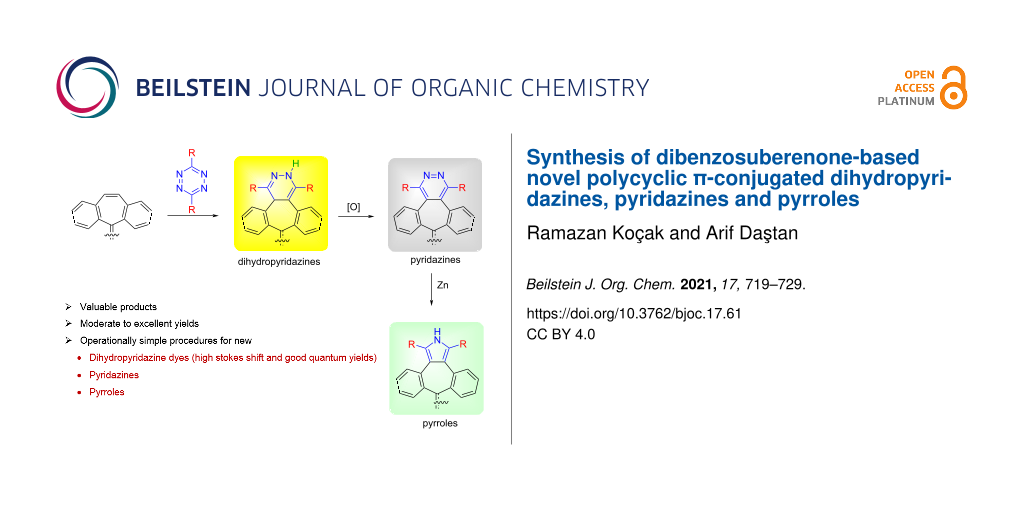
Abstract
The synthesis of novel polycyclic π-conjugated dihydropyridazines, pyridazines, and pyrroles was studied. Dihydropyridazine dyes were synthesized by inverse electron-demand Diels–Alder cycloaddition reactions between a dibenzosuberenone and tetrazines that bear various substituents. The pyridazines were synthesized in high yields by oxidation of dihydropyridazine-appended dibenzosuberenones with PIFA or NO. p-Quinone derivatives of pyridazines were also obtained by H-shift isomerization following the inverse electron-demand Diels–Alder reaction of tetrazines with p-quinone dibenzosuberenone. Then these pyridazines were converted to the corresponding pyrroles by reductive treatment with zinc. It was observed that all the dihydropyridazines obtained gave absorbance and emission at long wavelengths.

Graphical Abstract
Inroduction
Dibenzosuberone and dibenzosuberenone derivatives are commonly used for the synthesis of biologically active compounds having enzyme inhibition and antiviral activity [1,2], and are found in the structures of many commercially available antidepressant drugs [3-12]. In addition, dibenzosuberenone (1) and polyconjugated derivatives exhibit photophysical properties such as photosensitization [13], fluorescence, and aggregation-induced emission (AIE) [14-16].
π-Conjugated polycyclic hydrocarbons (CPHs) containing polycyclic heteroaromatic molecules (PHAs) and aza-polycyclic aromatic hydrocarbons (aza-PAHs) have been attracting considerable attention as they are widespread in natural products, as well as in pharmaceuticals, agrochemicals, and organic materials. Among the π-CPHs, pyridazines and pyrroles have important roles [17-22].
Although there are very few pyridazine ring-containing compounds isolated from nature (pyridazomycin, pyridazocidin, and azamerone (Figure 1)) [23-25], numerous pyridazine derivatives have been synthesized and used in a wide variety of biochemical and physicochemical applications. Examples of these applications are as drug ingredients [26-31]; for their analgesic, anticancer, antihypertensive, anti-Parkinson, anti-inflammatory, anticonvulsant, vasodilatory, antidiabetic, antitubercular, antifungal, and antibacterial activities [32-38], pH-sensing [39], OLEDs [40], chemiluminescent materials [41,42], metal complexes (Figure 1) [43-46], liquid crystal [47], and self-assembled supramolecular architectures [48].
![[1860-5397-17-61-1]](/bjoc/content/figures/1860-5397-17-61-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of dibenzosuberenone 1 and pyridazine and pyrrole derivatives.
Figure 1: Structures of dibenzosuberenone 1 and pyridazine and pyrrole derivatives.
On the other hand, many natural compounds that contain a pyrrole core, such as bilirubin, hemoglobin, chlorophyll a, and vitamin B12, are very important for life. In addition to being common in natural products and biological systems, the active ingredient of many top-selling drugs such as atorvastatin, sunitinib, and ketorolac, contains a pyrrole unit (Figure 1) [49].
Due to these unique features, the synthesis of new dihydropyridazines, pyridazines, and pyrroles, which have the potential to be used in many applications, is very important. inverse electron-demand Diels–Alder cycloaddition reactions of alkenes with tetrazines are commonly used for the synthesis of dihydropyridazines and pyridazines [50-54].
In our previous study, we made a discovery that would form the basis of a new class of dyestuffs with skeletons unlike those of classic organic dyestuffs. In that study, two highly fluorescent dibenzosuberenone-based dihydropyridazine dyes, 3a,b, were synthesized, and it was found that they can be used as a selective and sensitive sensor of fluoride anions (Scheme 1, Table 1) [55]. In another work, we reported the design, synthesis, and structural and photophysical characterization of a new series of 3,7-substituted dihydropyridazine dibenzosuberenone units with electron-withdrawing and electron-donating functional groups [56].
Herein, we report the examined impact of various electron-withdrawing and electron-donating functional groups at the 3- and 6-positions of s-tetrazine on inverse electron-demand Diels–Alder cycloaddition reactions with a dibenzosuberenone (1) and the photophysical properties of dihydropyridazines. The corresponding pyridazines and pyrroles were obtained from dihydropyridazines. Finally, we investigated the photophysical properties of dihydropyridazines.
Results and Discussion
Synthesis
In the first part of the study, we focused on the inverse electron-demand Diels–Alder cycloaddition reactions of dibenzosuberenone (1) with s-tetrazines 2a–l (Figure 2), which were synthesized according to the literature procedures (2j was synthesized by acetylation of 2i, while 2b and 2g were purchased) [57-63].
![[1860-5397-17-61-2]](/bjoc/content/figures/1860-5397-17-61-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of s-tetrazines 2a–l.
Figure 2: Structures of s-tetrazines 2a–l.
When dibenzosuberenone (1) and s-tetrazines 2a–j (1.1 equiv) were dissolved in toluene in a sealed tube and stirred at 100–125 °C for 2–48 h, a sequence of a [4 + 2]-Diels–Alder cycloaddition reaction, a retro Diels–Alder reaction of the resulting adduct, and a final 1,3-prototropic hydrogen shift took place to afford cycloadducts 3a,b [55] and 3c–f (87–96% yield) (Scheme 1 and Table 1).
