation in text: [1] -
Zhang, H.-p.; Shigemori, H.; Ishibashi, M.; Kosaka, T.; Pettit, G. R.; Kamano, Y.; Kobayashi, J. Tetrahedron 1994, 50, 10201–10206. doi:10.1016/s0040-4020(01)81752-x
Return to citation in text: [1] -
Baldwin, J. E.; Lynch, G. P.; Pitlik, J. J. Antibiot. 1991, 44, 1–24. doi:10.7164/antibiotics.44.1
Return to citation in text: [1] -
Boyd, D. B.; Elzey, T. K.; Hatfield, L. D.; Kinnick, M. D.; Morin, J. M., Jr. Tetrahedron Lett. 1986, 27, 3453–3456. doi:10.1016/s0040-4039(00)84820-0
Return to citation in text: [1] -
Coulton, S.; Southgate, R. J. Chem. Soc., Perkin Trans. 1 1992, 961–967. doi:10.1039/p19920000961
Return to citation in text: [1] -
Aszodi, J.; Rowlands, D. A.; Mauvais, P.; Collette, P.; Bonnefoy, A.; Lampilas, M. Bioorg. Med. Chem. Lett. 2004, 14, 2489–2492. doi:10.1016/j.bmcl.2004.03.004
Return to citation in text: [1] [2] -
Matsui, K.; Kan, Y.; Kikuchi, J.; Matsushima, K.; Takemura, M.; Maki, H.; Kozono, I.; Ueda, T.; Minagawa, K. J. Med. Chem. 2020, 63, 6090–6095. doi:10.1021/acs.jmedchem.0c00295
Return to citation in text: [1] -
Rivas, F.; Ling, T. Org. Prep. Proced. Int. 2016, 48, 254–295. doi:10.1080/00304948.2016.1165059
Return to citation in text: [1] -
Caruano, J.; Muccioli, G. G.; Robiette, R. Org. Biomol. Chem. 2016, 14, 10134–10156. doi:10.1039/c6ob01349j
Return to citation in text: [1] -
de Marigorta, E. M.; de los Santos, J. M.; Ochoa de Retana, A. M.; Vicario, J.; Palacios, F. Synthesis 2018, 50, 4539–4554. doi:10.1055/s-0037-1611014
Return to citation in text: [1] -
Gois, P. M. P.; Afonso, C. A. M. Eur. J. Org. Chem. 2004, 3773–3788. doi:10.1002/ejoc.200400237
Return to citation in text: [1] -
Kammerer, C.; Prestat, G.; Madec, D.; Poli, G. Acc. Chem. Res. 2014, 47, 3439–3447. doi:10.1021/ar500178n
Return to citation in text: [1] -
Ye, L.-W.; Shu, C.; Gagosz, F. Org. Biomol. Chem. 2014, 12, 1833–1845. doi:10.1039/c3ob42181c
Return to citation in text: [1] -
Kang, S.-W.; Kim, Y.-H.; Kim, H.-J.; Lee, J.-H.; Kim, S.-H. Bull. Korean Chem. Soc. 2009, 30, 691–694. doi:10.5012/bkcs.2009.30.3.691
Return to citation in text: [1] -
Holstein, P. M.; Dailler, D.; Vantourout, J.; Shaya, J.; Millet, A.; Baudoin, O. Angew. Chem., Int. Ed. 2016, 55, 2805–2809. doi:10.1002/anie.201511457
Return to citation in text: [1] -
Sofack-Kreutzer, J.; Martin, N.; Renaudat, A.; Jazzar, R.; Baudoin, O. Angew. Chem., Int. Ed. 2012, 51, 10399–10402. doi:10.1002/anie.201205403
Return to citation in text: [1] -
Pedroni, J.; Cramer, N. Angew. Chem., Int. Ed. 2015, 54, 11826–11829. doi:10.1002/anie.201505916
Return to citation in text: [1] -
Dalling, A. G.; Yamauchi, T.; McCreanor, N. G.; Cox, L.; Bower, J. F. Angew. Chem., Int. Ed. 2019, 58, 221–225. doi:10.1002/anie.201811460
Return to citation in text: [1] -
