Abstract
Pyrrole is an important aromatic heterocyclic scaffold found in many natural products and predominantly used in pharmaceuticals. Continuous efforts are being made to design and synthesize various pyrrole derivatives using different synthetic procedures. Among them, the Clauson–Kaas reaction is a very old and well-known method for synthesizing a large number of N-substituted pyrroles. In recent years, due to global warming and environmental concern, research laboratories and pharmaceutical industries around the world are searching for more environmentally friendly reaction conditions for synthesizing compounds. As a result, this review describes the use of various eco-friendly greener protocols to synthesize N-substituted pyrroles. This synthesis involves the reaction of various aliphatic/aromatic primary amines, and sulfonyl primary amines with 2,5-dimethoxytetrahydrofuran in the presence of numerous acid catalysts and transition metal catalysts. The goal of this review is to summarize the synthesis of various N-substituted pyrrole derivatives using a modified Clauson–Kaas reaction under diverse conventional and greener reaction conditions.

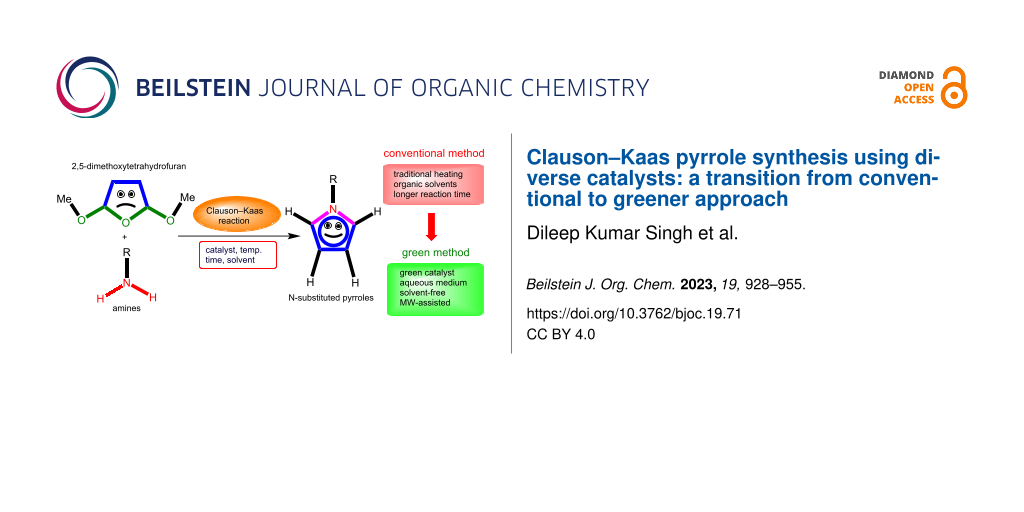
Graphical Abstract
Introduction
Heterocyclic compounds are the most explored molecules in organic chemistry in terms of their synthesis and various applications. Among the diverse heterocyclic compounds, N-containing heterocycles are found in many natural products [1-3] and biologically active molecules [4-7]. Pyrroles are a significant class of five-membered aromatic nitrogen-containing heterocyclic skeletons that have attracted much attention due to their broad spectrum of biological activity, such as anticancer [8-10], antiviral [11,12], antibacterial [13-15], antimalarial [16,17], anti-inflammatory [18,19], anti-oxidant [20-22], antifungal [23,24] and antibiotic [25,26] and as enzyme inhibitors [27,28]. Several pharmaceuticals, polymers and naturally occurring compounds, including heme, chlorophyll, vitamin B12, porphyrins, chlorins, and bacteriochlorins also contain pyrroles rings (Figure 1).
![[1860-5397-19-71-1]](/bjoc/content/figures/1860-5397-19-71-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Various pyrrole containing molecules.
Figure 1: Various pyrrole containing molecules.
For the synthesis of pyrrole derivatives, many classic methods have been used, including Knorr pyrrole synthesis [29,30], Paal–Knorr synthesis [31-33], Hantzsch pyrrole synthesis [34-36], Clauson–Kaas synthesis [37,38], Piloty–Robinson synthesis [39-41], and Barton–Zard reaction [42-44] (Scheme 1). Among these, the Clauson–Kaas pyrrole synthesis has received much attention because the synthesized pyrrole contains unsubstituted carbons that can be used for further functionalization. This methodology has been applied to diverse areas of chemistry, including natural product synthesis [45,46], medicinal chemistry [47,48], polymer chemistry [49,50] and porphyrin chemistry [51-53].
![[1860-5397-19-71-i1]](/bjoc/content/inline/1860-5397-19-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Various synthestic protocols for the synthesis of pyrroles.
Scheme 1: Various synthestic protocols for the synthesis of pyrroles.
In recent years, green chemistry has become a widely used method for organic synthesis in order to reduce energy consumption and the carbon footprint. The traditional heating techniques used to synthesize compounds are being replaced by contemporary green alternative energy systems, such as photocatalysis, microwave irradiation, ultrasonic irradiation, grinding, and ball milling processes. Furthermore, many solvent-free reactions and solid-supported reagents are becoming increasingly popular in organic synthesis. Moreover, in order to increase the product yield, different green catalysts were used in the synthesis of numerous organic and bioorganic molecules while reducing the amount of excess solvent and reaction time. The environmental impacts are manifold, including excessive utilization of organic solvents, high-temperature reactions, the production of hazardous byproducts, and by the use of traditional purification techniques. Therefore, green chemistry methods in Clauson–Kaas synthesis are desired in light of growing environmental concerns. As a result, this review will explain various green chemistry approaches to synthesize N-substituted pyrroles. Although, Wynn [54] summarized several Clauson–Kaas protocols for the synthesis of N-substituted pyrrole derivatives. However, a comprehensive review on Clauson–Kaas pyrrole synthesis has not yet been published. Therefore, a review on this topic was required, describing various reaction conditions used in the Clauson–Kaas reaction. At the end, this review is divided into two sections. The conventional Clauson-Kaas pyrrole synthesis procedure is explained in the first section, and the green protocol is covered in the second (Figure 2).
![[1860-5397-19-71-2]](/bjoc/content/figures/1860-5397-19-71-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A tree-diagram showing various conventional and green protocols for Clauson-Kaas pyrrole synthesis.
Figure 2: A tree-diagram showing various conventional and green protocols for Clauson-Kaas pyrrole synthesis.
The second section is further divided into three subsections that covers the various eco-friendly green processes used, such as, (i) reactions in water, (ii) solvent-free reactions, (iii) microwave-assisted reactions in water, solvent-free conditions and in other organic solvents.
Clauson–Kaas reaction and its mechanism
The Clauson–Kaas reaction refers to the synthesis of various N-substituted pyrroles via an acid-catalyzed reaction between aromatic or aliphatic primary amines and 2,5-dialkoxytetrahydrofuran. This reaction was originally discovered by N. Clauson–Kaas and Z. Tyle in 1952 [37] (Scheme 2a). Initially, acetic acid was used as a catalyst in this classic reaction; however, diverse modifications have been reported for this procedure using various Brønsted acid catalysts, metal catalysts, and nanoorganocatalysts. Various solvent systems, such as aqueous conditions, different organic solvents, solvent-free conditions, ionic liquids, and DESs, have been reported in modified Clauson–Kaas reactions at room temperature, under thermal and microwave-assisted conditions. In the Clauson–Kaas reaction mechanism proposed by Wang [55] (Scheme 2b), 2,5-dimethoxytetrahydrofuran (2) is first protonated with acetic acid, followed by ring opening to form carbocation B. In the following step, primary amine 1 nucleophilically attacks carbocation B to produce intermediate C, which, after proton rearrangement and the removal of methanol, produces intermediate E.
![[1860-5397-19-71-i2]](/bjoc/content/inline/1860-5397-19-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A general reaction of Clauson–Kaas pyrrole synthesis and proposed mechanism.
Scheme 2: A general reaction of Clauson–Kaas pyrrole synthesis and proposed mechanism.
In the next steps, the ring closure of intermediate E due to the lone pair of nitrogen gives F, which after the elimination of methanol and water in subsequent steps provides intermediate H. Finally, an aromatic N-substituted pyrrole 3 is produced by basic work-up of intermediate H (Scheme 2b).
Review
Conventional method for the Clauson–Kaas synthesis of N-substituted pyrroles
This section describes Clauson–Kaas pyrrole syntheses using traditional methods, such as Brønsted acid or Lewis acid-catalyzed reactions in various organic solvents at higher temeperatures. In 2000, the application of the Clauson–Kaas reaction was nicely explored by Sonnet et al. [56] for the preparation of diverse pyrrolizinones. The pyrrole derivatives 5 and 7 were synthesized by the classical Clauson–Kaas reaction by refluxing amines 4 or 6 with 2,5-dimethoxytetrahydrofuran (2) in acetic acid, affording ethyl arylpyrrolylpropionates 5 in 59–95% and pyrrole 7 in 78% yield. Representative examples of compounds 5 are shown in Scheme 3. Furthermore, these N-substituted pyrroles were used to prepare various aryl-substituted pyrrolizine and indolizine derivatives.
![[1860-5397-19-71-i3]](/bjoc/content/inline/1860-5397-19-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: AcOH-catalyzed synthesis of pyrroles 5 and 7.
Scheme 3: AcOH-catalyzed synthesis of pyrroles 5 and 7.
In 2014, Kumar and co-workers [57] synthesized various N-substituted pyrrole derivatives 9 using the Clauson–Kaas reaction to study their structure–reactivity relationship (SRR). These compounds were synthesized in 15–90% yields from the reaction between various aliphatic and aromatic amines 8 and 2,5-DMTHF (2) in the presence of NaOAc under an AcOH/H2O mixture at 75 °C in 2.5 h (Scheme 4). Further, these synthesized N-α-substituted compounds were subjected to electropolymerization studies.
![[1860-5397-19-71-i4]](/bjoc/content/inline/1860-5397-19-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N-substituted pyrroles 9.
Scheme 4: Synthesis of N-substituted pyrroles 9.
In another method described for the synthesis of N-substituted pyrroles 11, Fang et al. [58] used phosphorus pentoxide (P2O5) as a catalyst for the conversion of various aliphatic amines, aromatic amines, sulfonamides and primary amides into N-substituted pyrroles (Scheme 5). These pyrroles were synthesized in 46–100% yields by the modified Clauson–Kaas reaction between amines 10 and 2,5-DMTHF (2) in the presence of P2O5 under toluene at 110 °C. Since phosphorus pentoxide gives phosphoric acid esters upon reaction with alcohols and also has less acidic character, the authors hypothesized that it might be a good choice for the conversion of amines 10 into their corresponding pyrroles. The results were according to their expectation thus, they synthesized 13 derivatives of pyrroles in good yields in short reaction times (10–45 min). It has been observed that aromatic amines and amides take a longer time compared to the primary amines and sulfonamides.
![[1860-5397-19-71-i5]](/bjoc/content/inline/1860-5397-19-71-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: P2O5-catalyzed synthesis of N-substituted pyrroles 11.
Scheme 5: P2O5-catalyzed synthesis of N-substituted pyrroles 11.
In another study, Rochais et al. [59] reported in 2004 the synthesis of arylpyrrolo- and pyrazolopyrrolizinones, in which the Clauson–Kaas reaction was used as key step for the preparation of pyrrole derivatives 13. The condensation of amines 12 with 2,5-DMTHF (2) was carried out in the presence of p-chloropyridine hydrochloride as catalyst and dioxane as reaction solvent at 100 °C (Scheme 6). Finally, compounds 13 were used to prepare various arylpyrrolo- and pyrazolopyrrolizinones for diverse biological activity.
![[1860-5397-19-71-i6]](/bjoc/content/inline/1860-5397-19-71-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: p-Chloropyridine hydrochloride-catalyzed synthesis of pyrroles 13.