![[1860-5397-17-61-i1]](/bjoc/content/inline/1860-5397-17-61-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Inverse electron-demand Diels–Alder reactions of dibenzosuberenone (1) with tetrazines 2a–l.
Scheme 1: Inverse electron-demand Diels–Alder reactions of dibenzosuberenone (1) with tetrazines 2a–l.
Table 1: Synthesis of cycloadducts 3a–l.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-61-i10.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-61-i11.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-61-i12.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-61-i13.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-61-i14.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-61-i15.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-61-i16.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-61-i17.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-61-i18.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-61-i19.svg?max-width=637&scale=1.0)
aAll reactions were carried using the compounds 1 and 2a–l (1.1 equivalents) in toluene in a sealed tube (see the Experimental section in Supporting Information File 1 for details).
As shown by the reaction conditions in Table 1, the reactions of tetrazine derivatives with electron-withdrawing groups (EWGs) 2a–f (4-pyridyl, 3,5-dimethyl-1H-pyrazol-1-yl, CONH2, CN) resulted in the formation of the target molecules 3a–f in high yields. However, the reactions of tetrazine derivatives with electron-donating groups (EDGs) 2g–j (Ph, OMe, NH2, NHAc) did not work under the same reaction conditions.
Inverse electron-demand Diels–Alder reactions are cycloadditions between electron-rich dienophiles and electron-poor dienes. EDGs raise the electron density of dienes and, in parallel, raise the LUMOdiene–HOMOdienophile energy gap, and consequently the reactivity decreases. Although the tetrazine ring is an electron-poor diene, the dibenzosuberenone (1) dienophile is not electron-rich enough. Therefore, for such cycloaddition reactions, the tetrazine must be substituted by EWGs to decrease the electron density of the diene. Consequently, our results confirm that EDGs and EWGs play a crucial role in the reactivity.
It should be noted that, unlike the other tetrazine derivatives, dichlorotetrazine (2k) afforded the addition product 3k and the oxidation product 4k as a result of its reaction with dibenzosuberenone (1). When dibromotetrazine (2l) was used, on the other hand, the oxidation product 4l and an unexpected product, dibromobenzonorbornadiene 5l, were formed (Scheme 2).
![[1860-5397-17-61-i2]](/bjoc/content/inline/1860-5397-17-61-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Inverse electron-demand Diels–Alder reactions between dibenzosuberenone 1 and tetrazines 2ka and 2lb. a5.55 mmol 1, 3.70 mmol 2k, 10 mL toluene, 120 ˚C, 48 h. b4.85 mmol 1, 0.98 mmol 2l, 100 ˚C, overnight (solvent free).
Scheme 2: Inverse electron-demand Diels–Alder reactions between dibenzosuberenone 1 and tetrazines 2ka and 2lb...
The formation of the oxidized products 4k and 4l in these reactions was probably due to the properties of tetrazines because they are known to act, in some cases, as oxidizing agents in Diels–Alder reactions, depending on the structure of the diene and the reaction conditions [64]. A proposed reaction mechanism for the formation of the dibenzosuberenone derivatives 3 and 4 is illustrated in Scheme 3. Although 3k was isolated, 3l could not be detected in the reaction mixture due to its complete conversion to 4l, revealing its ease of oxidation compared to the chloro derivative 3k. In addition, dihydrotetrazines 6k and 6l could not be isolated, as probably compound 6k decomposed completely in the reaction medium and 6l decomposed following a bromine transfer to dibenzosuberenone (1, Scheme 3).
![[1860-5397-17-61-i3]](/bjoc/content/inline/1860-5397-17-61-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed reaction mechanism for the formation of dibenzosuberenone derivatives 3 and 4.
Scheme 3: Proposed reaction mechanism for the formation of dibenzosuberenone derivatives 3 and 4.
The formation of 5l was also an unexpected result because there has been no precedent reported to date, in which a tetrazine acts as a halogen source in the halogenation of a double bond. Therefore, the formation of 5l constitutes the first example of this unusual behavior. In the proposed mechanism illustrated in Scheme 4, tetrazine 6l first tautomerizes into 7, from which dibenzosuberenone (1) receives bromine to give 5l via bromonium 8. As tetrazine 9 was unstable under the current reaction conditions, it decomposed and could not be observed [65].
![[1860-5397-17-61-i4]](/bjoc/content/inline/1860-5397-17-61-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the formation of 5l.
Scheme 4: Proposed mechanism for the formation of 5l.
In the second part of the study, dihydropyridazines 3a–f were oxidized to pyridazines. In contrast to dihydropyridazineamide 3e, the reaction of dihydropyridazines 3a–d and 3f with PIFA ([bis(trifluoroacetoxy)iodo]benzene) afforded the corresponding pyridazine derivatives 4a–d and 4f in good yields (79–95%). As a result of the reaction of PIFA with dihydropyridazine 3e, the intended pyridazine compound could not be obtained. Alternatively, nitrogen monoxide (NO) gas was used as oxidizer and pyridazineamide 4e was obtained in high yield (83%, Scheme 5).
![[1860-5397-17-61-i5]](/bjoc/content/inline/1860-5397-17-61-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Oxidation of dihydropyridazines 3a–f. All reactions were carried in CH2Cl2 at room temperature (4e: 0 °C). a1.0 equivalent PIFA, 1 h. b1.5 equivalents PIFA, overnight. c1.2 equivalents PIFA, overnight. dNitrous gases were bubbled through a solution of 3e for 1 h. e1.0 equivalent PIFA, overnight.
Scheme 5: Oxidation of dihydropyridazines 3a–f. All reactions were carried in CH2Cl2 at room temperature (4e:...
In another important part of this study, pyridazines were converted into the corresponding pyrroles via a ring contraction under reductive conditions in presence of zinc dust in acetic acid according to the Boger procedure, which is a highly reliable synthetic approach [66-68]. For pyrrole conversions, methoxycarbonyl- and 2-pyridylpyridazine derivatives 4a and 4b were used.
When methoxycarbonylpyridazine 4a reacted with 5 equivalents of Zn in acetic acid at room temperature overnight the corresponding pyrrole 10aa was obtained. When using 10 equivalents of Zn instead of 5 equivalents, hydroxypyrrole 10ab was formed, in which the carbonyl group was also reduced to the hydroxy group. Then unsubstituted pyrrole 10ac was synthesized by the reaction of pyrrole 10ab with 4 equivalents of KOH under microwave irradiation (Scheme 6).