Yamazaki, S.; Niina, M.; Kakiuchi, K. Synthesis 2016, 48, 2889–2895. doi:10.1055/s-0035-1561643
Return to citation in text: [1] -
Li, Z.; Sharma, U. K.; Liu, Z.; Sharma, N.; Harvey, J. N.; Van der Eycken, E. V. Eur. J. Org. Chem. 2015, 3957–3962. doi:10.1002/ejoc.201500270
Return to citation in text: [1] -
Ghorai, M. K.; Tiwari, D. P. J. Org. Chem. 2010, 75, 6173–6181. doi:10.1021/jo101004x
Return to citation in text: [1] -
Goldberg, A. F. G.; O’Connor, N. R.; Craig, R. A., II; Stoltz, B. M. Org. Lett. 2012, 14, 5314–5317. doi:10.1021/ol302494n
Return to citation in text: [1] -
Murray, W. V.; Mishra, P. K.; Sun, S.; Maden, A. Tetrahedron Lett. 2002, 43, 7389–7392. doi:10.1016/s0040-4039(02)01711-2
Return to citation in text: [1] -
Bowman, W. R.; Bridge, C. F.; Brookes, P. J. Chem. Soc., Perkin Trans. 1 2000, 1–14. doi:10.1039/a808141g
Return to citation in text: [1] -
Bowman, W. R.; Cloonan, M. O.; Krintel, S. L. J. Chem. Soc., Perkin Trans. 1 2001, 2885–2902. doi:10.1039/a909340k
Return to citation in text: [1] -
Bowman, W. R.; Fletcher, A. J.; Potts, G. B. S. J. Chem. Soc., Perkin Trans. 1 2002, 2747–2762. doi:10.1039/b108582b
Return to citation in text: [1] -
Nagashima, H.; Wakamatsu, H.; Itoh, K. J. Chem. Soc., Chem. Commun. 1984, 652–653. doi:10.1039/c39840000652
Return to citation in text: [1] -
Nagashima, H.; Ara, K.-i.; Wakamatsu, H.; Itoh, K. J. Chem. Soc., Chem. Commun. 1985, 518–519. doi:10.1039/c39850000518
Return to citation in text: [1] -
Ishibashi, H.; Haruki, S.; Uchiyama, M.; Tamura, O.; Matsuo, J.-i. Tetrahedron Lett. 2006, 47, 6263–6266. doi:10.1016/j.tetlet.2006.05.146
Return to citation in text: [1] -
Parsons, A. F. C. R. Acad. Sci., Ser. IIc: Chim. 2001, 4, 391–400. doi:10.1016/s1387-1609(01)01255-5
Return to citation in text: [1] -
Ishibashi, H.; Sato, T.; Ikeda, M. Synthesis 2002, 695–713. doi:10.1055/s-2002-25759
Return to citation in text: [1] -
Fantinati, A.; Zanirato, V.; Marchetti, P.; Trapella, C. ChemistryOpen 2020, 9, 100–170. doi:10.1002/open.201900220
Return to citation in text: [1] -
Clark, A. J. Eur. J. Org. Chem. 2016, 2231–2243. doi:10.1002/ejoc.201501571
Return to citation in text: [1] -
Jansana, S.; Coussanes, G.; Diaba, F.; Bonjoch, J. Eur. J. Org. Chem. 2017, 2344–2352. doi:10.1002/ejoc.201700086
Return to citation in text: [1] -
Wang, C.; Russell, G. A. J. Org. Chem. 1999, 64, 2346–2352. doi:10.1021/jo9820770
Return to citation in text: [1] -
Borja-Miranda, A.; Sánchez-Chávez, A. C.; Polindara-García, L. A. Eur. J. Org. Chem. 2019, 2453–2471. doi:10.1002/ejoc.201801871
Return to citation in text: [1] -
Ueda, M.; Uenoyama, Y.; Terasoma, N.; Doi, S.; Kobayashi, S.; Ryu, I.; Murphy, J. A. Beilstein J. Org. Chem. 2013, 9, 1340–1345. doi:10.3762/bjoc.9.151
Return to citation in text: [1] -