Scheme 6: p-Chloropyridine hydrochloride-catalyzed synthesis of pyrroles 13.
In 2007, Török and co-workers [60] reported a convenient one-pot synthesis of N-sulfonyl-substituted pyrroles, indoles, and carbazole via a modified Clauson–Kaas reaction in a successive cyclization/annulation process from commercially available sulfonamides 14 in the presence of trifluomethanesulfonic acid (TfOH) as Brønsted-acid catalyst. This procedure produces only N-substituted products and preserves other positions open for further functionalization. The formation of the desired products depends on the amount of triflic acid used in the reaction. Using 0.05 equivalents triflic acid gives N-sulfonylpyrroles 15 in 80–92% yields, 1.0 equivalent of triflic acid provides N-sulfonylindole 16 in 75–91% yields and 3.5 equivalents of triflic acid gives N-sulfonylcarbazole 17 in 75–86% yields as shown in Scheme 7. This procedure provides the selectivities in a very short time and gives products in excellent yields.
![[1860-5397-19-71-i7]](/bjoc/content/inline/1860-5397-19-71-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: TfOH-catalyzed synthesis of N-sulfonylpyrroles 15, N-sulfonylindole 16, N-sulfonylcarbazole 17.
Scheme 7: TfOH-catalyzed synthesis of N-sulfonylpyrroles 15, N-sulfonylindole 16, N-sulfonylcarbazole 17.
Zuo et al. [61] reported the Clauson–Kaas synthesis of N-substituted pyrroles 19 using scandium triflate as the catalyst in good to excellent yields. To obtain the best reaction conditions, various Lewis acid catalysts (e.g., FeCl3 CuCl2, InCl3, Cu(OTf)2, Mg(OTf)2, Zn(OTf)2, Yb(OTf)3, Y(OTf)3, Bi(OTf)3, La(OTf)3 and Sc(OTf)3), different solvents (e.g., CH2Cl2, CHCl3, CH3CN, CH3NO2, n-hexane, and dioxane), temperatures (90–110 °C), and the catalyst loadings have been investigated. After optimizing the reaction conditions, 3 mol % of Sc(OTf)3 as catalyst, 1,4-dioxane as solvent, and a temperature of 100 °C were selected. Finally, using these reaction conditions, 16 examples of N-substituted pyrroles were synthesized by the reaction between various aromatic, sulfonyl- and aroylamines 18 with 2,5-DMTHF (2) in yields of 74–95% as shown in Scheme 8. Among various methods available for the synthesis of N-substituted pyrroles, this protocol requires less reaction time, mild reaction conditions, easy workup, and provides improved yields of the products. Furthermore, this method shows good functional group tolerance, because various aromatic amines, benzamides, and sulfonamides were successfully used as substrates in these transformations. In this study, it was found that the reactions of aromatic amines bearing electron-withdrawing groups or electron-donating groups proceed smoothly without showing any significant substituent effect. Also, there was no effect of the substituent on yields and reaction time.
![[1860-5397-19-71-i8]](/bjoc/content/inline/1860-5397-19-71-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Scandium triflate-catalyzed synthesis of N-substituted pyrroles 19.
Scheme 8: Scandium triflate-catalyzed synthesis of N-substituted pyrroles 19.
In 2011, Zhang and Shi [62] reported the Clauson–Kaas synthesis of N-substituted pyrroles 21 through the reaction between various substituted anilines, primary arylamides, and sufonylamides 20 and 2,5-DMTHF (2) in the presence of 10 mol % MgI2 etherate in MeCN at 80 °C (Scheme 9a). MgI2 etherate is a main-group Lewis acid catalyst that selectively activates electron-rich aromatic amines. This is a mild, efficient, and highly chemoselective procedure in which the iodine counterion and MeCN played key roles in the unique reactivity of this catalytic system. To optimize the reaction conditions, many catalysts, solvents, and temperatures were studied and finally, 10 mol % MgI2∙(OEt2)n as the catalyst, CH3CN as the solvent, and 80 °C were selected and used for the synthesis of 20 examples of N-substituted pyrroles in yields of 62–98%. In this study, it was observed that anilines with electron‐withdrawing groups (i.e., NO2, CF3, F, Cl, Br) deactivated arylamine and gave the corresponding pyrroles in moderate yields, while anilines with electron‐donating groups (i.e., OMe, Me) reacted much faster than aniline and furnished the pyrroles in excellent yields. In addition, the reaction of more sterically hindered aniline with 2,5-dimethoxytetrahydrofuran gave moderate yields and less sterically hindered aniline gave good yields. This reactivity of the aromatic amine depends on the electron density of the amino compounds.
![[1860-5397-19-71-i9]](/bjoc/content/inline/1860-5397-19-71-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: MgI2 etherate-catalyzed synthesis and proposed mechanism of N-arylpyrrole derivatives 21.
Scheme 9: MgI2 etherate-catalyzed synthesis and proposed mechanism of N-arylpyrrole derivatives 21.
In addition, the authors also performed the reaction of aliphatic amines with 2,5-DMTHF (2), and found that aliphatic amines are inert in the presence of MgI2 etherate. The proposed mechanism shown in Scheme 9b suggests the ability of MgI2 etherate to act as a Lewis acid activator. The iodine counterion is coordinated to the Lewis basic oxygen atom of the acetal group to give the more Lewis acidic cataonic Mg-coordinated intermediate A. Intermediate A upon nucleophilic reaction with amines 20 yields B, which upon subsequent removal of MeOH, dehydration and aromatization affords N-substituted pyrroles 21.
In 2013, Chatzopoulou [63] and co-workers reported a high-yielding Clauson–Kaas pyrrolyl-phenol synthesis using nicotinamide, which is a cheap and nontoxic catalyst and a vitamin and enzyme cofactor. The authors chose nicotinamide as the catalyst for this study because it has a pKa of 3.43 and could act as a chemical antioxidant. In some cases, aminophenol hydrochloride is the only form of the corresponding aminophenol that can be isolated, so use of aminophenol hydrochloride as the starting material is also an important aspect of this protocol. The N-substituted pyrrole derivatives 23 were synthesized in 63–77% yields by the Clauson–Kaas reaction between various aminophenol hydrochlorides 22 and 2,5-dimethoxytetrahydrofuran (2) in the presence of an equimolar amount of nicotinamide in 1,4-dioxane under reflux conditions (Scheme 10).
![[1860-5397-19-71-i10]](/bjoc/content/inline/1860-5397-19-71-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Nicotinamide catalyzed synthesis of pyrroles 23.
Scheme 10: Nicotinamide catalyzed synthesis of pyrroles 23.
Green chemistry approach for the Clauson–Kaas synthesis of N-substituted pyrroles
This section describes the Clauson-Kaas pyrrole syntheses using different greener methods in the presence of various Brønsted acids or Lewis acids. These include reactions in aqueous media, under solvent-free conditions, and under microwave irradiation.
(1) Reactions in water
In recent years, organic reactions in aqueous medium have grown in popularity due to their low cost and environmental friendliness. The use of water as a solvent medium for the synthesis of pyrrole derivatives using various catalysts is described here. Ghafuri and Emami [64] reported a simple, efficient, and green Clauson–Kaas method for the construction of N-substituted pyrrole derivatives 25 in 70–98% yields through the reaction between aryl-/alkyl-, sulfonyl-, and acylamines 24 and 2,5-dimethoxytetrahydrofuran (2) in the presence of 4 mol % ZrOCl2∙8H2O as a catalyst in water at 60 °C (Scheme 11a). Among many catalysts used for the synthesis of N-substituted pyrroles, Zr(IV) compounds, especially ZrOCl2∙8H2O have received more attention because of its high coordination ability, low toxicity, low cost and high activity. In addition, this procedure has many other advantages such as simple experimental work-up, easy availability of reagents, high to excellent yields, and environmental friendliness of the catalyst. To optimize the reaction conditions, the authors screened different solvents, catalyst loading, and temperature. The best result was obtained when the model reaction was run at 60 °C for 30 min in the presence of 5 mol % of ZrOCl2∙8H2O catalyst in water as a solvent. Among the different solvents used, water turned out to be the best solvent because it takes a short time and gives a higher yield of product compared to other organic solvents. In addition, the use of water as the green solvent for this conversion contributed more to the development of a green reaction. The proposed mechanism is depicted in Scheme 11b, in which the methoxy group of 2,5-DMTHF (2) is opened by the deprotection in acidic medium and forms an unstable intermediate that easily forms activated dialdehyde B. The amine 24 reacts with activated dialdehyde and provides the corresponding pyrrole 25 through the removal of water molecules.
![[1860-5397-19-71-i11]](/bjoc/content/inline/1860-5397-19-71-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: ZrOCl2∙8H2O catalyzed synthesis and proposed mechanism of pyrrole derivatives 25.
Scheme 11: ZrOCl2∙8H2O catalyzed synthesis and proposed mechanism of pyrrole derivatives 25.
Smith and co-workers [65] reported a modified one-pot, two-step Clauson–Kaas procedure for the synthesis of various acid- or heat-sensitive N-substituted pyrrole derivatives 27. In this method, 2,5-dimethoxytetrahydrofuran undergoes mild hydrolysis in water to provide an activated species that reacts with various primary amines 26 in the presence of acetate buffer solution at ambient temperature to afford various N-substituted pyrroles 27 in 89–94% yields (Scheme 12). A major advantage of this protocol is that in the case of chiral amines, pyrrole formation proceeds without detectable epimerization.
![[1860-5397-19-71-i12]](/bjoc/content/inline/1860-5397-19-71-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: AcONa catalyzed synthesis of N-substituted pyrroles 27.
Scheme 12: AcONa catalyzed synthesis of N-substituted pyrroles 27.
In 2013 Azizi et al. [66] have demonstrated a simple and environmentally friendly protocol for the synthesis of N-substituted pyrroles 29 in the yields of 85–97% using squaric acid as catalyst. In this reaction, various amines 28 and 2,5-DMTHF (2) were reacted in greener solvents like water, deep eutectic solvent (DEC) and polyethylene glycol (PEG) under thermal or ultrasonic irradiation (Scheme 13a).
![[1860-5397-19-71-i13]](/bjoc/content/inline/1860-5397-19-71-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Squaric acid-catalyzed synthesis and proposed mechanism of N-substituted pyrroles 29.
Scheme 13: Squaric acid-catalyzed synthesis and proposed mechanism of N-substituted pyrroles 29.
The function of squaric acid as a catalyst was not clear, but the authors suggested that the Brønsted acidity of squaric acid affects the reactivity and selectivity of this process. The tentative mechanism of this protocol was proposed (Scheme 13b), in which a reversible acid–base reaction of aniline 28 with squaric acid afforded anilinium squarate salt A. Further, a catalytic amount of squaric acid hydrolyzes 2,5-dimethoxytetrahydrofuran (2) to give a 1,4-dicarbonyl compound B in water. Finally, N-phenylpyrrole 29 was obtained by condensation of activated 1,4-dicarbonyl compound with aniline.
In 2013, Zhang and co-workers [67] reported the preparation of a new recyclable magnetic nanoparticle-supported antimony catalyst (γ-Fe2O3@SiO2-Sb-IL) and its application in the filtration-free, Clauson–Kaas synthesis of N-substituted pyrroles. This catalyst is fairly easy to make, air stable, and magnetically recoverable by simple magnetic decantation. In addition, the catalytic activity of the catalyst remains unaltered after six consecutive cycles (Figure 3). Using this catalyst, various nitrogen-substituted pyrrole derivatives 31 were synthesized in 55–96% yields through the reaction between various amines 30 and 2,5-dimethoxytetrahydrofuran (2) under aqueous conditions (Scheme 14). Among various solvents used to optimize the reaction conditions, H2O proved to be the best solvent for these transformations.