![[1860-5397-17-61-i6]](/bjoc/content/inline/1860-5397-17-61-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of pyrrole 10a. a1.34 mmol 4a, Zinc (for 10aa: 6.68 mmol, for 10ab: 13.36 mmol), 10 mL glacial acetic acid, room temperature, overnight. b0.55 mmol 10ab, 2.20 mmol KOH, 5 mL THF/CH3OH/H2O (2:2:1) solvent mixture, 150 °C, 200 W, 2 h.
Scheme 6: Synthesis of pyrrole 10a. a1.34 mmol 4a, Zinc (for 10aa: 6.68 mmol, for 10ab: 13.36 mmol), 10 mL gl...
The reaction of 2-pyridylpyridazine 4b with Zn did not work at room temperature. Under reflux conditions compound 10ba was obtained, which contained the corresponding pyrrole and acetate structures. The acetate is formed by reducing the carbonyl group to alcohol and then reacting this alcohol with acetic acid. After hydrolysis of the acetate group with sodium hydroxide, alcohol derivate 10bb was formed. Oxidation of alcohol 10bb with MnO2 led to the formation of the corresponding ketone 10bc. Eventually, pyrrole derivative 10bc, having a carbonyl group, was obtained by these reactions (Scheme 7).
![[1860-5397-17-61-i7]](/bjoc/content/inline/1860-5397-17-61-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of pyrrole 10b. a1.21 mmol 4b, 12.10 mmol Zinc, 118 °C, 2 h. b1.13 mmol 10ba, 1.69 mmol KOH, 10 mL H2O/EtOH (1:3), room temperature, 4 h. c1.25 mmol 10bb, 12.45 mmol MnO2, 10 mL CH2Cl2, room temperature, 3 h.
Scheme 7: Synthesis of pyrrole 10b. a1.21 mmol 4b, 12.10 mmol Zinc, 118 °C, 2 h. b1.13 mmol 10ba, 1.69 mmol K...
In order to increase the conjugation of dibenzosuberenone 1 for the photophysical aspect, the p-quinone methide derivative of dibenzosuberenone 11 was synthesized according to the method in the literature [69]. The products expected from the reaction of p-quinone methide 11 with tetrazines 2a,b are dihydropyridazines 12a,b, but this molecule could not be obtained. Instead, surprisingly, products 13a,b were obtained, of which the dihydropyridazine part was oxidized to pyridazine and the p-quinone methides part was reduced to phenol. After the phenolic part of 13a,b was oxidized to p-quinone methides with PIFA, 14a and 14b were synthesized in 87% and 91% yields, respectively. Moreover, by submitting 13a,b to reductive conditions in presence of Zn, the pyridazine part of 13a,b was converted to pyrrole 15a,b. Finally, the phenolic parts of 15a,b were oxidized to p-quinone methides 16a,b with PIFA in excellent yield (89–97%, Scheme 8).
![[1860-5397-17-61-i8]](/bjoc/content/inline/1860-5397-17-61-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of p-quinone methides 13–16. a1.77 mmol 11, 1.77 mmol 2, 5 mL toluene, 80 °C (13a: overnight, 13b: 3 days). b1.11 mmol 13, 1.33 mmol PIFA, 20 mL CH2Cl2, room temperature, overnight. c1.1 mmol 13, 11.1 mmol Zinc, 10 mL glacial acetic acid, room temperature, overnight. d1.02 mmol 15a, 1.22 mmol PIFA, 20 mL CH2Cl2, at room temperature, overnight. e1.05 mmol 15b, 1.05 mmol DDQ, 20 mL CH2Cl2, room temperature, 30 min.
Scheme 8: Synthesis of p-quinone methides 13–16. a1.77 mmol 11, 1.77 mmol 2, 5 mL toluene, 80 °C (13a: overni...
For the formation of unexpected compound 13, we propose two different mechanisms. First, following the formation of phenolic tautomer 12A by tautomerization A with a [1,7]-H shift 13 would be formed. Alternatively, in the second mechanism, 13 is thought to occur by tautomerization B and a [1,5]-H shift (Scheme 9).
![[1860-5397-17-61-i9]](/bjoc/content/inline/1860-5397-17-61-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Proposed mechanism for the formation of 13.
Scheme 9: Proposed mechanism for the formation of 13.
Photophysical properties
In our previous work, we examined the photophysical and fluoride sensing properties of dihydropyridazine fluorescent dyes 3a,b in detail. In the present study, the effects of functional groups with different conjugations on maximum absorbance (λmax, abs) and emission (λmax, ems), and wavelengths of dihydropyridazines 3c–f and 3k were investigated. Compounds 3c,d, with high conjugation, show the highest absorption and emission maxima values and compounds 3f and 3k, with low conjugation, show the lowest absorption and emission maxima values (Figure 3, Figure 4, and Figure 5). All these molecules (3c–f and 3k) have Stokes shifts greater than 100 nm. The fluorescence quantum yields of 3c–f and 3k were calculated by comparison with a well-known reference, quinine sulfate, in 0.5 M H2SO4 solution (ФF = 0.546) as the standard dye (Table 2). When the UV–vis and fluorescence spectra of pyridazines and pyrroles were examined, it was seen that generally, they did not have effective absorbance or emission intensity. Although all π-conjugated pyridazines and pyrroles known in the literature did not show high emission, along with other photophysical properties the excellent coordination ability and especially their importance in biological systems always make these compounds valuable.
Table 2: Some photophysical properties of cycloadducts 3c–3f and 3k.
| Compound | λems/nma ( λexc/nm) | λabs/nm [a] | Stokes shift (nm) | Quantum yields (ФF) |
| 3c | 534 (400) | 427 | 107 | 0.78 |
| 3d | 539 (375) | 408 | 131 | 0.60 |
| 3e | 515 (360) | 393 | 122 | 0.53 |
| 3f | 487 (350) | 378 | 109 | 0.28 |
| 3k | 503 (350) | 378 | 125 | 0.16 |
ac = 5 μM (CH3CN).
![[1860-5397-17-61-3]](/bjoc/content/figures/1860-5397-17-61-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of 3c–f and 3k in CH3CN at rt (c = 5 μM).
Figure 3: UV–vis spectra of 3c–f and 3k in CH3CN at rt (c = 5 μM).
![[1860-5397-17-61-4]](/bjoc/content/figures/1860-5397-17-61-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fluorescence spectra of 3c–f and 3k in CH3CN at rt (c = 5 μM).
Figure 4: Fluorescence spectra of 3c–f and 3k in CH3CN at rt (c = 5 μM).
![[1860-5397-17-61-5]](/bjoc/content/figures/1860-5397-17-61-5.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 5: Ambient (top) and fluorescence (bottom, under 365 nm UV light) images of 3c–f and 3k in CH3CN.