Bella, A. F.; Jackson, L. V.; Walton, J. C. Org. Biomol. Chem. 2004, 2, 421–428. doi:10.1039/b313932h
Return to citation in text: [1] -
Lizos, D. E.; Murphy, J. A. Org. Biomol. Chem. 2003, 1, 117–122. doi:10.1039/b208114h
Return to citation in text: [1] -
Gesmundo, N. J.; Grandjean, J.-M. M.; Nicewicz, D. A. Org. Lett. 2015, 17, 1316–1319. doi:10.1021/acs.orglett.5b00316
Return to citation in text: [1] -
Okitsu, T.; Saito, M.; Tamura, O.; Ishibashi, H. Tetrahedron 2005, 61, 9180–9187. doi:10.1016/j.tet.2005.06.116
Return to citation in text: [1] -
Klychnikov, M. K.; Pohl, R.; Císařová, I.; Jahn, U. Eur. J. Org. Chem. 2020, 2854–2866. doi:10.1002/ejoc.202000126
Return to citation in text: [1] [2] [3] -
Šimek, M.; Bártová, K.; Pohl, R.; Císařová, I.; Jahn, U. Angew. Chem., Int. Ed. 2020, 59, 6160–6165. doi:10.1002/anie.201916188
Return to citation in text: [1] [2] -
Moser, W. H. Tetrahedron 2001, 57, 2065–2084. doi:10.1016/s0040-4020(00)01089-9
Return to citation in text: [1] -
Kirschning, A.; Gille, F.; Wolling, M. Brook Rearrangement as the Key Step in Domino Reactions. In Applications of Domino Transformations in Organic Synthesis 1; Snyder, S. A.; Schaumann, E., Eds.; Science of Synthesis; Thieme: Stuttgart, Germany, 2016; pp 355–448. doi:10.1055/sos-sd-219-00200
Return to citation in text: [1] -
Deng, Y.; Smith, A. B., III. Acc. Chem. Res. 2020, 53, 988–1000. doi:10.1021/acs.accounts.0c00076
Return to citation in text: [1] -
Studer, A. Angew. Chem., Int. Ed. 2000, 39, 1108–1111. doi:10.1002/(sici)1521-3773(20000317)39:6<1108::aid-anie1108>3.0.co;2-a
Return to citation in text: [1] -
Leifert, D.; Studer, A. Angew. Chem., Int. Ed. 2020, 59, 74–108. doi:10.1002/anie.201903726
Return to citation in text: [1] -
Montgomery, T. D.; Smith, A. B., III. Org. Lett. 2017, 19, 6216–6219. doi:10.1021/acs.orglett.7b03142
Return to citation in text: [1] -
De Riggi, I.; Gastaldi, S.; Surzur, J.-M.; Bertrand, M. P.; Virgili, A. J. Org. Chem. 1992, 57, 6118–6125. doi:10.1021/jo00049a014
Return to citation in text: [1] -
Nagashima, H.; Wakamatsu, H.; Ozaki, N.; Ishii, T.; Watanabe, M.; Tajima, T.; Itoh, K. J. Org. Chem. 1992, 57, 1682–1689. doi:10.1021/jo00032a016
Return to citation in text: [1] -
Miyabe, H.; Takemoto, Y. Chem. – Eur. J. 2007, 13, 7280–7286. doi:10.1002/chem.200700864
Return to citation in text: [1] -
Motoyama, Y.; Hanada, S.; Shimamoto, K.; Nagashima, H. Tetrahedron 2006, 62, 2779–2788. doi:10.1016/j.tet.2006.01.011
Return to citation in text: [1] -
Pattarozzi, M.; Roncaglia, F.; Giangiordano, V.; Davoli, P.; Prati, F.; Ghelfi, F. Synthesis 2010, 694–700. doi:10.1055/s-0029-1218583
Return to citation in text: [1] -
Parsons, A. F.; Taylor, R. J. K. J. Chem. Soc., Perkin Trans. 1 1994, 1945–1951. doi:10.1039/p19940001945
Return to citation in text: [1] -