![[1860-5397-19-71-3]](/bjoc/content/figures/1860-5397-19-71-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Reusability of catalyst γ-Fe2O3@SiO2-Sb-IL in six cycles.
Figure 3: Reusability of catalyst γ-Fe2O3@SiO2-Sb-IL in six cycles.
![[1860-5397-19-71-i14]](/bjoc/content/inline/1860-5397-19-71-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Magnetic nanoparticle-supported antimony catalyst used in the synthesis of N-substituted pyrroles 31.
Scheme 14: Magnetic nanoparticle-supported antimony catalyst used in the synthesis of N-substituted pyrroles 31...
Azizi and co-workers [68] described a one step, economical, and green method for the synthesis of N-substituted pyrroles using iron(III) chloride as a Lewis acid catalyst. These nitrogen-substituted pyrroles 33 were obtained in 74–98% yields by the reaction between various alkyl-, aryl-, sulfonyl- and aroylamines 32 with 2,5-DMTHF (2) in the presence of 2 mol % FeCl3∙7H2O as catalyst under H2O at 60 °C (Scheme 15). Among the different solvents used to optimize the reaction conditions, H2O turned out to be a better and greener solvent compared to other organic solvents (e.g., MeCN, C6H6, CH2Cl2, THF, EtOH, EtOAc).
![[1860-5397-19-71-i15]](/bjoc/content/inline/1860-5397-19-71-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Iron(III) chloride-catalyzed synthesis of N-substituted pyrroles 33.
Scheme 15: Iron(III) chloride-catalyzed synthesis of N-substituted pyrroles 33.
Deng et al. [69] brilliantly described an expedient copper-catalyzed Clauson–Kaas synthesis of a wide range of N-substituted pyrroles 35 in excellent yields of 71–96% by the reaction between various amines 34 and 2,5-dimethoxytetrahydrofuran (2) in water at reflux conditions (Scheme 16a). This protocol requires an easy workup, an inexpensive catalyst, mild reaction conditions, and simple operation in a more environmentally benign environment. Among various catalysts (CuO, CuSO4, Cu(OAc)2, CuF2, CuBr2, CuCl, CuI, Cu2O, CuCl2), solvents (THF, EtOAc, EtOH, CH2Cl2, CH3CN, H2O) and amount of catalyst loading (5, 10, 15 mol %) to optimize the reaction conditions, CuCl2 as the catalyst, H2O as solvent, and 10 mol % catalyst loading were chosen for the synthesis of various N-substituted pyrrole products. Interestingly, the Clauson–Kaas reaction of 2,5-DMTHF (2) with m-phenylenediamine or p-phenylenediamine proceed readily to afford the corresponding N-substituted monopyrroles and bispyrroles in good yields and high selectivity. To control the selectivity for the synthesis of mono- and bispyrrole-containing compounds, two different reaction conditions have been used. One equivalent of 2,5-DMTHF (2) was used for the synthesis of monopyrroles under reflux conditions for 2 hours, whereas two equivalents of 2,5-DMTHF were needed for the synthesis of bispyrrole under 8 hours reflux conditions. During this study, it was found that electron-donating groups on the phenyl ring of aromatic amines favor the formation of the corresponding product in excellent yields, whereas the electron-withdrawing groups of the aromatic amine slightly decreases the reactivity of the substrate and requires more time for the formation of products. The authors proposed a tentative mechanism for the formation of N-substituted pyrroles as shown in Scheme 16b. In the first step, intermediate B is formed by the hydrolysis of 2,5-DMTHF (2), which provide intermediate C after the removal of methanol in the presence of CuCl2. In the next steps, nucleophilic addition reaction of amines 34 with intermediate C, dehydration, and intramolecular aromatization affords N-substituted pyrroles 35.
![[1860-5397-19-71-i16]](/bjoc/content/inline/1860-5397-19-71-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Copper-catalyzed Clauson–Kaas synthesis and mechanism of pyrroles 35.
Scheme 16: Copper-catalyzed Clauson–Kaas synthesis and mechanism of pyrroles 35.
In 2018, Patil and Kumar [70] demonstrated a mild and environmentally friendly biomimetic Clauson–Kaas synthesis of N-substituted pyrroles 37 through the reaction between various arylamines 36 and 2,5-DMTHF (2) using a sustainable catalyst β-cyclodextrin-SO3H in the nontoxic green solvent H2O without the formation of side products (Scheme 17a). In this protocol, cyclodextrin (CD) plays a dual role as an acid catalyst as well as a phase-transfer agent that facilitates the smooth conversion of reactants into products. Most importantly, this catalyst can be easily recovered and used again for up to five cycles (Figure 4), with almost no effect on product yield and no loss of catalytic activity. Moreover, a plausible mechanism for the constructions of N-arylpyrroles is shown in Scheme 17b. In the first step, the acetal group of 2,5-DMTHF (2) is protonated by β-CD-SO3H to produce intermediate A. Intermediate A is nucleophilically attacked by a water molecule to form intermediate B. Further, dehydration and protonation leads to the formation of intermediate E. Nucleophilic attack of amine 36 on E then affords intermediate F. Finally, protonation, cyclization, and dehydration afford N-substituted pyrrole 37. The authors also demonstrated the synthesis of polygonatine, an alkaloid natural product, using this protocol.
![[1860-5397-19-71-i17]](/bjoc/content/inline/1860-5397-19-71-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: β-CD-SO3H-catalyzed synthesis and proposed mechanism of pyrroles 37.
Scheme 17: β-CD-SO3H-catalyzed synthesis and proposed mechanism of pyrroles 37.
![[1860-5397-19-71-4]](/bjoc/content/figures/1860-5397-19-71-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Recyclability of β-cyclodextrin-SO3H.
Figure 4: Recyclability of β-cyclodextrin-SO3H.
(2) Reactions under solvent-free conditions
In recent years, interest in organic reactions that do not use toxic organic solvents has increased due to environmental considerations. Ramesh et al. [71] in 2012 reported an efficient, economical, greener solvent-free process for synthesizing N-substituted pyrroles 39 with a yield of 66–94% by the reaction between different aromatic amines 38 and 2,5-dimethoxytetrahydrofuran (2) without using any catalyst or solvent (Scheme 18a). This method is also effective for aniline having electron withdrawing and electron donating substituents on the aromatic ring. However, this protocol did not produce a product with aliphatic amines and satirically hindered o-substituted anilines. A plausible mechanism for the formation of N-arylpyrroles is depicted in Scheme 18b.
![[1860-5397-19-71-i18]](/bjoc/content/inline/1860-5397-19-71-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Solvent-free and catalyst-free synthesis and plausible mechanism of N-substituted pyrroles 39.
Scheme 18: Solvent-free and catalyst-free synthesis and plausible mechanism of N-substituted pyrroles 39.
In 2014, Hosseini-Sarvari and co-workers [72] described a new and greener Clauson–Kaas method for the synthesis of N-substituted pyrroles 41 in 80–98 % yields by condensing 2,5-DMTHF (2) with amines 40 in the presence of the novel heterogeneous catalyst nano-sulfated TiO2 under solvent-free conditions (Scheme 19). This process has many advantages such as ease of operation, short reaction times, high to excellent product yields, and stable, readily available and green catalysts. Moreover, this protocol is also useful for the preparation of N-substituted compounds with β-lactam fragments.
![[1860-5397-19-71-i19]](/bjoc/content/inline/1860-5397-19-71-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Nano-sulfated TiO2-catalyzed synthesis of N-substituted pyrroles 41.
Scheme 19: Nano-sulfated TiO2-catalyzed synthesis of N-substituted pyrroles 41.
The authors also proposed a mechanism for the synthesis of nano-sulfated titania-catalyzed N-substituted pyrroles as shown in Figure 5b. In general, both Lewis and Brønsted acid sites are present in sulfated metallic oxides, as shown in Figure 5a. The acidity of these Brønsted acid sites is increased by the presence of adjacent strong Lewis acid sites, and the acidity of these Lewis acid sites is due to the inductive effect of sulfate on the metallic cation. Therefore, this nano-sulfated titanium dioxide acts as a new type of Lewis acid catalyst. Intermediate A was first formed by reaction of the catalyst with 2,5-DMTHF (2). Further, a nucleophilic attack of amines 40 with intermediate A, MeOH removal, dehydration, and aromatization steps produce N-substituted pyrroles 41 in good to excellent yields.
![[1860-5397-19-71-5]](/bjoc/content/figures/1860-5397-19-71-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Plausible mechanism for the formation of N-substituted pyrroles catalyzed by nano-sulfated TiO2 catalyst.
Figure 5: Plausible mechanism for the formation of N-substituted pyrroles catalyzed by nano-sulfated TiO2 cat...
In 2019, Wani et al. [73] used the alkaline-earth metal-based catalyst Ca(NO3)2∙4H2O for a mild, transition metal-free and greener synthesis of various N-substituted pyrroles 43 under solvent-free conditions (Scheme 20a). For the optimizations of the reaction conditions, various catalysts (Ca(NO3)2∙4H2O, UO2(NO3)2∙6H2O, Bi(NO3)2∙5H2O) and solvents (H2O, CH3CN, EtOH, DMSO, DMF, MeOH, CH2Cl2, no solvent) were investigated. As a result, 40 mol % Ca(NO3)2∙4H2O was selected as the best catalyst under solvent-free conditions and used to prepare 16 examples of various N-aryl-substituted pyrrole derivatives in yields of 55–85%. The authors also proposed a mechanism for the preparation of these N-substituted pyrroles as shown in Scheme 20b.
![[1860-5397-19-71-i20]](/bjoc/content/inline/1860-5397-19-71-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Copper nitrate-catalyzed Clauson–Kaas synthesis and mechanism of N-substituted pyrroles 43.
Scheme 20: Copper nitrate-catalyzed Clauson–Kaas synthesis and mechanism of N-substituted pyrroles 43.
In this mechanism, calcium nitrate first activates the decomposition process of 2,5-DMTHF (2) from A to B. Further, a nucleophilic attack of amines 42 on B generates imine intermediate C. Finally, intramolecular nucleophilic attack by enamine D on the aldehyde group, dehydration and aromatization steps led to product 43 upon catalyst regeneration.
Recently, Ryabchuk et al. [74] used the 3d-metal cobalt catalyst Co/NGr-C@SiO2-L under solvent-free conditions to synthesize various N-aryl-substituted pyrroles 45 in 50–88% yields from the corresponding nitroarenes 44 via the Clauson–Kaas reaction involving benign reducing agents H2 or HCOOH or CO/H2O mixtures (Scheme 21). The main advantage of this heterogeneous Co catalyst is that it can be used up to 10 times without significant loss of activity and the active cobalt hydride species selectively reduces nitroarenes to their corresponding amines during pyrrole synthesis. This protocol has a high functional group tolerance and has been used to manufacture many major pharmaceutical compounds.
![[1860-5397-19-71-i21]](/bjoc/content/inline/1860-5397-19-71-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of N-substituted pyrroles 45 by using Co catalyst Co/NGr-C@SiO2-L.
Scheme 21: Synthesis of N-substituted pyrroles 45 by using Co catalyst Co/NGr-C@SiO2-L.
Very recently, Afsina et al. [75] reported a solvent-free greener protocol for the synthesis of N-substituted pyrroles via the modified Clauson–Kaas method using a zinc catalyst. The synthesis of pyrrole derivatives 47 were achieved in moderate to excellent yields without using any co-catalyst, ligands or bases by stirring various aniline derivatives 46 with 2,5-dimethoxytetrahydrofuran (2) in the presence of 5 mol % of Zn(OTf)2 for 8 hours at 70 °C. Many zinc catalysts (Zn(OTf)2, Et2Zn, ZnI2, Zn powder, anhydrous ZnCl2, Zn(NO3)2⋅6H2O, ZnSO4⋅H2O, Zn(OAc)2⋅2H2O and ZnO), reaction temperature, time, and catalyst loading was screened for the optimization. After optimizations, 5 mol % Zn(OTf)2 as catalyst, reaction time for 8 h, and temperature of 70 °C were selected as optimal reaction conditions, and used for the preparations of 19 examples of N-aryl-substittuted pyrroles in 15–94% yields (Scheme 22).