Figure 5: Ambient (top) and fluorescence (bottom, under 365 nm UV light) images of 3c–f and 3k in CH3CN.
Conclusion
Novel polycyclic π-conjugated dibenzosuberenone-based dihydropyridazine dyes were synthesized by inverse electron-demand Diels–Alder cycloaddition reactions between dibenzosuberenone (1) and tetrazines bearing various substituents. These products showed long absorption wavelengths and emission bands, large Stokes shifts, and good fluorescence quantum yields. The dihydropyridazines were oxidized into pyridazines and then converted to pyrroles. Moreover, p-quinone methide derivatives of dibenzosuberenone-based pyridazines and pyrroles were synthesized. We continue intensively to perform various photochemical and biochemical studies on these compounds, which have the potential to be used in chemosensors, light harvesting, organic optoelectronics, metal coordination complexes and other photophysical applications and in biological systems.
Supporting Information
| Supporting Information File 1: Experimental procedures, copies of 1H NMR, 13C NMR, and HRMS(Q-TOF) spectra. | ||
| Format: PDF | Size: 5.5 MB | Download |
References
-
Koeberle, S. C.; Fischer, S.; Schollmeyer, D.; Schattel, V.; Grütter, C.; Rauh, D.; Laufer, S. A. J. Med. Chem. 2012, 55, 5868–5877. doi:10.1021/jm300327h
Return to citation in text: [1] -
Murray, M. A.; Babe, L. M. Antiviral Res. 1999, 44, 123–131. doi:10.1016/s0166-3542(99)00061-3
Return to citation in text: [1] -
Silver, H.; Blacker, M.; Weller, M. P. I.; Lerer, B. Biol. Psychiatry 1989, 25, 502–504. doi:10.1016/0006-3223(89)90206-0
Return to citation in text: [1] -
Andersen, J. M.; Sugerman, K. S.; Lockhart, J. R.; Weinberg, W. A. Pediatrics 1997, 100, 977–981. doi:10.1542/peds.100.6.977
Return to citation in text: [1] -
Wilens, T. E.; Biederman, J.; Geist, D. E.; Steingard, R.; Spencer, T. J. Am. Acad. Child Adolesc. Psychiatry 1993, 32, 343–349. doi:10.1097/00004583-199303000-00015
Return to citation in text: [1] -
Prince, J. B.; Wilens, T. E.; Biederman, J.; Spencer, T. J.; Millstein, R.; Polisner, D. A.; Bostic, J. Q. J. Child Adolesc. Psychopharmacol. 2000, 10, 193–204. doi:10.1089/10445460050167304
Return to citation in text: [1] -
Thompson, C. Br. J. Psychiatry 2001, 178, 99–100. doi:10.1192/bjp.178.2.99
Return to citation in text: [1] -
Buschmann, H.; Díaz, J. L.; Holenz, J.; Párraga, A.; Torrens, A.; Vela, J. M. Antidepressants, antipsychotics, anxiolytics from chemistry and pharmacology to clinical application; Wiley-VCH: Weinheim, Germany, 2007; Vol. 1 & 2. doi:10.1002/9783527619337
Return to citation in text: [1] -
Sittig, M. Pharmaceutical manufacturing encyclopedia; Noyes Publications: Park Ridge, NJ, USA, 1988.
Return to citation in text: [1] -
Paykel, E. S. Handbook of affective disorders; The Guilford Press: New York, NY, USA, 1992.
Return to citation in text: [1] -
Vaugeois, J.-M.; Corera, A. T.; Deslandes, A.; Costentin, J. Pharmacol., Biochem. Behav. 1999, 63, 285–290. doi:10.1016/s0091-3057(98)00242-1
Return to citation in text: [1] -
Chou, R.; Peterson, K.; Helfand, M. J. Pain Symptom Manage. 2004, 28, 140–175. doi:10.1016/j.jpainsymman.2004.05.002
Return to citation in text: [1] -
Bume, D. D.; Pitts, C. R.; Jokhai, R. T.; Lectka, T. Tetrahedron 2016, 72, 6031–6036. doi:10.1016/j.tet.2016.08.018
Return to citation in text: [1] -
Wang, Z.; Shao, H.; Ye, J.; Tang, L.; Lu, P. J. Phys. Chem. B 2005, 109, 19627–19633. doi:10.1021/jp053113j
Return to citation in text: [1] -
Shao, H.; Chen, X.; Wang, Z.; Lu, P. J. Phys. Chem. B 2007, 111, 10386–10396. doi:10.1021/jp073767n
Return to citation in text: [1] -
Brosius, V.; Müller, M.; Borstelmann, J.; Rominger, F.; Freudenberg, J.; Bunz, U. H. F. J. Org. Chem. 2020, 85, 296–300. doi:10.1021/acs.joc.9b02756
Return to citation in text: [1] -
Hirai, M.; Tanaka, N.; Sakai, M.; Yamaguchi, S. Chem. Rev. 2019, 119, 8291–8331. doi:10.1021/acs.chemrev.8b00637
Return to citation in text: [1] -
Shankar, M.; Ghosh, K.; Mukherjee, K.; Rit, R. K.; Sahoo, A. K. Org. Lett. 2016, 18, 6416–6419. doi:10.1021/acs.orglett.6b03314
Return to citation in text: [1] -
Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076
Return to citation in text: [1] -
Huang, R.; Phan, H.; Herng, T. S.; Hu, P.; Zeng, W.; Dong, S.-q.; Das, S.; Shen, Y.; Ding, J.; Casanova, D.; Wu, J. J. Am. Chem. Soc. 2016, 138, 10323–10330. doi:10.1021/jacs.6b06188
Return to citation in text: [1] -
Kethe, A.; Li, A.; Klumpp, D. A. Tetrahedron 2012, 68, 3357–3360. doi:10.1016/j.tet.2012.02.047
Return to citation in text: [1] -
Zheng, X.; Su, R.; Wang, Z.; Wang, T.; Bin, Z.; She, Z.; Gao, G.; You, J. Org. Lett. 2019, 21, 797–801. doi:10.1021/acs.orglett.8b04059
Return to citation in text: [1] -
Bockholt, H.; Beale, J. M.; Rohr, J. Angew. Chem., Int. Ed. Engl. 1994, 33, 1648–1651. doi:10.1002/anie.199416481
Return to citation in text: [1] -
Gerwick, B. C.; Fields, S. S.; Graupner, P. R.; Gray, J. A.; Chapin, E. L.; Cleveland, J. A.; Heim, D. R. Weed Sci. 1997, 45, 654–657.