Sato, T.; Wada, Y.; Nishimoto, M.; Ishibashi, H.; Ikeda, M. J. Chem. Soc., Perkin Trans. 1 1989, 879–886. doi:10.1039/p19890000879
Return to citation in text: [1] -
Dokli, I.; Pohl, R.; Klepetářová, B.; Jahn, U. Chem. Commun. 2019, 55, 3931–3934. doi:10.1039/c9cc00945k
Return to citation in text: [1] -
Amatov, T.; Pohl, R.; Cisařová, I.; Jahn, U. Org. Lett. 2017, 19, 1152–1155. doi:10.1021/acs.orglett.7b00187
Return to citation in text: [1] -
Xu, F.; Zhu, L.; Zhu, S.; Yan, X.; Xu, H.-C. Chem. – Eur. J. 2014, 20, 12740–12744. doi:10.1002/chem.201404078
Return to citation in text: [1] -
Gui, J.; Tian, H.; Tian, W. Org. Lett. 2013, 15, 4802–4805. doi:10.1021/ol402193b
Return to citation in text: [1]
| 38. | Aszodi, J.; Rowlands, D. A.; Mauvais, P.; Collette, P.; Bonnefoy, A.; Lampilas, M. Bioorg. Med. Chem. Lett. 2004, 14, 2489–2492. doi:10.1016/j.bmcl.2004.03.004 |
| 85. | Motoyama, Y.; Hanada, S.; Shimamoto, K.; Nagashima, H. Tetrahedron 2006, 62, 2779–2788. doi:10.1016/j.tet.2006.01.011 |
| 86. | Pattarozzi, M.; Roncaglia, F.; Giangiordano, V.; Davoli, P.; Prati, F.; Ghelfi, F. Synthesis 2010, 694–700. doi:10.1055/s-0029-1218583 |
| 87. | Parsons, A. F.; Taylor, R. J. K. J. Chem. Soc., Perkin Trans. 1 1994, 1945–1951. doi:10.1039/p19940001945 |
| 88. | Sato, T.; Wada, Y.; Nishimoto, M.; Ishibashi, H.; Ikeda, M. J. Chem. Soc., Perkin Trans. 1 1989, 879–886. doi:10.1039/p19890000879 |
| 89. | Dokli, I.; Pohl, R.; Klepetářová, B.; Jahn, U. Chem. Commun. 2019, 55, 3931–3934. doi:10.1039/c9cc00945k |
| 90. | Amatov, T.; Pohl, R.; Cisařová, I.; Jahn, U. Org. Lett. 2017, 19, 1152–1155. doi:10.1021/acs.orglett.7b00187 |
| 91. | Xu, F.; Zhu, L.; Zhu, S.; Yan, X.; Xu, H.-C. Chem. – Eur. J. 2014, 20, 12740–12744. doi:10.1002/chem.201404078 |
| 92. | Gui, J.; Tian, H.; Tian, W. Org. Lett. 2013, 15, 4802–4805. doi:10.1021/ol402193b |
| 4. | Taylor, A. P.; Robinson, R. P.; Fobian, Y. M.; Blakemore, D. C.; Jones, L. H.; Fadeyi, O. Org. Biomol. Chem. 2016, 14, 6611–6637. doi:10.1039/c6ob00936k |
| 5. | Pyne, S. G.; Davis, A. S.; Gates, N. J.; Hartley, J. P.; Lindsay, K. B.; Machan, T.; Tang, M. Synlett 2004, 2670–2680. doi:10.1055/s-2004-834801 |
| 1. | Mal, D.; Shome, B.; Dinda, B. K. Pyrrole and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 187–220. doi:10.1002/9783527634880.ch6 |
| 2. | Kiuru, P.; Yli-Kauhaluoma, J. Pyridine and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 267–297. doi:10.1002/9783527634880.ch8 |
| 3. | Martins, P.; Jesus, J.; Santos, S.; Raposo, L. R.; Roma-Rodrigues, C.; Baptista, P. V.; Fernandes, A. R. Molecules 2015, 20, 16852–16891. doi:10.3390/molecules200916852 |
| 4. | Taylor, A. P.; Robinson, R. P.; Fobian, Y. M.; Blakemore, D. C.; Jones, L. H.; Fadeyi, O. Org. Biomol. Chem. 2016, 14, 6611–6637. doi:10.1039/c6ob00936k |