![[1860-5397-19-71-i22]](/bjoc/content/inline/1860-5397-19-71-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Zinc-catalyzed synthesis of N-arylpyrroles 47.
Scheme 22: Zinc-catalyzed synthesis of N-arylpyrroles 47.
In another report, Aydogan and co-workers demonstrated a silica sulfuric acid (SSA) catalyzed solvent-free Clauson–Kaas synthesis of N-substituted pyrrole in 60–80% yields in very short reaction times [76] (Scheme 23). To optimize the reaction conditions, several green protocols were used, among these sulfuric acid-immobilised on silica gel (SSA) catalyst under solvent-free conditions was chosen for the synthesis of these N-substituted pyrrole derivatives.
![[1860-5397-19-71-i23]](/bjoc/content/inline/1860-5397-19-71-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Silica sulfuric acid-catalyzed synthesis of pyrrole derivatives 49.
Scheme 23: Silica sulfuric acid-catalyzed synthesis of pyrrole derivatives 49.
In 2012, Bandyopadhyay et al. [77] reported the solvent-free and eco-friendly synthesis of various N-substituted pyrroles 51 in excellent 76–99% yields using bismuth nitrate as catalyst (Scheme 24). In this reaction, various aliphatic, aromatic, polyaromatic and heteropolyaromatic amines 50 were reacted with 2,5-DMTHF (2) in 5 mol % Bi(NO3)3·5H2O in solvent-free conditions under ultrasound irradiation at room temperature. The main advantages of this procedure are its simple isolation process, excellent product yield without using expensive or sensitive solvents and reagents/instruments. Some of these synthesized N-substituted pyrrole derivatives were evaluated for in vitro cytotoxicity for various cancer cell lines.
![[1860-5397-19-71-i24]](/bjoc/content/inline/1860-5397-19-71-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Bismuth nitrate-catalyzed synthesis of pyrroles 51.
Scheme 24: Bismuth nitrate-catalyzed synthesis of pyrroles 51.
In 2014, Wang et al. [78] demonstrated a modified Clauson–Kaas reaction to synthesize various N-substituted pyrroles 53 in 75–95% yields by reacting various aromatic amines 52 with 2,5-DMTHF (2) under a L-(+)-tartaric acid-choline chloride based deep eutectic solvent as green medium (Scheme 25a). Deep eutectic solvents (DESs) are mainly synthesized by mixing quaternary ammonium salts and hydrogen-bond donors. In this study, various combinations of salts and hydrogen bond donating groups were investigated. Among these, the DES prepared by mixing quaternary ammonium salts choline chloride (ChCl) and the hydrogen-bond donor L-(+)-tartaric acid gives the best result. Moreover, L-(+)-tartaric acid–ChCl acts as both solvent and catalyst for pyrrole synthesis. This process has many advantages because it is a metal-free, easy to use, and environmentally friendly method that also gives good product yields with a wide range of substrates. The deep eutectic solvent used in this protocol is cheap, reusable, non-toxic, and biodegradable. In this study, it was found that aniline bearing an electron-donating group produced slightly higher yields than aromatic amines with an electron-withdrawing group. The authors also proposed a tentative mechanism for this reaction, in which intermediate A was first formed by deprotection of 2,5-DMTHF (2), and then A reacted with amines 52 to produce N-substituted pyrroles 53 by nucleophilic addition, subsequent expulsion of methanol, followed by dehydration and aromatization steps (Scheme 25b). The authors suggested that the acidity of DESs may play an important role in the removal of the methoxy groups, and the hydrogen-bonding interaction between DES and the amino group enhance the nucleophlicity of the amines.
![[1860-5397-19-71-i25]](/bjoc/content/inline/1860-5397-19-71-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: L-(+)-tartaric acid-choline chloride-catalyzed Clauson–Kaas synthesis and plausible mechanism of pyrroles 53.
Scheme 25: L-(+)-tartaric acid-choline chloride-catalyzed Clauson–Kaas synthesis and plausible mechanism of py...
(3) Microwave-assisted reactions
Microwave-assisted heating offers a number of advantages over conventional heating, such as greater precision, excellent product yields, and very rapid reaction. This section describes the modified Clauson–Kaas synthesis of different pyrroles in water, solvent-free conditions, and in different organic solvents.
(i) Microwave-assisted reactions in water: In 2009, Ketcha and colleagues [79] nicely utilized the Clauson–Kaas reaction for the environmentally friendly synthesis of various N-substituted pyrroles 55 through the microwave-assisted reaction of various primary amines 54 with 2,5-dimethoxytetrahydrofuran (2). In this protocol, without the use of any additional catalyst, the products were obtained in 12–74% yields in water and 59–96% yields in acetic acid, respectively (Scheme 26). This reaction has been successfully used for common amines using acetic acid and water, but benzylamines and benzamides show no reaction under aqueous conditions. In the case of amino acid ester hydrochlorides, the reaction to give pyrrole can be carried out without the need for the two-phase conditions by lowering the temperature from 170 °C to 120 °C.
![[1860-5397-19-71-i26]](/bjoc/content/inline/1860-5397-19-71-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Microwave-assisted synthesis of N-substituted pyrroles 55 in AcOH or water.
Scheme 26: Microwave-assisted synthesis of N-substituted pyrroles 55 in AcOH or water.
Varma and co-workers [80,81] reported the synthesis of various N-substituted pyrrole derivatives 57 in good yields using a nano-ferric-supported glutathione organocatalyst (Scheme 27, Figure 6). This organoccatalyst was prepared by a post-functionalization method using a benign and naturally occurring glutathione and magnetic ferrite nanoparticles by sonication in water at room temperature. Furthermore, using this organocatalyst, various N-substituted pyrroles were prepared by reacting various amines 56 with 2,5-DMTHF (2) in water at 140 °C under microwave conditions. The main advantages of this protocol are the environmentally friendly reaction medium, recoverable catalysts, high yields, and the absence of toxic solvents.
![[1860-5397-19-71-i27]](/bjoc/content/inline/1860-5397-19-71-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of pyrrole derivatives 57 using a nano-organocatalyst.
Scheme 27: Synthesis of pyrrole derivatives 57 using a nano-organocatalyst.
![[1860-5397-19-71-6]](/bjoc/content/figures/1860-5397-19-71-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Nano-ferric supported glutathione organocatalyst.
Figure 6: Nano-ferric supported glutathione organocatalyst.
In 2009, Wilson et al. [82] described a simple and efficient synthesis of N-substituted pyrrole derivatives without the use of any catalysts under microwave conditions. In this procedure, various arylsulfonamides or anilines 58 are heated with 2,5-dimethoxytetrahydrofuran (2) in H2O under microwave irradiation conditions to produce the corresponding N-substituted pyrrole derivatives 59 in 81–99% yields (Scheme 28). A variety of amine nucleophiles were examined in this method. As a result, it was found that nucleophiles with low pKa values, such as sulfonamides, reacted very well, while reactions with nucleophiles with high pKa values, such as benzylamine, did not work well. Compared with other classical methods, this protocol is easy to use, cheap and environmentally friendly.
![[1860-5397-19-71-i28]](/bjoc/content/inline/1860-5397-19-71-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Microwave-assisted synthesis of N-substituted pyrroles 59 in water.
Scheme 28: Microwave-assisted synthesis of N-substituted pyrroles 59 in water.
(ii) Microwave-assisted reactions under solvent-free conditions: Banik et al. [83] described the synthesis of diverse N-substituted pyrroles using a microwave-assisted and iodine-catalyzed protocol. These pyrrole derivatives were prepared in 75–98% yields by reacting various amines 60 and 2,5-DMTHF 2 under solvent-free conditions in the presence of 5 mol % molecular iodine as catalyst (Scheme 29a). These synthesized products were tested against various cancer cells in vitro. In the proposed mechanism, deprotection of the methoxy group of 2,5-DMTHF (2) in the presence of iodine under MW irradiation afford intermediate A, which is converted to dialdehyde B. Finally, N-substituted pyrroles 61 are produced by nucleophilic addition of amines with dialdehyde, followed by dehydration and aromatization steps (Scheme 29b).
![[1860-5397-19-71-i29]](/bjoc/content/inline/1860-5397-19-71-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Iodine-catalyzed synthesis and proposed mechanism of pyrroles 61.
Scheme 29: Iodine-catalyzed synthesis and proposed mechanism of pyrroles 61.
Jafari et al. [84] used the heterogeneous catalyst H3PW12O40/SiO2 for the Clauson–Kaas synthesis of N-substituted pyrrole derivatives 63 (Scheme 30) by the reaction of amines 62 with 2,5-dimethoxytetrahydrofuran (2) in petroleum ether at reflux conditions in 60–93% yields (method 1) and MW-assisted solvent-free conditions in 90–96% yields (method 2). The optimization of the reaction conditions was performed in search of suitable conditions for this condensation reaction. Various acid catalysts (SiO2, HPA, HPA/SiO2, catalyst loadings (1 mol %, 2 mol %, 2.5 mol %, 0.3 g), solvent-systems (petroleum ether 40/60, toluene, n-hexane, acetonitrile) and reaction conditions (room temperature, 60 °C, reflux, and MW (power 5, 8 or 10), were studied. Among these, the optimized reaction conditions for method 1 are 2.5 mol % HPA/SiO2 as catalyst in refluxing petroleum ether, whereas the optimized conditions for method 2 are 2.5 mol % HPA/SiO2 as catalyst, solventless under MW irradiation. The main advantages of this protocol is its greener, eco-friendly, stable and reusable heterogeneous catalyst, smooth and selective reaction under solvent-free conditions, excellent yields of products, easy work-up and simple handling of reaction.
![[1860-5397-19-71-i30]](/bjoc/content/inline/1860-5397-19-71-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: H3PW12O40/SiO2-catalyzed synthesis of N-substituted pyrroles 63.
Scheme 30: H3PW12O40/SiO2-catalyzed synthesis of N-substituted pyrroles 63.
Mahmoudi and Jafari [85] reported the preparation of a new on magnetic nanoparticles sulfonic acid-supported catalyst with maghemite coating as a magnetically recyclable catalyst Fe3O4@-γ-Fe2O3-SO3H. This heterogeneous catalyst is used for the synthesis of N-substituted pyrrole derivatives 65 in 90–95% yields using a solvent-free reaction of amines 64 with 2,5-DMTHF (2) under microwave irradiation conditions (Scheme 31). This method has many advantages, including simpler, less expensive, and faster catalyst preparation, easy removal of the catalyst from the reaction mixture, and use of the catalyst for up to nine consecutive runs without loss of catalytic activity.
![[1860-5397-19-71-i31]](/bjoc/content/inline/1860-5397-19-71-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Fe3O4@-γ-Fe2O3-SO3H-catalyzed synthesis of pyrroles 65.
Scheme 31: Fe3O4@-γ-Fe2O3-SO3H-catalyzed synthesis of pyrroles 65.