Return to citation in text: [1] -
Wermuth, C. G. Med. Chem. Commun. 2011, 2, 935–941. doi:10.1039/c1md00074h
Return to citation in text: [1] -
Worms, P.; Kan, J.-P.; Steinberg, R.; Terranova, J.-P.; Perio, A.; Biziere, K. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989, 340, 411–418. doi:10.1007/bf00167042
Return to citation in text: [1] -
Biziere, K.; Worms, P.; Kan, J.; Mandel, P.; Garattini, S.; Roncucci, R. Drugs Exp. Clin. Res. 1985, 11, 831–840.
Return to citation in text: [1] -
McTavish, D.; Young, R. A.; Clissold, S. P. Drugs 1990, 40, 543–560. doi:10.2165/00003495-199040040-00005
Return to citation in text: [1] -
Iizawa, Y.; Okonogi, K.; Hayashi, R.; Iwahi, T.; Yamazaki, T.; Imada, A. Antimicrob. Agents Chemother. 1993, 37, 100–105. doi:10.1128/aac.37.1.100
Return to citation in text: [1] -
Aleeva, G. N.; Molodavkin, G. M.; Voronina, T. A. Bull. Exp. Biol. Med. 2009, 148, 54–56. doi:10.1007/s10517-009-0638-4
Return to citation in text: [1] -
Rezaei, Z.; Sharbaf, F. R.; Pourmojieb, M.; Youefzadeh-Fard, Y.; Motevalian, M.; Khazaeipour, Z.; Esmaeili, S. Acta Med. Iran. 2011, 701–706.
Return to citation in text: [1] -
Bansal, R.; Kumar, D.; Carron, R.; de la Calle, C. Eur. J. Med. Chem. 2009, 44, 4441–4447. doi:10.1016/j.ejmech.2009.06.006
Return to citation in text: [1] -
Islam, M.; Siddiqui, A. A.; Rajesh, R. Acta Pol. Pharm. 2008, 65, 353–362.
Return to citation in text: [1] -
Banerjee, P.; Sharma, P.; Nema, R. Int. J. ChemTech Res. 2009, 1, 522–525.
Return to citation in text: [1] -
Rathish, I. G.; Javed, K.; Bano, S.; Ahmad, S.; Alam, M. S.; Pillai, K. K. Eur. J. Med. Chem. 2009, 44, 2673–2678. doi:10.1016/j.ejmech.2008.12.013
Return to citation in text: [1] -
Rathish, I. G.; Javed, K.; Ahmad, S.; Bano, S.; Alam, M. S.; Akhter, M.; Pillai, K. K.; Ovais, S.; Samim, M. Eur. J. Med. Chem. 2012, 49, 304–309. doi:10.1016/j.ejmech.2012.01.026
Return to citation in text: [1] -
Siddiqui, A. A.; Mishra, R.; Shaharyar, M.; Husain, A.; Rashid, M.; Pal, P. Bioorg. Med. Chem. Lett. 2011, 21, 1023–1026. doi:10.1016/j.bmcl.2010.12.028
Return to citation in text: [1] -
Al-Harbi, N. O.; Bahashwan, S. A.; Shadid, K. A. J. Am. Sci. 2010, 6, 353–357.
Return to citation in text: [1] -
Do, J.; Kim, Y.; Attias, A.-J.; Kreher, D.; Kim, E. J. Nanosci. Nanotechnol. 2010, 10, 6874–6878. doi:10.1166/jnn.2010.2955
Return to citation in text: [1] -
Lincker, F.; Kreher, D.; Attias, A.-J.; Do, J.; Kim, E.; Hapiot, P.; Lemaître, N.; Geffroy, B.; Ulrich, G.; Ziessel, R. Inorg. Chem. 2010, 49, 3991–4001. doi:10.1021/ic901925w
Return to citation in text: [1] -
Atılgan, N.; Algı, F.; Önal, A. M.; Cihaner, A. Tetrahedron 2009, 65, 5776–5781. doi:10.1016/j.tet.2009.05.019
Return to citation in text: [1] -
Asil, D.; Cihaner, A.; Önal, A. M. Electrochim. Acta 2009, 54, 6740–6746. doi:10.1016/j.electacta.2009.06.053
Return to citation in text: [1] -
Yu, Q.; Zhang, A.-S.; Hu, T.-L.; Bu, X.-H. Solid State Sci. 2010, 12, 1484–1489. doi:10.1016/j.solidstatesciences.2010.06.013
Return to citation in text: [1] -
Panigati, M.; Donghi, D.; D'Alfonso, G.; Mercandelli, P.; Sironi, A.; D'Alfonso, L. Inorg. Chem. 2006, 45, 10909–10921. doi:10.1021/ic061467z
Return to citation in text: [1] -
Donghi, D.; D’Alfonso, G.; Mauro, M.; Panigati, M.; Mercandelli, P.; Sironi, A.; Mussini, P.; D’Alfonso, L. Inorg. Chem. 2008, 47, 4243–4255. doi:10.1021/ic7023692
Return to citation in text: [1] -
Beckmann, U.; Brooker, S. Coord. Chem. Rev. 2003, 245, 17–29. doi:10.1016/s0010-8545(03)00030-4
Return to citation in text: [1] -
Park, Y. S.; Kim, D.; Lee, H.; Moon, B. Org. Lett. 2006, 8, 4699–4702. doi:10.1021/ol061711q
Return to citation in text: [1] -
Cuccia, L. A.; Lehn, J.-M.; Homo, J.-C.; Schmutz, M. Angew. Chem., Int. Ed. 2000, 39, 233–237. doi:10.1002/(sici)1521-3773(20000103)39:1<233::aid-anie233>3.0.co;2-r
Return to citation in text: [1] -
Joule, J. A.; Mills, K. Heterocyclic chemistry; John Wiley & Sons: Chichester, UK, 2008.