| 22. | He, H.; Yang, H. Y.; Bigelis, R.; Solum, E. H.; Greenstein, M.; Carter, G. T. Tetrahedron Lett. 2002, 43, 1633–1636. doi:10.1016/s0040-4039(02)00099-0 |
| 45. | Ye, L.-W.; Shu, C.; Gagosz, F. Org. Biomol. Chem. 2014, 12, 1833–1845. doi:10.1039/c3ob42181c |
| 46. | Kang, S.-W.; Kim, Y.-H.; Kim, H.-J.; Lee, J.-H.; Kim, S.-H. Bull. Korean Chem. Soc. 2009, 30, 691–694. doi:10.5012/bkcs.2009.30.3.691 |
| 47. | Holstein, P. M.; Dailler, D.; Vantourout, J.; Shaya, J.; Millet, A.; Baudoin, O. Angew. Chem., Int. Ed. 2016, 55, 2805–2809. doi:10.1002/anie.201511457 |
| 48. | Sofack-Kreutzer, J.; Martin, N.; Renaudat, A.; Jazzar, R.; Baudoin, O. Angew. Chem., Int. Ed. 2012, 51, 10399–10402. doi:10.1002/anie.201205403 |
| 49. | Pedroni, J.; Cramer, N. Angew. Chem., Int. Ed. 2015, 54, 11826–11829. doi:10.1002/anie.201505916 |
| 50. | Dalling, A. G.; Yamauchi, T.; McCreanor, N. G.; Cox, L.; Bower, J. F. Angew. Chem., Int. Ed. 2019, 58, 221–225. doi:10.1002/anie.201811460 |
| 17. | Takao, K.-i.; Aoki, S.-y.; Tadano, K.-i. J. Synth. Org. Chem., Jpn. 2007, 65, 460–469. doi:10.5059/yukigoseikyokaishi.65.460 |
| 18. | Lebedev, A. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2007, 43, 673–684. doi:10.1007/s10593-007-0110-1 |
| 19. | Nagao, Y.; Matsunaga, H.; Kumagai, T.; Inoue, Y.; Miwa, Y.; Taga, T. J. Chem. Soc., Chem. Commun. 1992, 437–439. doi:10.1039/c39920000437 |
| 20. | Ishibashi, H.; Sasaki, M.; Taniguchi, T. Tetrahedron 2008, 64, 7771–7773. doi:10.1016/j.tet.2008.06.005 |
| 21. | Yin, X.; Ma, K.; Dong, Y.; Dai, M. Org. Lett. 2020, 22, 5001–5004. doi:10.1021/acs.orglett.0c01570 |
| 51. | Yamazaki, S.; Niina, M.; Kakiuchi, K. Synthesis 2016, 48, 2889–2895. doi:10.1055/s-0035-1561643 |
| 52. | Li, Z.; Sharma, U. K.; Liu, Z.; Sharma, N.; Harvey, J. N.; Van der Eycken, E. V. Eur. J. Org. Chem. 2015, 3957–3962. doi:10.1002/ejoc.201500270 |
| 53. | Ghorai, M. K.; Tiwari, D. P. J. Org. Chem. 2010, 75, 6173–6181. doi:10.1021/jo101004x |
| 54. | Goldberg, A. F. G.; O’Connor, N. R.; Craig, R. A., II; Stoltz, B. M. Org. Lett. 2012, 14, 5314–5317. doi:10.1021/ol302494n |
| 10. | Bhat, C.; Tilve, S. G. RSC Adv. 2014, 4, 5405–5452. doi:10.1039/c3ra44193h |
| 11. | O’Hagan, D. Nat. Prod. Rep. 2000, 17, 435–446. doi:10.1039/a707613d |
| 12. | Pinder, A. R. Nat. Prod. Rep. 1986, 3, 171–180. doi:10.1039/np9860300171 |
| 13. | Moreira, R.; Pereira, D. M.; Valentão, P.; Andrade, P. B. Int. J. Mol. Sci. 2018, 19, 1668. doi:10.3390/ijms19061668 |
| 14. | Robertson, J.; Stevens, K. Nat. Prod. Rep. 2017, 34, 62–89. doi:10.1039/c5np00076a |
| 15. | Ratmanova, N. K.; Andreev, I. A.; Leontiev, A. V.; Momotova, D.; Novoselov, A. M.; Ivanova, O. A.; Trushkov, I. V. Tetrahedron 2020, 76, 131031. doi:10.1016/j.tet.2020.131031 |