Recently, Rohit et al. [86] described a modified Clauson–Kaas synthesis of N-substituted pyrrole with up to 89% yields by microwave irradiation at 120 °C in neat conditions by reacting amines 66 with 2,5-DMTHF (2) in the presence of Mn(II) catalyst Mn(NO3)2.4H2O (Scheme 32a). Numerous Mn catalysts (e.g., MnCl2∙4H2O, MnSO4∙H2O, Mn(NO3)2∙4H2O, Mn(OAc)2∙4H2O, MnBr(CO)5, MnO2), catalyst loading (e.g., 0, 5, 10, 20 mol %), reaction temperatures (90–130 °C) and reaction times (e.g., 10, 20, 25 min) were investigated in order to optimize the reaction conditions. Among these, 10 mol % Mn(NO3)2∙4H2O was selected as the best catalyst for the synthesis of the pyrrole derivative 67, with yields of 19–89% under MW in solvent-free conditions at 120 °C for 20 min. According to the results of this study, it was discovered that amines with electron-donating substituents produce higher yields of the final product than those with electron-withdrawing substituents. The authors also put forth a plausible mechanism for this protocol, in which Mn first coordinates with an OMe group of 2,5-DMTHF (2) and helps in the removal of the methoxy group to yield intermediate B. The electron-deficient carbon of intermediate B is then attacked by the amine in a nucleophilic reaction to produce intermediate C, which is then followed by the removal of the second OMe group to produce intermediate D. Further, the lone pair of N attacks the carbonyl carbon to form the 5-membered ring E bearing the iminium ion. Finally, N-substituted amines 67 were obtained after deprotonation/protonation, dehydration, and aromatization steps as shown in Scheme 32b.
![[1860-5397-19-71-i32]](/bjoc/content/inline/1860-5397-19-71-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Mn(NO3)2·4H2O-catalyzed synthesis and proposed mechanism of pyrroles 67.
Scheme 32: Mn(NO3)2·4H2O-catalyzed synthesis and proposed mechanism of pyrroles 67.
In another report, Ozaki et al. [87] used the Clauson–Kaas approach to synthesize sulfonic acids/sulfonates from various primary sulfonamides via sulfonylpyrroles. First, various sulfonylpyrroles 69 were prepared from primary sulfonamides 68 by reaction with 2,5-DMTHF (2) using two methods as shown in Scheme 33. In method 1, amides 68 and 2 were heated in toluene at 100 °C for 30–60 min in the presence of 5 mol % p-TsOH∙H2O. In contrast, method 2 involves a greener protocol in which amides 68 and 2 are reacted under microwave condition at 150 °C for 30–60 min without the use of additives or solvents. Furthermore, these sulfonylpyrroles were converted to the corresponding sulfonic acid/sulfonates. Using this method, various arylsulfonamides, alkylsulfonamides and diverse drug molecules bearing sulfonamides were easily converted to sulfonylpyrroles and then sulfonic acids. This cheap and easily accessible protocol may be useful in pharmaceutical and industrial applications.
![[1860-5397-19-71-i33]](/bjoc/content/inline/1860-5397-19-71-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: p-TsOH∙H2O-catalyzed (method 1) and MW-assisted (method 2) synthesis of N-sulfonylpyrroles 69.
Scheme 33: p-TsOH∙H2O-catalyzed (method 1) and MW-assisted (method 2) synthesis of N-sulfonylpyrroles 69.
Aydogan and Yolacan [88] reported the Clauson–Kaas reaction for the preparation of acidic ionic liquid 1-hexyl-3-methylimidazolium hydrogen sulfate ([hmim][HSO4])-catalyzed N-substituted pyrrole derivatives 71 under microwave irradiation (Scheme 34). To optimize of reaction conditions, the catalytic activity of various acids and ionic liquids such as [hmim][HSO4], CH3COOH, [hmim][H2PO4], and [bmim][BF4] was investigated. Among these, [hmim][HSO4] proved to be the best catalyst providing higher yields in shorter time. This protocol provides a greener, environmental friendly, and a cleaner route for the synthesis of acid-sensitive pyrrole derivatives without decomposition in a short time.
![[1860-5397-19-71-i34]](/bjoc/content/inline/1860-5397-19-71-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: ([hmim][HSO4]-catalyzed Clauson–Kaas synthesis of pyrroles 71.
Scheme 34: ([hmim][HSO4]-catalyzed Clauson–Kaas synthesis of pyrroles 71.
Abid et al. [89] described the synthesis of N-substituted pyrrole derivatives in good yields using K-10 montmorillonite as an effective solid acid catalyst under microwave irradiation. K-10 monomorillonite is a widely used solid acid catalyst, which features strong acidity, larger surface area and high stability. Pyrrole derivatives 73 were prepared in 83–95 % yields by condensation reactions between various alkyl-, aryl-, heteroaryl-, and sulfonylamines 72 and 2,5-dimethoxytetrahydrofuran (2) under MW conditions at 100 °C (Scheme 35). This solvent-free protocol has many advantages, including better selectivity and excellent product yields, mild reaction conditions, and ease of product isolation.
![[1860-5397-19-71-i35]](/bjoc/content/inline/1860-5397-19-71-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Synthesis of N-substituted pyrroles 73 using K-10 montmorillonite catalyst.
Scheme 35: Synthesis of N-substituted pyrroles 73 using K-10 montmorillonite catalyst.
(iii) Microwave-assisted reactions under organic solvents: Silveira and co-workers [90] developed the Clauson–Kaas synthesis of N-substituted pyrroles catalyzed by CeCl3∙7H2O. In this reaction, N-arylpyrroles 75 were synthesized from various aniline derivatives 74 by the reaction with 2,5-dimethoxytetrahydrofuran (2) in both microwave irradiation and conventional heating in acetonitrile (Scheme 36). Furthermore, these N-arylpyrroles were converted to the corresponding thiocyanated derivatives and then to sulfur-containing heterocycles. Moreover, this procedure provides N-substituted pyrroles from various aromatic amines in good to excellent yields under both reflux and MW conditions. In contrast, aliphatic amines such as cyclohexylamine and benzylamine give only traces of products even under prolonged reflux or MW irradiation times. In addition, it was also found that aniline bearing electron-donating substituents are less reactive and take more time to complete the reactions.
![[1860-5397-19-71-i36]](/bjoc/content/inline/1860-5397-19-71-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: CeCl3∙7H2O-catalyzed Clauson–Kaas synthesis of pyrroles 75.
Scheme 36: CeCl3∙7H2O-catalyzed Clauson–Kaas synthesis of pyrroles 75.
Rivera et al. [91] synthesized N-substituted pyrrole derivatives 77 in good to excellent yields using a microwave-assisted and bismuth nitrate Bi(NO3)3∙5H2O-catalyzed reaction between various amines 76 and 2,5-dimethoxytetrahydrofuran (2) in neat, H2O or THF (Scheme 37). This approach provides different pyrrole derivatives without the use of sensitive or expensive reagents and also has numerous applications in other areas of research. In this study, it was discovered that amines with less nucleophilicity also produced good yields of the product. In contrast, for the reaction with polyaromatic amines, THF was found to be the best solvent because these compounds are insoluble in water and provides poor yields of the product in solvent-free conditions.
![[1860-5397-19-71-i37]](/bjoc/content/inline/1860-5397-19-71-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Synthesis of N-substituted pyrroles 77 using Bi(NO3)3∙5H2O.
Scheme 37: Synthesis of N-substituted pyrroles 77 using Bi(NO3)3∙5H2O.
Gullapelli et al. [92] described the Clauson–Kaas synthesis of N-arylpyrroles 79 under microwave irradiation using oxone (2KHSO5∙KHSO4∙K2SO4) as a catalyst (Scheme 38a). Many solvents (EtOH, CH3CN, THF, DMF, H2O, neat), reaction times (10–22 minutes), and amount of oxone were investigated in order to stabilize the best reaction conditions. Among these, CH3CN as the best solvent system, 0.09 g of oxone, and a temperature of 110 ± 10 °C were chosen as optimized reaction conditions and used for the synthesis of N-arylpyrroles from various amines 78 via the reaction with 2,5-dimethoxytetrahydrofuran (2). Oxone is a mild, inexpensive, nontoxic, stable, and transition-metal-free catalyst that is very easy to handle during this transformation and provided high yields of the product. The authors also proposed a mechanism for this protocol as shown in Scheme 38b.
![[1860-5397-19-71-i38]](/bjoc/content/inline/1860-5397-19-71-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Oxone-catalyzed synthesis and proposed mechanism of N-substituted pyrroles 79.
Scheme 38: Oxone-catalyzed synthesis and proposed mechanism of N-substituted pyrroles 79.
Conclusion
The synthesis of pyrrole derivatives has piqued the interest of many synthetic researchers as they offer promising applications in pharmaceutical, polymer, and natural product chemistry. The introduction of a pyrrole moiety into a molecule alters its diverse properties and increases the possibility for further functionalization. The first section of this review successfully describes the use of the Clauson–Kaas reaction for the synthesis of N-substituted pyrroles using conventional methods such as Brønsted acid and Lewis acid-catalyzed reactions under heating conditions. However, due to environmental concerns, the second part of this review focuses on greener Clauson–Kaas reaction protocols. Various Brønsted acids, Lewis acids, transition metal catalysts, and organocatalysts have been used in water as a green solvent, under solvent-free conditions or in microwave-assisted reactions to synthesize various N-substituted pyrrole derivatives. Because of the high functional group tolerance of this method, various primary aliphatic/aromatic amines, benzamides, and sulfonamides have been used successfully as substrates in these transformations. It was discovered that, in general, arylamines with electron-withdrawing groups deactivated the arylamines and produced the corresponding pyrroles in only modest yields, whereas arylamines with electron-donating groups reacted much faster and produced the pyrroles in excellent yields. In addition, reactions of sterically hindered anilines with 2,5-dimethoxytetrahydrofuran give moderate yields, and reactions with less sterically hindered anilines give good yields of products. The Clauson–Kaas method for the synthesis of pyrroles has a wide range of applications, including the synthesis of various pharmaceutically active molecules, polymer synthesis, total synthesis of natural products, porphyrin functionalization, and the synthesis of various pyrrole-containing heterocyclic molecules. We believe that this review will be useful and will encourage researchers to apply the modified Clauson–Kaas reaction to different areas of chemistry.
Table 1: List of abbreviations.
| Abbreviation | Name |
| AcOH | acetic acid |
| β-CD | β-cyclodextrin |
| Co-NP | cobalt nanoparticle |
| DES | deep eutectic solvent |
| DMAP | 4-(dimethylamino)pyridine |
| DMF | N,N-dimethylformamide |
| 2,5-DMTHF | 2,5-dimethoxytetrahydrofuran |
| EtOH | ethanol |
| [Hmim][HSO4] | 1-methylimidazolium hydrogen sulfate |
| HPA | hydroxypropyl acrylate |
| MeCN | acetonitrile |
| MW | microwave |
| NaOAc | sodium acetate |
| OPV | organic photovoltic |
| PEG | polyethylene glycol |
| p-TsOH | p-toluenesulfonic acid |
| SRR | structure–reactivity relationship |
| SSA | silica sulfuric acid |
| TEA | triethylamine |
| TfOH | trifluoromethanesulfonic acid |
| THF | tetrahydrofuran |
Acknowledgements
The authors are thankful to Prof. M. Nath, Department of Chemistry, University of Delhi for his valuable suggestions in writing this review. DKS and RK are also grateful to Bipin Bihari College, Jhansi and R. D. S. College, Muzaffarpur, respectively, for providing facilities for this work.