Return to citation in text: [1] -
Saracoglu, N. Tetrahedron 2007, 63, 4199–4236. doi:10.1016/j.tet.2007.02.051
Return to citation in text: [1] -
Schoch, J.; Wiessler, M.; Jäschke, A. J. Am. Chem. Soc. 2010, 132, 8846–8847. doi:10.1021/ja102871p
Return to citation in text: [1] -
Devaraj, N. K.; Upadhyay, R.; Haun, J. B.; Hilderbrand, S. A.; Weissleder, R. Angew. Chem., Int. Ed. 2009, 48, 7013–7016. doi:10.1002/anie.200903233
Return to citation in text: [1] -
Kocak, R.; Akın, E. T.; Kalın, P.; Talaz, O.; Saracoglu, N.; Dastan, A.; Gülcin, I.; Durdagi, S. J. Heterocycl. Chem. 2016, 53, 2049–2056. doi:10.1002/jhet.2558
Return to citation in text: [1] -
Kocak, R.; Dastan, A.; Saracoglu, N. J. Heterocycl. Chem. 2018, 55, 1489–1493. doi:10.1002/jhet.3180
Return to citation in text: [1] -
Koçak, R.; Yıldız, D.; Bozkaya, U.; Daştan, A.; Bozdemir, Ö. A. Tetrahedron Lett. 2017, 58, 2981–2985. doi:10.1016/j.tetlet.2017.06.059
Return to citation in text: [1] [2] [3] [4] -
Erdoğan, M.; Daştan, A. Tetrahedron 2020, 76, 131271. doi:10.1016/j.tet.2020.131271
Return to citation in text: [1] -
Robins, L. I.; Carpenter, R. D.; Fettinger, J. C.; Haddadin, M. J.; Tinti, D. S.; Kurth, M. J. J. Org. Chem. 2006, 71, 2480–2485. doi:10.1021/jo052577a
Return to citation in text: [1] -
Farha, O. K.; Malliakas, C. D.; Kanatzidis, M. G.; Hupp, J. T. J. Am. Chem. Soc. 2010, 132, 950–952. doi:10.1021/ja909519e
Return to citation in text: [1] -
Li, Y.; Asadi, A.; Perrin, D. M. J. Fluorine Chem. 2009, 130, 377–382. doi:10.1016/j.jfluchem.2008.12.006
Return to citation in text: [1] -
Vo, H.-L.; Arthur, J. L.; Capdevila-Cortada, M.; Lapidus, S. H.; Stephens, P. W.; Novoa, J. J.; Arif, A. M.; Nagi, R. K.; Bartl, M. H.; Miller, J. S. J. Org. Chem. 2014, 79, 8189–8201. doi:10.1021/jo5014004
Return to citation in text: [1] -
Ishmetova, R. I.; Latosh, N. I.; Ganebnykh, I. N.; Ignatenko, N. K.; Tolshchina, S. G.; Rusinov, G. L. Russ. J. Org. Chem. 2009, 45, 1102–1107. doi:10.1134/s1070428009070197
Return to citation in text: [1] -
Glidewell, C.; Lightfoot, P.; Royles, B. J. L.; Smith, D. M. J. Chem. Soc., Perkin Trans. 2 1997, 1167–1174. doi:10.1039/a607646g
Return to citation in text: [1] -
Abdo, M.; Brown, S. P.; Courter, J. R.; Tucker, M. J.; Hochstrasser, R. M.; Smith, A. B., III. Org. Lett. 2012, 14, 3518–3521. doi:10.1021/ol301490h
Return to citation in text: [1] -
Benson, S. C.; Palabrica, C. A.; Snyder, J. K. J. Org. Chem. 1987, 52, 4610–4614. doi:10.1021/jo00229a034
Return to citation in text: [1] -
Degtyarenko, A. S.; Solntsev, P. V.; Krautscheid, H.; Rusanov, E. B.; Chernega, A. N.; Domasevitch, K. V. New J. Chem. 2008, 32, 1910–1918. doi:10.1039/b801231h
Return to citation in text: [1] -
Bach, N. J.; Kornfeld, E. C.; Jones, N. D.; Chaney, M. O.; Dorman, D. E.; Paschal, J. W.; Clemens, J. A.; Smalstig, E. B. J. Med. Chem. 1980, 23, 481–491. doi:10.1021/jm00179a003
Return to citation in text: [1] -
Boger, D. L.; Coleman, R. S.; Panek, J. S.; Yohannes, D. J. Org. Chem. 1984, 49, 4405–4409. doi:10.1021/jo00197a015
Return to citation in text: [1] -
Boger, D. L.; Patel, M. J. Org. Chem. 1988, 53, 1405–1415. doi:10.1021/jo00242a013
Return to citation in text: [1] -
Taljaard, B.; Taljaard, J. H.; Imrie, C.; Caira, M. R. Eur. J. Org. Chem. 2005, 2607–2619. doi:10.1002/ejoc.200400754
Return to citation in text: [1]
| 1. | Koeberle, S. C.; Fischer, S.; Schollmeyer, D.; Schattel, V.; Grütter, C.; Rauh, D.; Laufer, S. A. J. Med. Chem. 2012, 55, 5868–5877. doi:10.1021/jm300327h |
| 2. | Murray, M. A.; Babe, L. M. Antiviral Res. 1999, 44, 123–131. doi:10.1016/s0166-3542(99)00061-3 |
| 17. | Hirai, M.; Tanaka, N.; Sakai, M.; Yamaguchi, S. Chem. Rev. 2019, 119, 8291–8331. doi:10.1021/acs.chemrev.8b00637 |
| 18. | Shankar, M.; Ghosh, K.; Mukherjee, K.; Rit, R. K.; Sahoo, A. K. Org. Lett. 2016, 18, 6416–6419. doi:10.1021/acs.orglett.6b03314 |
| 19. | Stępień, M.; Gońka, E.; Żyła, M.; Sprutta, N. Chem. Rev. 2017, 117, 3479–3716. doi:10.1021/acs.chemrev.6b00076 |
| 20. | Huang, R.; Phan, H.; Herng, T. S.; Hu, P.; Zeng, W.; Dong, S.-q.; Das, S.; Shen, Y.; Ding, J.; Casanova, D.; Wu, J. J. Am. Chem. Soc. 2016, 138, 10323–10330. doi:10.1021/jacs.6b06188 |
| 21. | Kethe, A.; Li, A.; Klumpp, D. A. Tetrahedron 2012, 68, 3357–3360. doi:10.1016/j.tet.2012.02.047 |
| 22. | Zheng, X.; Su, R.; Wang, Z.; Wang, T.; Bin, Z.; She, Z.; Gao, G.; You, J. Org. Lett. 2019, 21, 797–801. doi:10.1021/acs.orglett.8b04059 |