| 16. | Bronner, S. M.; Im, G.-Y. J.; Garg, N. K. Indoles and Indolizidines. In Heterocycles in Natural Product Synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 221–265. doi:10.1002/9783527634880.ch7 |
| 35. | Baldwin, J. E.; Lynch, G. P.; Pitlik, J. J. Antibiot. 1991, 44, 1–24. doi:10.7164/antibiotics.44.1 |
| 36. | Boyd, D. B.; Elzey, T. K.; Hatfield, L. D.; Kinnick, M. D.; Morin, J. M., Jr. Tetrahedron Lett. 1986, 27, 3453–3456. doi:10.1016/s0040-4039(00)84820-0 |
| 37. | Coulton, S.; Southgate, R. J. Chem. Soc., Perkin Trans. 1 1992, 961–967. doi:10.1039/p19920000961 |
| 38. | Aszodi, J.; Rowlands, D. A.; Mauvais, P.; Collette, P.; Bonnefoy, A.; Lampilas, M. Bioorg. Med. Chem. Lett. 2004, 14, 2489–2492. doi:10.1016/j.bmcl.2004.03.004 |
| 39. | Matsui, K.; Kan, Y.; Kikuchi, J.; Matsushima, K.; Takemura, M.; Maki, H.; Kozono, I.; Ueda, T.; Minagawa, K. J. Med. Chem. 2020, 63, 6090–6095. doi:10.1021/acs.jmedchem.0c00295 |
| 5. | Pyne, S. G.; Davis, A. S.; Gates, N. J.; Hartley, J. P.; Lindsay, K. B.; Machan, T.; Tang, M. Synlett 2004, 2670–2680. doi:10.1055/s-2004-834801 |
| 6. | Milen, M.; Abranyi-Balogh, P.; Keglevich, G. Curr. Org. Synth. 2014, 11, 889–901. doi:10.2174/1570179411666140818210247 |
| 7. | Felpin, F.-X.; Lebreton, J. Eur. J. Org. Chem. 2003, 3693–3712. doi:10.1002/ejoc.200300193 |
| 8. | Li, J.; Ye, Y.; Zhang, Y. Org. Chem. Front. 2018, 5, 864–892. doi:10.1039/c7qo01077j |
| 9. | Shibano, M.; Tsukamoto, D.; Masuda, A.; Tanaka, Y.; Kusano, G. Chem. Pharm. Bull. 2001, 49, 1362–1365. doi:10.1248/cpb.49.1362 |
| 40. | Rivas, F.; Ling, T. Org. Prep. Proced. Int. 2016, 48, 254–295. doi:10.1080/00304948.2016.1165059 |
| 41. | Caruano, J.; Muccioli, G. G.; Robiette, R. Org. Biomol. Chem. 2016, 14, 10134–10156. doi:10.1039/c6ob01349j |
| 42. | de Marigorta, E. M.; de los Santos, J. M.; Ochoa de Retana, A. M.; Vicario, J.; Palacios, F. Synthesis 2018, 50, 4539–4554. doi:10.1055/s-0037-1611014 |
| 43. | Gois, P. M. P.; Afonso, C. A. M. Eur. J. Org. Chem. 2004, 3773–3788. doi:10.1002/ejoc.200400237 |
| 44. | Kammerer, C.; Prestat, G.; Madec, D.; Poli, G. Acc. Chem. Res. 2014, 47, 3439–3447. doi:10.1021/ar500178n |
| 26. | Shibasaki, M.; Kanai, M.; Fukuda, N. Chem. – Asian J. 2007, 2, 20–38. doi:10.1002/asia.200600310 |
| 28. | Lee, Y. S.; Kim, S. H.; Jung, S. H.; Lee, S. J.; Park, H.-K. Heterocycles 1994, 37, 303–309. doi:10.3987/com-93-s2 |
| 25. | Bloch, P.; Tamm, C.; Bollinger, P.; Petcher, T. J.; Weber, H. P. Helv. Chim. Acta 1976, 59, 133–137. doi:10.1002/hlca.19760590114 |
| 29. | Matsumoto, T.; Nakamura, S.; Nakashima, S.; Ohta, T.; Yano, M.; Tsujihata, J.; Tsukioka, J.; Ogawa, K.; Fukaya, M.; Yoshikawa, M.; Matsuda, H. J. Nat. Med. 2016, 70, 376–383. doi:10.1007/s11418-015-0963-z |