References
-
Gao, B.; Yang, B.; Feng, X.; Li, C. Nat. Prod. Rep. 2022, 39, 139–162. doi:10.1039/d1np00017a
Return to citation in text: [1] -
Joule, J. A. Natural Products Containing Nitrogen Heterocycles—Some Highlights 1990–2015. In Advances in Heterocyclic Chemistry; Scriven, E. F. V.; Ramsden, C. A., Eds.; Academic Press, 2016; Vol. 119, pp 81–106. doi:10.1016/bs.aihch.2015.10.005
Return to citation in text: [1] -
Singh, N.; Singh, S.; Kohli, S.; Singh, A.; Asiki, H.; Rathee, G.; Chandra, R.; Anderson, E. A. Org. Chem. Front. 2021, 8, 5550–5573. doi:10.1039/d0qo01574a
Return to citation in text: [1] -
Heravi, M. M.; Zadsirjan, V. RSC Adv. 2020, 10, 44247–44311. doi:10.1039/d0ra09198g
Return to citation in text: [1] -
Gupta, S. S.; Kumari, S.; Kumar, I.; Sharma, U. Chem. Heterocycl. Compd. 2020, 56, 433–444. doi:10.1007/s10593-020-02678-5
Return to citation in text: [1] -
Kim, J.; Park, M.; Choi, J.; Singh, D. K.; Kwon, H. J.; Kim, S. H.; Kim, I. Bioorg. Med. Chem. Lett. 2019, 29, 1350–1356. doi:10.1016/j.bmcl.2019.03.044
Return to citation in text: [1] -
Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K. K.; Jonnalagadda, S. B. Molecules 2020, 25, 1909. doi:10.3390/molecules25081909
Return to citation in text: [1] -
Paludetto, M.-N.; Bijani, C.; Puisset, F.; Bernardes-Génisson, V.; Arellano, C.; Robert, A. J. Med. Chem. 2018, 61, 7849–7860. doi:10.1021/acs.jmedchem.8b00812
Return to citation in text: [1] -
Mateev, E.; Georgieva, M.; Zlatkov, A. J. Pharm. Pharm. Sci. 2022, 25, 24–40. doi:10.18433/jpps32417
Return to citation in text: [1] -
Kuznietsova, H.; Dziubenko, N.; Byelinska, I.; Hurmach, V.; Bychko, A.; Lynchak, O.; Milokhov, D.; Khilya, O.; Rybalchenko, V. J. Drug Targeting 2020, 28, 547–563. doi:10.1080/1061186x.2019.1703189
Return to citation in text: [1] -
Jiang, S.; Lu, H.; Liu, S.; Zhao, Q.; He, Y.; Debnath, A. K. Antimicrob. Agents Chemother. 2004, 48, 4349–4359. doi:10.1128/aac.48.11.4349-4359.2004
Return to citation in text: [1] -
Pegklidou, K.; Papastavrou, N.; Gkizis, P.; Komiotis, D.; Balzarini, J.; Nicolaou, I. Med. Chem. 2015, 11, 602–608. doi:10.2174/1573406411666150313161225
Return to citation in text: [1] -
Aatif, M.; Raza, M. A.; Javed, K.; Nashre-ul-Islam, S. M.; Farhan, M.; Alam, M. W. Antibiotics (Basel, Switz.) 2022, 11, 1750. doi:10.3390/antibiotics11121750
Return to citation in text: [1] -
Williamson, N. R.; Simonsen, H. T.; Ahmed, R. A. A.; Goldet, G.; Slater, H.; Woodley, L.; Leeper, F. J.; Salmond, G. P. C. Mol. Microbiol. 2005, 56, 971–989. doi:10.1111/j.1365-2958.2005.04602.x
Return to citation in text: [1] -
Green, K. D.; Pang, A. H.; Thamban Chandrika, N.; Garzan, A.; Baughn, A. D.; Tsodikov, O. V.; Garneau-Tsodikova, S. ACS Infect. Dis. 2022, 8, 757–767. doi:10.1021/acsinfecdis.1c00450
Return to citation in text: [1] -
Palmer, M. J.; Deng, X.; Watts, S.; Krilov, G.; Gerasyuto, A.; Kokkonda, S.; El Mazouni, F.; White, J.; White, K. L.; Striepen, J.; Bath, J.; Schindler, K. A.; Yeo, T.; Shackleford, D. M.; Mok, S.; Deni, I.; Lawong, A.; Huang, A.; Chen, G.; Wang, W.; Jayaseelan, J.; Katneni, K.; Patil, R.; Saunders, J.; Shahi, S. P.; Chittimalla, R.; Angulo-Barturen, I.; Jiménez-Díaz, M. B.; Wittlin, S.; Tumwebaze, P. K.; Rosenthal, P. J.; Cooper, R. A.; Aguiar, A. C. C.; Guido, R. V. C.; Pereira, D. B.; Mittal, N.; Winzeler, E. A.; Tomchick, D. R.; Laleu, B.; Burrows, J. N.; Rathod, P. K.; Fidock, D. A.; Charman, S. A.; Phillips, M. A. J. Med. Chem. 2021, 64, 6085–6136. doi:10.1021/acs.jmedchem.1c00173
Return to citation in text: [1] -
Kancharla, P.; Li, Y.; Yeluguri, M.; Dodean, R. A.; Reynolds, K. A.; Kelly, J. X. J. Med. Chem. 2021, 64, 8739–8754. doi:10.1021/acs.jmedchem.1c00748
Return to citation in text: [1] -
Battilocchio, C.; Poce, G.; Alfonso, S.; Porretta, G. C.; Consalvi, S.; Sautebin, L.; Pace, S.; Rossi, A.; Ghelardini, C.; Di Cesare Mannelli, L.; Schenone, S.; Giordani, A.; Di Francesco, L.; Patrignani, P.; Biava, M. Bioorg. Med. Chem. 2013, 21, 3695–3701. doi:10.1016/j.bmc.2013.04.031
Return to citation in text: [1] -
Said Fatahala, S.; Hasabelnaby, S.; Goudah, A.; Mahmoud, G. I.; Helmy Abd-El Hameed, R. Molecules 2017, 22, 461. doi:10.3390/molecules22030461
Return to citation in text: [1] -
Narule, M. N.; Gaidhane, M. K.; Gaidhane, P. K. J. Pharm. Res. (Mohali, India) 2013, 6, 626–632. doi:10.1016/j.jopr.2013.04.046
Return to citation in text: [1] -
Mallikarjuna Reddy, G.; Camilo, A., Jr.; Raul Garcia, J. Bioorg. Chem. 2021, 106, 104465. doi:10.1016/j.bioorg.2020.104465
Return to citation in text: [1] -
Fatahala, S. S.; Shalaby, E. A.; Kassab, S. E.; Mohamed, M. S. Anti-Cancer Agents Med. Chem. 2015, 15, 517–526. doi:10.2174/1871520615666150105113946
Return to citation in text: [1] -
Di Santo, R.; Tafi, A.; Costi, R.; Botta, M.; Artico, M.; Corelli, F.; Forte, M.; Caporuscio, F.; Angiolella, L.; Palamara, A. T. J. Med. Chem. 2005, 48, 5140–5153. doi:10.1021/jm048997u
Return to citation in text: [1] -
Zhang, S.-G.; Liang, C.-G.; Sun, Y.-Q.; Teng, P.; Wang, J.-Q.; Zhang, W.-H. Mol. Diversity 2019, 23, 915–925. doi:10.1007/s11030-019-09920-z
Return to citation in text: [1] -
McArthur, K. A.; Mitchell, S. S.; Tsueng, G.; Rheingold, A.; White, D. J.; Grodberg, J.; Lam, K. S.; Potts, B. C. M. J. Nat. Prod. 2008, 71, 1732–1737. doi:10.1021/np800286d
Return to citation in text: [1] -
Ding, X.-B.; Furkert, D. P.; Brimble, M. A. Chem. Commun. 2016, 52, 12638–12641. doi:10.1039/c6cc07532k
Return to citation in text: [1] -
Kocaoğlu, E.; Talaz, O.; Çavdar, H.; Şentürk, M.; Supuran, C. T.; Ekinci, D. J. Enzyme Inhib. Med. Chem. 2019, 34, 51–54. doi:10.1080/14756366.2018.1520228
Return to citation in text: [1] -
Williams, I. S.; Joshi, P.; Gatchie, L.; Sharma, M.; Satti, N. K.; Vishwakarma, R. A.; Chaudhuri, B.; Bharate, S. B. Bioorg. Med. Chem. Lett. 2017, 27, 3683–3687. doi:10.1016/j.bmcl.2017.07.010
Return to citation in text: [1] -
Knorr, L. Ber. Dtsch. Chem. Ges. 1884, 17, 1635–1642. doi:10.1002/cber.18840170220
Return to citation in text: [1] -
Alberola, A.; González Ortega, A.; Luisa Sádaba, M.; Sañudo, C. Tetrahedron 1999, 55, 6555–6566. doi:10.1016/s0040-4020(99)00289-6
Return to citation in text: [1] -
Paal, C. Ber. Dtsch. Chem. Ges. 1884, 17, 2756–2767. doi:10.1002/cber.188401702228
Return to citation in text: [1] -
Knorr, L. Ber. Dtsch. Chem. Ges. 1884, 17, 2863–2870. doi:10.1002/cber.188401702254
Return to citation in text: [1] -
Balakrishna, A.; Aguiar, A.; Sobral, P. J. M.; Wani, M. Y.; Almeida e Silva, J.; Sobral, A. J. F. N. Catal. Rev.: Sci. Eng. 2019, 61, 84–110. doi:10.1080/01614940.2018.1529932
Return to citation in text: [1] -
Hantzsch, A. Ber. Dtsch. Chem. Ges. 1890, 23, 1474–1476. doi:10.1002/cber.189002301243
Return to citation in text: [1] -
Leonardi, M.; Estévez, V.; Villacampa, M.; Menéndez, J. C. Synthesis 2019, 51, 816–828. doi:10.1055/s-0037-1610320
Return to citation in text: [1] -
Trautwein, A. W.; Süßmuth, R. D.; Jung, G. Bioorg. Med. Chem. Lett. 1998, 8, 2381–2384. doi:10.1016/s0960-894x(98)00430-2
Return to citation in text: [1] -
Clauson-Kaas, N.; Tyle, Z. Acta Chem. Scand. 1952, 6, 667–670. doi:10.3891/acta.chem.scand.06-0667
Return to citation in text: [1] [2] -
Josey, A. D.; Jenner, E. L. J. Org. Chem. 1962, 27, 2466–2470. doi:10.1021/jo01054a042
Return to citation in text: [1] -
Wang, Z. Piloty-Robinson Pyrrole Synthesis. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, 2010; pp 2217–2220. doi:10.1002/9780470638859.conrr500
Return to citation in text: [1] -
Piloty, O. Ber. Dtsch. Chem. Ges. 1910, 43, 489–498. doi:10.1002/cber.19100430182
Return to citation in text: [1] -
Robinson, G. M.; Robinson, R. J. Chem. Soc., Trans. 1918, 113, 639–645. doi:10.1039/ct9181300639
Return to citation in text: [1] -
Barton, D. H. R.; Zard, S. Z. J. Chem. Soc., Chem. Commun. 1985, 1098–1100. doi:10.1039/c39850001098
Return to citation in text: [1] -
He, X.-L.; Zhao, H.-R.; Song, X.; Jiang, B.; Du, W.; Chen, Y.-C. ACS Catal. 2019, 9, 4374–4381. doi:10.1021/acscatal.9b00767
Return to citation in text: [1] -
Ono, N. Heterocycles 2008, 75, 243. doi:10.3987/rev-07-622
Return to citation in text: [1] -
Yoshimitsu, T.; Ino, T.; Tanaka, T. Org. Lett. 2008, 10, 5457–5460. doi:10.1021/ol802225g