| 49. | Joule, J. A.; Mills, K. Heterocyclic chemistry; John Wiley & Sons: Chichester, UK, 2008. |
| 14. | Wang, Z.; Shao, H.; Ye, J.; Tang, L.; Lu, P. J. Phys. Chem. B 2005, 109, 19627–19633. doi:10.1021/jp053113j |
| 15. | Shao, H.; Chen, X.; Wang, Z.; Lu, P. J. Phys. Chem. B 2007, 111, 10386–10396. doi:10.1021/jp073767n |
| 16. | Brosius, V.; Müller, M.; Borstelmann, J.; Rominger, F.; Freudenberg, J.; Bunz, U. H. F. J. Org. Chem. 2020, 85, 296–300. doi:10.1021/acs.joc.9b02756 |
| 50. | Saracoglu, N. Tetrahedron 2007, 63, 4199–4236. doi:10.1016/j.tet.2007.02.051 |
| 51. | Schoch, J.; Wiessler, M.; Jäschke, A. J. Am. Chem. Soc. 2010, 132, 8846–8847. doi:10.1021/ja102871p |
| 52. | Devaraj, N. K.; Upadhyay, R.; Haun, J. B.; Hilderbrand, S. A.; Weissleder, R. Angew. Chem., Int. Ed. 2009, 48, 7013–7016. doi:10.1002/anie.200903233 |
| 53. | Kocak, R.; Akın, E. T.; Kalın, P.; Talaz, O.; Saracoglu, N.; Dastan, A.; Gülcin, I.; Durdagi, S. J. Heterocycl. Chem. 2016, 53, 2049–2056. doi:10.1002/jhet.2558 |
| 54. | Kocak, R.; Dastan, A.; Saracoglu, N. J. Heterocycl. Chem. 2018, 55, 1489–1493. doi:10.1002/jhet.3180 |
| 13. | Bume, D. D.; Pitts, C. R.; Jokhai, R. T.; Lectka, T. Tetrahedron 2016, 72, 6031–6036. doi:10.1016/j.tet.2016.08.018 |
| 47. | Park, Y. S.; Kim, D.; Lee, H.; Moon, B. Org. Lett. 2006, 8, 4699–4702. doi:10.1021/ol061711q |
| 3. | Silver, H.; Blacker, M.; Weller, M. P. I.; Lerer, B. Biol. Psychiatry 1989, 25, 502–504. doi:10.1016/0006-3223(89)90206-0 |
| 4. | Andersen, J. M.; Sugerman, K. S.; Lockhart, J. R.; Weinberg, W. A. Pediatrics 1997, 100, 977–981. doi:10.1542/peds.100.6.977 |
| 5. | Wilens, T. E.; Biederman, J.; Geist, D. E.; Steingard, R.; Spencer, T. J. Am. Acad. Child Adolesc. Psychiatry 1993, 32, 343–349. doi:10.1097/00004583-199303000-00015 |
| 6. | Prince, J. B.; Wilens, T. E.; Biederman, J.; Spencer, T. J.; Millstein, R.; Polisner, D. A.; Bostic, J. Q. J. Child Adolesc. Psychopharmacol. 2000, 10, 193–204. doi:10.1089/10445460050167304 |
| 7. | Thompson, C. Br. J. Psychiatry 2001, 178, 99–100. doi:10.1192/bjp.178.2.99 |
| 8. | Buschmann, H.; Díaz, J. L.; Holenz, J.; Párraga, A.; Torrens, A.; Vela, J. M. Antidepressants, antipsychotics, anxiolytics from chemistry and pharmacology to clinical application; Wiley-VCH: Weinheim, Germany, 2007; Vol. 1 & 2. doi:10.1002/9783527619337 |
| 9. | Sittig, M. Pharmaceutical manufacturing encyclopedia; Noyes Publications: Park Ridge, NJ, USA, 1988. |
| 10. | Paykel, E. S. Handbook of affective disorders; The Guilford Press: New York, NY, USA, 1992. |
| 11. | Vaugeois, J.-M.; Corera, A. T.; Deslandes, A.; Costentin, J. Pharmacol., Biochem. Behav. 1999, 63, 285–290. doi:10.1016/s0091-3057(98)00242-1 |
| 12. | Chou, R.; Peterson, K.; Helfand, M. J. Pain Symptom Manage. 2004, 28, 140–175. doi:10.1016/j.jpainsymman.2004.05.002 |
| 48. | Cuccia, L. A.; Lehn, J.-M.; Homo, J.-C.; Schmutz, M. Angew. Chem., Int. Ed. 2000, 39, 233–237. doi:10.1002/(sici)1521-3773(20000103)39:1<233::aid-anie233>3.0.co;2-r |
| 39. | Do, J.; Kim, Y.; Attias, A.-J.; Kreher, D.; Kim, E. J. Nanosci. Nanotechnol. 2010, 10, 6874–6878. doi:10.1166/jnn.2010.2955 |
| 41. | Atılgan, N.; Algı, F.; Önal, A. M.; Cihaner, A. Tetrahedron 2009, 65, 5776–5781. doi:10.1016/j.tet.2009.05.019 |
| 42. | Asil, D.; Cihaner, A.; Önal, A. M. Electrochim. Acta 2009, 54, 6740–6746. doi:10.1016/j.electacta.2009.06.053 |
| 32. | Bansal, R.; Kumar, D.; Carron, R.; de la Calle, C. Eur. J. Med. Chem. 2009, 44, 4441–4447. doi:10.1016/j.ejmech.2009.06.006 |
| 33. | Islam, M.; Siddiqui, A. A.; Rajesh, R. Acta Pol. Pharm. 2008, 65, 353–362. |
| 34. | Banerjee, P.; Sharma, P.; Nema, R. Int. J. ChemTech Res. 2009, 1, 522–525. |
| 35. | Rathish, I. G.; Javed, K.; Bano, S.; Ahmad, S.; Alam, M. S.; Pillai, K. K. Eur. J. Med. Chem. 2009, 44, 2673–2678. doi:10.1016/j.ejmech.2008.12.013 |
| 36. | Rathish, I. G.; Javed, K.; Ahmad, S.; Bano, S.; Alam, M. S.; Akhter, M.; Pillai, K. K.; Ovais, S.; Samim, M. Eur. J. Med. Chem. 2012, 49, 304–309. doi:10.1016/j.ejmech.2012.01.026 |
| 37. | Siddiqui, A. A.; Mishra, R.; Shaharyar, M.; Husain, A.; Rashid, M.; Pal, P. Bioorg. Med. Chem. Lett. 2011, 21, 1023–1026. doi:10.1016/j.bmcl.2010.12.028 |
| 38. | Al-Harbi, N. O.; Bahashwan, S. A.; Shadid, K. A. J. Am. Sci. 2010, 6, 353–357. |
| 43. | Yu, Q.; Zhang, A.-S.; Hu, T.-L.; Bu, X.-H. Solid State Sci. 2010, 12, 1484–1489. doi:10.1016/j.solidstatesciences.2010.06.013 |