| 30. | Inoue, T.; Konishi, T.; Kiyosawa, S.; Fujiwara, Y. Chem. Pharm. Bull. 1994, 42, 154–155. doi:10.1248/cpb.42.154 |
| 31. | Ujihara, Y.; Nakayama, K.; Sengoku, T.; Takahashi, M.; Yoda, H. Org. Lett. 2012, 14, 5142–5145. doi:10.1021/ol3024506 |
| 32. | Kim, K.-H.; Lee, I.-K.; Park, K.-M.; Kim, W.-K.; Lee, K.-R. Bull. Korean Chem. Soc. 2008, 29, 1591–1593. doi:10.5012/bkcs.2008.29.8.1591 |
| 33. | Zhang, Y.; Zhao, X.-c.; Xie, Y.-g.; Fan, C.; Huang, Y.-y.; Yan, S.-k.; Zhang, Y.; Jin, H.-z.; Zhang, W.-d. Fitoterapia 2017, 118, 80–86. doi:10.1016/j.fitote.2017.03.001 |
| 34. | Zhang, H.-p.; Shigemori, H.; Ishibashi, M.; Kosaka, T.; Pettit, G. R.; Kamano, Y.; Kobayashi, J. Tetrahedron 1994, 50, 10201–10206. doi:10.1016/s0040-4020(01)81752-x |
| 24. | Jang, J.-H.; Asami, Y.; Jang, J.-P.; Kim, S.-O.; Moon, D. O.; Shin, K.-S.; Hashizume, D.; Muroi, M.; Saito, T.; Oh, H.; Kim, B. Y.; Osada, H.; Ahn, J. S. J. Am. Chem. Soc. 2011, 133, 6865–6867. doi:10.1021/ja1110688 |
| 74. | Klychnikov, M. K.; Pohl, R.; Císařová, I.; Jahn, U. Eur. J. Org. Chem. 2020, 2854–2866. doi:10.1002/ejoc.202000126 |
| 23. | Gelderblom, W. C. A.; Marasas, W. F. O.; Steyn, P. S.; Thiel, P. G.; van der Merwe, K. J.; van Rooyen, P. H.; Vleggaar, R.; Wessels, P. L. J. Chem. Soc., Chem. Commun. 1984, 122–124. doi:10.1039/c39840000122 |
| 27. | Pilli, R. A.; Rosso, G. B.; de Oliveira, M. d. C. F. Nat. Prod. Rep. 2010, 27, 1908–1937. doi:10.1039/c005018k |
| 62. | Parsons, A. F. C. R. Acad. Sci., Ser. IIc: Chim. 2001, 4, 391–400. doi:10.1016/s1387-1609(01)01255-5 |
| 63. | Ishibashi, H.; Sato, T.; Ikeda, M. Synthesis 2002, 695–713. doi:10.1055/s-2002-25759 |
| 64. | Fantinati, A.; Zanirato, V.; Marchetti, P.; Trapella, C. ChemistryOpen 2020, 9, 100–170. doi:10.1002/open.201900220 |
| 65. | Clark, A. J. Eur. J. Org. Chem. 2016, 2231–2243. doi:10.1002/ejoc.201501571 |
| 66. | Jansana, S.; Coussanes, G.; Diaba, F.; Bonjoch, J. Eur. J. Org. Chem. 2017, 2344–2352. doi:10.1002/ejoc.201700086 |
| 55. | Murray, W. V.; Mishra, P. K.; Sun, S.; Maden, A. Tetrahedron Lett. 2002, 43, 7389–7392. doi:10.1016/s0040-4039(02)01711-2 |
| 56. | Bowman, W. R.; Bridge, C. F.; Brookes, P. J. Chem. Soc., Perkin Trans. 1 2000, 1–14. doi:10.1039/a808141g |
| 57. | Bowman, W. R.; Cloonan, M. O.; Krintel, S. L. J. Chem. Soc., Perkin Trans. 1 2001, 2885–2902. doi:10.1039/a909340k |
| 58. | Bowman, W. R.; Fletcher, A. J.; Potts, G. B. S. J. Chem. Soc., Perkin Trans. 1 2002, 2747–2762. doi:10.1039/b108582b |
| 59. | Nagashima, H.; Wakamatsu, H.; Itoh, K. J. Chem. Soc., Chem. Commun. 1984, 652–653. doi:10.1039/c39840000652 |
| 60. | Nagashima, H.; Ara, K.-i.; Wakamatsu, H.; Itoh, K. J. Chem. Soc., Chem. Commun. 1985, 518–519. doi:10.1039/c39850000518 |