Return to citation in text: [1] -
Ma, K.; Yin, X.; Dai, M. Angew. Chem. 2018, 130, 15429–15432. doi:10.1002/ange.201809114
Return to citation in text: [1] -
Plasencia, C.; Grande, F.; Oshima, T.; Cao, X.; Yamada, R.; Sanchez, T.; Aiello, F.; Garofalo, A.; Neamati, N. Cancer Biol. Ther. 2009, 8, 458–465. doi:10.4161/cbt.8.5.7741
Return to citation in text: [1] -
Butini, S.; Brindisi, M.; Gemma, S.; Minetti, P.; Cabri, W.; Gallo, G.; Vincenti, S.; Talamonti, E.; Borsini, F.; Caprioli, A.; Stasi, M. A.; Di Serio, S.; Ros, S.; Borrelli, G.; Maramai, S.; Fezza, F.; Campiani, G.; Maccarrone, M. J. Med. Chem. 2012, 55, 6898–6915. doi:10.1021/jm300689c
Return to citation in text: [1] -
Kiskan, B.; Gacal, B.; Asan, M.; Gunaydin, E. C.; Yilmaz, I.; Yagci, Y. Polym. Bull. 2011, 67, 609–621. doi:10.1007/s00289-010-0412-9
Return to citation in text: [1] -
Gursoy, S. S.; Uygun (GOK), A.; Tilki, T. J. Macromol. Sci., Part A: Pure Appl. Chem. 2010, 47, 681–688. doi:10.1080/10601325.2010.483376
Return to citation in text: [1] -
Singh, D. K.; Nath, M. Beilstein J. Org. Chem. 2014, 10, 808–813. doi:10.3762/bjoc.10.76
Return to citation in text: [1] -
Singh, D. K.; Nath, M. Org. Biomol. Chem. 2015, 13, 1836–1845. doi:10.1039/c4ob02370f
Return to citation in text: [1] -
Berionni Berna, B.; Nardis, S.; Galloni, P.; Savoldelli, A.; Stefanelli, M.; Fronczek, F. R.; Smith, K. M.; Paolesse, R. Org. Lett. 2016, 18, 3318–3321. doi:10.1021/acs.orglett.6b01314
Return to citation in text: [1] -
Wynn, T. A. Pyrroles and Pyrrolidines. In Name Reactions in Heterocyclic Chemistry II; Li, J. J.; Corey, E. J., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp 41–82. doi:10.1002/9781118092828.ch2
Return to citation in text: [1] -
Wang, Z. Clauson-Kaas Reaction. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, 2010; pp 665–668. doi:10.1002/9780470638859.conrr146
Return to citation in text: [1] -
Sonnet, P.; Dallemagne, P.; Guillon, J.; Enguehard, C.; Stiebing, S.; Tanguy, J.; Bureau, R.; Rault, S.; Auvray, P.; Moslemi, S.; Sourdaine, P.; Séralini, G.-E. Bioorg. Med. Chem. 2000, 8, 945–955. doi:10.1016/s0968-0896(00)00024-9
Return to citation in text: [1] -
Kumar, S.; Krishnakanth, S.; Mathew, J.; Pomerantz, Z.; Lellouche, J.-P.; Ghosh, S. J. Phys. Chem. C 2014, 118, 2570–2579. doi:10.1021/jp411098y
Return to citation in text: [1] -
Fang, Y.; Leysen, D.; Ottenheijm, H. C. J. Synth. Commun. 1995, 25, 1857–1861. doi:10.1080/00397919508015431
Return to citation in text: [1] -
Rochais, C.; Lisowski, V.; Dallemagne, P.; Rault, S. Tetrahedron Lett. 2004, 45, 6353–6355. doi:10.1016/j.tetlet.2004.06.047
Return to citation in text: [1] -
Abid, M.; Teixeira, L.; Török, B. Tetrahedron Lett. 2007, 48, 4047–4050. doi:10.1016/j.tetlet.2007.04.021
Return to citation in text: [1] -
Zuo, B.; Chen, J.; Liu, M.; Ding, J.; Wu, H.; Su, W. J. Chem. Res. 2009, 14–16. doi:10.3184/030823409x393628
Return to citation in text: [1] -
Zhang, X.; Shi, J. Tetrahedron 2011, 67, 898–903. doi:10.1016/j.tet.2010.12.018
Return to citation in text: [1] -
Chatzopoulou, M.; Kotsampasakou, E.; Demopoulos, V. J. Synth. Commun. 2013, 43, 2949–2954. doi:10.1080/00397911.2012.753460
Return to citation in text: [1] -
Ghafuri, H.; Emami, A. J. Appl. Chem. Res. 2015, 9, 55–60.
Return to citation in text: [1] -
Gourlay, B. S.; Molesworth, P. P.; Ryan, J. H.; Smith, J. A. Tetrahedron Lett. 2006, 47, 799–801. doi:10.1016/j.tetlet.2005.11.104
Return to citation in text: [1] -
Azizi, N.; Davoudpour, A.; Eskandari, F.; Batebi, E. Monatsh. Chem. 2013, 144, 405–409. doi:10.1007/s00706-012-0841-2
Return to citation in text: [1] -
Ma, F.-P.; Li, P.-H.; Li, B.-L.; Mo, L.-P.; Liu, N.; Kang, H.-J.; Liu, Y.-N.; Zhang, Z.-H. Appl. Catal., A 2013, 457, 34–41. doi:10.1016/j.apcata.2013.03.005
Return to citation in text: [1] -
Azizi, N.; Khajeh-Amiri, A.; Ghafuri, H.; Bolourtchian, M.; Saidi, M. Synlett 2009, 2245–2248. doi:10.1055/s-0029-1217799
Return to citation in text: [1] -
Deng, H.-J.; Fang, Y.-J.; Chen, G.-W.; Liu, M.-C.; Wu, H.-Y.; Chen, J.-X. Appl. Organomet. Chem. 2012, 26, 164–167. doi:10.1002/aoc.1864
Return to citation in text: [1] -
Patil, R. N.; Kumar, A. V. ChemistrySelect 2018, 3, 9812–9818. doi:10.1002/slct.201801559
Return to citation in text: [1] -
Ramesh, K.; Murthy, S. N.; Nageswar, Y. V. D. Synth. Commun. 2012, 42, 2471–2477. doi:10.1080/00397911.2011.560744
Return to citation in text: [1] -
Hosseini-Sarvari, M.; Najafvand-Derikvandi, S.; Jarrahpour, A.; Heiran, R. Chem. Heterocycl. Compd. 2014, 49, 1732–1739. doi:10.1007/s10593-014-1425-3
Return to citation in text: [1] -
Wani, R. R.; Chaudhari, H. K.; Takale, B. S. J. Heterocycl. Chem. 2019, 56, 1337–1340. doi:10.1002/jhet.3507
Return to citation in text: [1] -
Ryabchuk, P.; Leischner, T.; Kreyenschulte, C.; Spannenberg, A.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2020, 59, 18679–18685. doi:10.1002/anie.202007613
Return to citation in text: [1] -
Afsina, C. M. A.; Rohit, K. R.; Anilkumar, G. Results Chem. 2022, 4, 100350. doi:10.1016/j.rechem.2022.100350
Return to citation in text: [1] -
Khammas, A. J.; Yolacan, C.; Aydogan, F. Russ. J. Gen. Chem. 2018, 88, 2680–2683. doi:10.1134/s1070363218120332
Return to citation in text: [1] -
Bandyopadhyay, D.; Mukherjee, S.; Granados, J. C.; Short, J. D.; Banik, B. K. Eur. J. Med. Chem. 2012, 50, 209–215. doi:10.1016/j.ejmech.2012.01.055
Return to citation in text: [1] -
Wang, P.; Ma, F.-P.; Zhang, Z.-H. J. Mol. Liq. 2014, 198, 259–262. doi:10.1016/j.molliq.2014.07.015
Return to citation in text: [1] -
Ketcha, D. M.; Miles, K. C.; Mays, S. M.; Southerland, B. K.; Auvil, T. J. ARKIVOC 2010, No. xiv, 181–190. doi:10.3998/ark.5550190.0010.e17
Return to citation in text: [1] -
Polshettiwar, V.; Varma, R. S. Tetrahedron 2010, 66, 1091–1097. doi:10.1016/j.tet.2009.11.015
Return to citation in text: [1] -
Polshettiwar, V.; Baruwati, B.; Varma, R. S. Chem. Commun. 2009, 1837–1839. doi:10.1039/b900784a
Return to citation in text: [1] -
Wilson, M. A.; Filzen, G.; Welmaker, G. S. Tetrahedron Lett. 2009, 50, 4807–4809. doi:10.1016/j.tetlet.2009.06.079
Return to citation in text: [1] -
Bandyopadhyay, D.; Mukherjee, S.; Banik, B. K. Molecules 2010, 15, 2520–2525. doi:10.3390/molecules15042520
Return to citation in text: [1] -
Jafari, A. A.; Mahmoudi, H.; Mirjalili, B. F. J. Iran. Chem. Soc. 2011, 8, 851–856. doi:10.1007/bf03245915
Return to citation in text: [1] -
Mahmoudi, H.; Jafari, A. A. ChemCatChem 2013, 5, 3743–3749. doi:10.1002/cctc.201300623
Return to citation in text: [1] -
Rohit, K. R.; Meera, G.; Anilkumar, G. J. Heterocycl. Chem. 2022, 59, 194–200. doi:10.1002/jhet.4372
Return to citation in text: [1] -
Ozaki, T.; Yorimitsu, H.; Perry, G. J. P. Tetrahedron 2022, 117–118, 132830. doi:10.1016/j.tet.2022.132830
Return to citation in text: [1] -
Aydogan, F.; Yolacan, C. J. Chem. 2013, 2013, 976724. doi:10.1155/2013/976724
Return to citation in text: [1] -
Abid, M.; Landge, S. M.; Török, B. Org. Prep. Proced. Int. 2006, 38, 495–500. doi:10.1080/00304940609356444
Return to citation in text: [1] -
Silveira, C. C.; Fortes, M. P.; Mendes, S. R. Curr. Org. Chem. 2012, 16, 1540–1548. doi:10.2174/138527212800672538
Return to citation in text: [1] -
Rivera, S.; Bandyopadhyay, D.; Banik, B. K. Tetrahedron Lett. 2009, 50, 5445–5448. doi:10.1016/j.tetlet.2009.06.002
Return to citation in text: [1] -
Gullapelli, K.; Brahmeshwari, G.; Ravichander, M. Bull. Chem. Soc. Ethiop. 2019, 33, 143–148. doi:10.4314/bcse.v33i1.14
Return to citation in text: [1]
| 59. | Rochais, C.; Lisowski, V.; Dallemagne, P.; Rault, S. Tetrahedron Lett. 2004, 45, 6353–6355. doi:10.1016/j.tetlet.2004.06.047 |
| 60. | Abid, M.; Teixeira, L.; Török, B. Tetrahedron Lett. 2007, 48, 4047–4050. doi:10.1016/j.tetlet.2007.04.021 |
| 61. | Zuo, B.; Chen, J.; Liu, M.; Ding, J.; Wu, H.; Su, W. J. Chem. Res. 2009, 14–16. doi:10.3184/030823409x393628 |
| 68. | Azizi, N.; Khajeh-Amiri, A.; Ghafuri, H.; Bolourtchian, M.; Saidi, M. Synlett 2009, 2245–2248. doi:10.1055/s-0029-1217799 |
| 69. | Deng, H.-J.; Fang, Y.-J.; Chen, G.-W.; Liu, M.-C.; Wu, H.-Y.; Chen, J.-X. Appl. Organomet. Chem. 2012, 26, 164–167. doi:10.1002/aoc.1864 |