| 44. | Panigati, M.; Donghi, D.; D'Alfonso, G.; Mercandelli, P.; Sironi, A.; D'Alfonso, L. Inorg. Chem. 2006, 45, 10909–10921. doi:10.1021/ic061467z |
| 45. | Donghi, D.; D’Alfonso, G.; Mauro, M.; Panigati, M.; Mercandelli, P.; Sironi, A.; Mussini, P.; D’Alfonso, L. Inorg. Chem. 2008, 47, 4243–4255. doi:10.1021/ic7023692 |
| 46. | Beckmann, U.; Brooker, S. Coord. Chem. Rev. 2003, 245, 17–29. doi:10.1016/s0010-8545(03)00030-4 |
| 26. | Worms, P.; Kan, J.-P.; Steinberg, R.; Terranova, J.-P.; Perio, A.; Biziere, K. Naunyn-Schmiedeberg's Arch. Pharmacol. 1989, 340, 411–418. doi:10.1007/bf00167042 |
| 27. | Biziere, K.; Worms, P.; Kan, J.; Mandel, P.; Garattini, S.; Roncucci, R. Drugs Exp. Clin. Res. 1985, 11, 831–840. |
| 28. | McTavish, D.; Young, R. A.; Clissold, S. P. Drugs 1990, 40, 543–560. doi:10.2165/00003495-199040040-00005 |
| 29. | Iizawa, Y.; Okonogi, K.; Hayashi, R.; Iwahi, T.; Yamazaki, T.; Imada, A. Antimicrob. Agents Chemother. 1993, 37, 100–105. doi:10.1128/aac.37.1.100 |
| 30. | Aleeva, G. N.; Molodavkin, G. M.; Voronina, T. A. Bull. Exp. Biol. Med. 2009, 148, 54–56. doi:10.1007/s10517-009-0638-4 |
| 31. | Rezaei, Z.; Sharbaf, F. R.; Pourmojieb, M.; Youefzadeh-Fard, Y.; Motevalian, M.; Khazaeipour, Z.; Esmaeili, S. Acta Med. Iran. 2011, 701–706. |
| 23. | Bockholt, H.; Beale, J. M.; Rohr, J. Angew. Chem., Int. Ed. Engl. 1994, 33, 1648–1651. doi:10.1002/anie.199416481 |
| 24. | Gerwick, B. C.; Fields, S. S.; Graupner, P. R.; Gray, J. A.; Chapin, E. L.; Cleveland, J. A.; Heim, D. R. Weed Sci. 1997, 45, 654–657. |
| 25. | Wermuth, C. G. Med. Chem. Commun. 2011, 2, 935–941. doi:10.1039/c1md00074h |
| 40. | Lincker, F.; Kreher, D.; Attias, A.-J.; Do, J.; Kim, E.; Hapiot, P.; Lemaître, N.; Geffroy, B.; Ulrich, G.; Ziessel, R. Inorg. Chem. 2010, 49, 3991–4001. doi:10.1021/ic901925w |
| 57. | Robins, L. I.; Carpenter, R. D.; Fettinger, J. C.; Haddadin, M. J.; Tinti, D. S.; Kurth, M. J. J. Org. Chem. 2006, 71, 2480–2485. doi:10.1021/jo052577a |
| 58. | Farha, O. K.; Malliakas, C. D.; Kanatzidis, M. G.; Hupp, J. T. J. Am. Chem. Soc. 2010, 132, 950–952. doi:10.1021/ja909519e |
| 59. | Li, Y.; Asadi, A.; Perrin, D. M. J. Fluorine Chem. 2009, 130, 377–382. doi:10.1016/j.jfluchem.2008.12.006 |
| 60. | Vo, H.-L.; Arthur, J. L.; Capdevila-Cortada, M.; Lapidus, S. H.; Stephens, P. W.; Novoa, J. J.; Arif, A. M.; Nagi, R. K.; Bartl, M. H.; Miller, J. S. J. Org. Chem. 2014, 79, 8189–8201. doi:10.1021/jo5014004 |
| 61. | Ishmetova, R. I.; Latosh, N. I.; Ganebnykh, I. N.; Ignatenko, N. K.; Tolshchina, S. G.; Rusinov, G. L. Russ. J. Org. Chem. 2009, 45, 1102–1107. doi:10.1134/s1070428009070197 |
| 62. | Glidewell, C.; Lightfoot, P.; Royles, B. J. L.; Smith, D. M. J. Chem. Soc., Perkin Trans. 2 1997, 1167–1174. doi:10.1039/a607646g |
| 63. | Abdo, M.; Brown, S. P.; Courter, J. R.; Tucker, M. J.; Hochstrasser, R. M.; Smith, A. B., III. Org. Lett. 2012, 14, 3518–3521. doi:10.1021/ol301490h |
| 55. | Koçak, R.; Yıldız, D.; Bozkaya, U.; Daştan, A.; Bozdemir, Ö. A. Tetrahedron Lett. 2017, 58, 2981–2985. doi:10.1016/j.tetlet.2017.06.059 |
| 56. | Erdoğan, M.; Daştan, A. Tetrahedron 2020, 76, 131271. doi:10.1016/j.tet.2020.131271 |
| 69. | Taljaard, B.; Taljaard, J. H.; Imrie, C.; Caira, M. R. Eur. J. Org. Chem. 2005, 2607–2619. doi:10.1002/ejoc.200400754 |
| 65. | Degtyarenko, A. S.; Solntsev, P. V.; Krautscheid, H.; Rusanov, E. B.; Chernega, A. N.; Domasevitch, K. V. New J. Chem. 2008, 32, 1910–1918. doi:10.1039/b801231h |
| 66. | Bach, N. J.; Kornfeld, E. C.; Jones, N. D.; Chaney, M. O.; Dorman, D. E.; Paschal, J. W.; Clemens, J. A.; Smalstig, E. B. J. Med. Chem. 1980, 23, 481–491. doi:10.1021/jm00179a003 |
| 67. | Boger, D. L.; Coleman, R. S.; Panek, J. S.; Yohannes, D. J. Org. Chem. 1984, 49, 4405–4409. doi:10.1021/jo00197a015 |
| 68. | Boger, D. L.; Patel, M. J. Org. Chem. 1988, 53, 1405–1415. doi:10.1021/jo00242a013 |
| 55. | Koçak, R.; Yıldız, D.; Bozkaya, U.; Daştan, A.; Bozdemir, Ö. A. Tetrahedron Lett. 2017, 58, 2981–2985. doi:10.1016/j.tetlet.2017.06.059 |
| 64. | Benson, S. C.; Palabrica, C. A.; Snyder, J. K. J. Org. Chem. 1987, 52, 4610–4614. doi:10.1021/jo00229a034 |
| 55. | Koçak, R.; Yıldız, D.; Bozkaya, U.; Daştan, A.; Bozdemir, Ö. A. Tetrahedron Lett. 2017, 58, 2981–2985. doi:10.1016/j.tetlet.2017.06.059 |
| 55. | Koçak, R.; Yıldız, D.; Bozkaya, U.; Daştan, A.; Bozdemir, Ö. A. Tetrahedron Lett. 2017, 58, 2981–2985. doi:10.1016/j.tetlet.2017.06.059 |
© 2021 Koçak and Daştan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)