| 61. | Ishibashi, H.; Haruki, S.; Uchiyama, M.; Tamura, O.; Matsuo, J.-i. Tetrahedron Lett. 2006, 47, 6263–6266. doi:10.1016/j.tetlet.2006.05.146 |
| 74. | Klychnikov, M. K.; Pohl, R.; Císařová, I.; Jahn, U. Eur. J. Org. Chem. 2020, 2854–2866. doi:10.1002/ejoc.202000126 |
| 75. | Šimek, M.; Bártová, K.; Pohl, R.; Císařová, I.; Jahn, U. Angew. Chem., Int. Ed. 2020, 59, 6160–6165. doi:10.1002/anie.201916188 |
| 83. | Nagashima, H.; Wakamatsu, H.; Ozaki, N.; Ishii, T.; Watanabe, M.; Tajima, T.; Itoh, K. J. Org. Chem. 1992, 57, 1682–1689. doi:10.1021/jo00032a016 |
| 84. | Miyabe, H.; Takemoto, Y. Chem. – Eur. J. 2007, 13, 7280–7286. doi:10.1002/chem.200700864 |
| 81. | Montgomery, T. D.; Smith, A. B., III. Org. Lett. 2017, 19, 6216–6219. doi:10.1021/acs.orglett.7b03142 |
| 82. | De Riggi, I.; Gastaldi, S.; Surzur, J.-M.; Bertrand, M. P.; Virgili, A. J. Org. Chem. 1992, 57, 6118–6125. doi:10.1021/jo00049a014 |
| 76. | Moser, W. H. Tetrahedron 2001, 57, 2065–2084. doi:10.1016/s0040-4020(00)01089-9 |
| 77. | Kirschning, A.; Gille, F.; Wolling, M. Brook Rearrangement as the Key Step in Domino Reactions. In Applications of Domino Transformations in Organic Synthesis 1; Snyder, S. A.; Schaumann, E., Eds.; Science of Synthesis; Thieme: Stuttgart, Germany, 2016; pp 355–448. doi:10.1055/sos-sd-219-00200 |
| 78. | Deng, Y.; Smith, A. B., III. Acc. Chem. Res. 2020, 53, 988–1000. doi:10.1021/acs.accounts.0c00076 |
| 79. | Studer, A. Angew. Chem., Int. Ed. 2000, 39, 1108–1111. doi:10.1002/(sici)1521-3773(20000317)39:6<1108::aid-anie1108>3.0.co;2-a |
| 80. | Leifert, D.; Studer, A. Angew. Chem., Int. Ed. 2020, 59, 74–108. doi:10.1002/anie.201903726 |
| 67. | Wang, C.; Russell, G. A. J. Org. Chem. 1999, 64, 2346–2352. doi:10.1021/jo9820770 |
| 68. | Borja-Miranda, A.; Sánchez-Chávez, A. C.; Polindara-García, L. A. Eur. J. Org. Chem. 2019, 2453–2471. doi:10.1002/ejoc.201801871 |
| 69. | Ueda, M.; Uenoyama, Y.; Terasoma, N.; Doi, S.; Kobayashi, S.; Ryu, I.; Murphy, J. A. Beilstein J. Org. Chem. 2013, 9, 1340–1345. doi:10.3762/bjoc.9.151 |
| 70. | Bella, A. F.; Jackson, L. V.; Walton, J. C. Org. Biomol. Chem. 2004, 2, 421–428. doi:10.1039/b313932h |
| 71. | Lizos, D. E.; Murphy, J. A. Org. Biomol. Chem. 2003, 1, 117–122. doi:10.1039/b208114h |
| 72. | Gesmundo, N. J.; Grandjean, J.-M. M.; Nicewicz, D. A. Org. Lett. 2015, 17, 1316–1319. doi:10.1021/acs.orglett.5b00316 |
| 73. | Okitsu, T.; Saito, M.; Tamura, O.; Ishibashi, H. Tetrahedron 2005, 61, 9180–9187. doi:10.1016/j.tet.2005.06.116 |
| 74. | Klychnikov, M. K.; Pohl, R.; Císařová, I.; Jahn, U. Eur. J. Org. Chem. 2020, 2854–2866. doi:10.1002/ejoc.202000126 |
| 75. | Šimek, M.; Bártová, K.; Pohl, R.; Císařová, I.; Jahn, U. Angew. Chem., Int. Ed. 2020, 59, 6160–6165. doi:10.1002/anie.201916188 |
© 2021 Klychnikov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)