| 66. | Azizi, N.; Davoudpour, A.; Eskandari, F.; Batebi, E. Monatsh. Chem. 2013, 144, 405–409. doi:10.1007/s00706-012-0841-2 |
| 67. | Ma, F.-P.; Li, P.-H.; Li, B.-L.; Mo, L.-P.; Liu, N.; Kang, H.-J.; Liu, Y.-N.; Zhang, Z.-H. Appl. Catal., A 2013, 457, 34–41. doi:10.1016/j.apcata.2013.03.005 |
| 65. | Gourlay, B. S.; Molesworth, P. P.; Ryan, J. H.; Smith, J. A. Tetrahedron Lett. 2006, 47, 799–801. doi:10.1016/j.tetlet.2005.11.104 |
| 62. | Zhang, X.; Shi, J. Tetrahedron 2011, 67, 898–903. doi:10.1016/j.tet.2010.12.018 |
| 63. | Chatzopoulou, M.; Kotsampasakou, E.; Demopoulos, V. J. Synth. Commun. 2013, 43, 2949–2954. doi:10.1080/00397911.2012.753460 |
| 70. | Patil, R. N.; Kumar, A. V. ChemistrySelect 2018, 3, 9812–9818. doi:10.1002/slct.201801559 |
| 71. | Ramesh, K.; Murthy, S. N.; Nageswar, Y. V. D. Synth. Commun. 2012, 42, 2471–2477. doi:10.1080/00397911.2011.560744 |
| 72. | Hosseini-Sarvari, M.; Najafvand-Derikvandi, S.; Jarrahpour, A.; Heiran, R. Chem. Heterocycl. Compd. 2014, 49, 1732–1739. doi:10.1007/s10593-014-1425-3 |
| 79. | Ketcha, D. M.; Miles, K. C.; Mays, S. M.; Southerland, B. K.; Auvil, T. J. ARKIVOC 2010, No. xiv, 181–190. doi:10.3998/ark.5550190.0010.e17 |
| 80. | Polshettiwar, V.; Varma, R. S. Tetrahedron 2010, 66, 1091–1097. doi:10.1016/j.tet.2009.11.015 |
| 81. | Polshettiwar, V.; Baruwati, B.; Varma, R. S. Chem. Commun. 2009, 1837–1839. doi:10.1039/b900784a |
| 77. | Bandyopadhyay, D.; Mukherjee, S.; Granados, J. C.; Short, J. D.; Banik, B. K. Eur. J. Med. Chem. 2012, 50, 209–215. doi:10.1016/j.ejmech.2012.01.055 |
| 78. | Wang, P.; Ma, F.-P.; Zhang, Z.-H. J. Mol. Liq. 2014, 198, 259–262. doi:10.1016/j.molliq.2014.07.015 |
| 75. | Afsina, C. M. A.; Rohit, K. R.; Anilkumar, G. Results Chem. 2022, 4, 100350. doi:10.1016/j.rechem.2022.100350 |
| 76. | Khammas, A. J.; Yolacan, C.; Aydogan, F. Russ. J. Gen. Chem. 2018, 88, 2680–2683. doi:10.1134/s1070363218120332 |
| 73. | Wani, R. R.; Chaudhari, H. K.; Takale, B. S. J. Heterocycl. Chem. 2019, 56, 1337–1340. doi:10.1002/jhet.3507 |
| 74. | Ryabchuk, P.; Leischner, T.; Kreyenschulte, C.; Spannenberg, A.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2020, 59, 18679–18685. doi:10.1002/anie.202007613 |
| 83. | Bandyopadhyay, D.; Mukherjee, S.; Banik, B. K. Molecules 2010, 15, 2520–2525. doi:10.3390/molecules15042520 |
| 84. | Jafari, A. A.; Mahmoudi, H.; Mirjalili, B. F. J. Iran. Chem. Soc. 2011, 8, 851–856. doi:10.1007/bf03245915 |
| 82. | Wilson, M. A.; Filzen, G.; Welmaker, G. S. Tetrahedron Lett. 2009, 50, 4807–4809. doi:10.1016/j.tetlet.2009.06.079 |
| 1. | Gao, B.; Yang, B.; Feng, X.; Li, C. Nat. Prod. Rep. 2022, 39, 139–162. doi:10.1039/d1np00017a |
| 2. | Joule, J. A. Natural Products Containing Nitrogen Heterocycles—Some Highlights 1990–2015. In Advances in Heterocyclic Chemistry; Scriven, E. F. V.; Ramsden, C. A., Eds.; Academic Press, 2016; Vol. 119, pp 81–106. doi:10.1016/bs.aihch.2015.10.005 |
| 3. | Singh, N.; Singh, S.; Kohli, S.; Singh, A.; Asiki, H.; Rathee, G.; Chandra, R.; Anderson, E. A. Org. Chem. Front. 2021, 8, 5550–5573. doi:10.1039/d0qo01574a |
| 13. | Aatif, M.; Raza, M. A.; Javed, K.; Nashre-ul-Islam, S. M.; Farhan, M.; Alam, M. W. Antibiotics (Basel, Switz.) 2022, 11, 1750. doi:10.3390/antibiotics11121750 |
| 14. | Williamson, N. R.; Simonsen, H. T.; Ahmed, R. A. A.; Goldet, G.; Slater, H.; Woodley, L.; Leeper, F. J.; Salmond, G. P. C. Mol. Microbiol. 2005, 56, 971–989. doi:10.1111/j.1365-2958.2005.04602.x |
| 15. | Green, K. D.; Pang, A. H.; Thamban Chandrika, N.; Garzan, A.; Baughn, A. D.; Tsodikov, O. V.; Garneau-Tsodikova, S. ACS Infect. Dis. 2022, 8, 757–767. doi:10.1021/acsinfecdis.1c00450 |
| 37. | Clauson-Kaas, N.; Tyle, Z. Acta Chem. Scand. 1952, 6, 667–670. doi:10.3891/acta.chem.scand.06-0667 |
| 38. | Josey, A. D.; Jenner, E. L. J. Org. Chem. 1962, 27, 2466–2470. doi:10.1021/jo01054a042 |
| 91. | Rivera, S.; Bandyopadhyay, D.; Banik, B. K. Tetrahedron Lett. 2009, 50, 5445–5448. doi:10.1016/j.tetlet.2009.06.002 |
| 11. | Jiang, S.; Lu, H.; Liu, S.; Zhao, Q.; He, Y.; Debnath, A. K. Antimicrob. Agents Chemother. 2004, 48, 4349–4359. doi:10.1128/aac.48.11.4349-4359.2004 |
| 12. | Pegklidou, K.; Papastavrou, N.; Gkizis, P.; Komiotis, D.; Balzarini, J.; Nicolaou, I. Med. Chem. 2015, 11, 602–608. doi:10.2174/1573406411666150313161225 |
| 39. | Wang, Z. Piloty-Robinson Pyrrole Synthesis. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, 2010; pp 2217–2220. doi:10.1002/9780470638859.conrr500 |
| 40. | Piloty, O. Ber. Dtsch. Chem. Ges. 1910, 43, 489–498. doi:10.1002/cber.19100430182 |
| 41. | Robinson, G. M.; Robinson, R. J. Chem. Soc., Trans. 1918, 113, 639–645. doi:10.1039/ct9181300639 |
| 8. | Paludetto, M.-N.; Bijani, C.; Puisset, F.; Bernardes-Génisson, V.; Arellano, C.; Robert, A. J. Med. Chem. 2018, 61, 7849–7860. doi:10.1021/acs.jmedchem.8b00812 |
| 9. | Mateev, E.; Georgieva, M.; Zlatkov, A. J. Pharm. Pharm. Sci. 2022, 25, 24–40. doi:10.18433/jpps32417 |
| 10. | Kuznietsova, H.; Dziubenko, N.; Byelinska, I.; Hurmach, V.; Bychko, A.; Lynchak, O.; Milokhov, D.; Khilya, O.; Rybalchenko, V. J. Drug Targeting 2020, 28, 547–563. doi:10.1080/1061186x.2019.1703189 |
| 31. | Paal, C. Ber. Dtsch. Chem. Ges. 1884, 17, 2756–2767. doi:10.1002/cber.188401702228 |
| 32. | Knorr, L. Ber. Dtsch. Chem. Ges. 1884, 17, 2863–2870. doi:10.1002/cber.188401702254 |
| 33. | Balakrishna, A.; Aguiar, A.; Sobral, P. J. M.; Wani, M. Y.; Almeida e Silva, J.; Sobral, A. J. F. N. Catal. Rev.: Sci. Eng. 2019, 61, 84–110. doi:10.1080/01614940.2018.1529932 |
| 89. | Abid, M.; Landge, S. M.; Török, B. Org. Prep. Proced. Int. 2006, 38, 495–500. doi:10.1080/00304940609356444 |
| 4. | Heravi, M. M.; Zadsirjan, V. RSC Adv. 2020, 10, 44247–44311. doi:10.1039/d0ra09198g |
| 5. | Gupta, S. S.; Kumari, S.; Kumar, I.; Sharma, U. Chem. Heterocycl. Compd. 2020, 56, 433–444. doi:10.1007/s10593-020-02678-5 |
| 6. | Kim, J.; Park, M.; Choi, J.; Singh, D. K.; Kwon, H. J.; Kim, S. H.; Kim, I. Bioorg. Med. Chem. Lett. 2019, 29, 1350–1356. doi:10.1016/j.bmcl.2019.03.044 |
| 7. | Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K. K.; Jonnalagadda, S. B. Molecules 2020, 25, 1909. doi:10.3390/molecules25081909 |
| 34. | Hantzsch, A. Ber. Dtsch. Chem. Ges. 1890, 23, 1474–1476. doi:10.1002/cber.189002301243 |
| 35. | Leonardi, M.; Estévez, V.; Villacampa, M.; Menéndez, J. C. Synthesis 2019, 51, 816–828. doi:10.1055/s-0037-1610320 |
| 36. | Trautwein, A. W.; Süßmuth, R. D.; Jung, G. Bioorg. Med. Chem. Lett. 1998, 8, 2381–2384. doi:10.1016/s0960-894x(98)00430-2 |
| 90. | Silveira, C. C.; Fortes, M. P.; Mendes, S. R. Curr. Org. Chem. 2012, 16, 1540–1548. doi:10.2174/138527212800672538 |
| 23. | Di Santo, R.; Tafi, A.; Costi, R.; Botta, M.; Artico, M.; Corelli, F.; Forte, M.; Caporuscio, F.; Angiolella, L.; Palamara, A. T. J. Med. Chem. 2005, 48, 5140–5153. doi:10.1021/jm048997u |
| 24. | Zhang, S.-G.; Liang, C.-G.; Sun, Y.-Q.; Teng, P.; Wang, J.-Q.; Zhang, W.-H. Mol. Diversity 2019, 23, 915–925. doi:10.1007/s11030-019-09920-z |
| 27. | Kocaoğlu, E.; Talaz, O.; Çavdar, H.; Şentürk, M.; Supuran, C. T.; Ekinci, D. J. Enzyme Inhib. Med. Chem. 2019, 34, 51–54. doi:10.1080/14756366.2018.1520228 |
| 28. | Williams, I. S.; Joshi, P.; Gatchie, L.; Sharma, M.; Satti, N. K.; Vishwakarma, R. A.; Chaudhuri, B.; Bharate, S. B. Bioorg. Med. Chem. Lett. 2017, 27, 3683–3687. doi:10.1016/j.bmcl.2017.07.010 |
| 87. | Ozaki, T.; Yorimitsu, H.; Perry, G. J. P. Tetrahedron 2022, 117–118, 132830. doi:10.1016/j.tet.2022.132830 |
| 20. | Narule, M. N.; Gaidhane, M. K.; Gaidhane, P. K. J. Pharm. Res. (Mohali, India) 2013, 6, 626–632. doi:10.1016/j.jopr.2013.04.046 |
| 21. | Mallikarjuna Reddy, G.; Camilo, A., Jr.; Raul Garcia, J. Bioorg. Chem. 2021, 106, 104465. doi:10.1016/j.bioorg.2020.104465 |
| 22. | Fatahala, S. S.; Shalaby, E. A.; Kassab, S. E.; Mohamed, M. S. Anti-Cancer Agents Med. Chem. 2015, 15, 517–526. doi:10.2174/1871520615666150105113946 |
| 29. | Knorr, L. Ber. Dtsch. Chem. Ges. 1884, 17, 1635–1642. doi:10.1002/cber.18840170220 |
| 30. | Alberola, A.; González Ortega, A.; Luisa Sádaba, M.; Sañudo, C. Tetrahedron 1999, 55, 6555–6566. doi:10.1016/s0040-4020(99)00289-6 |
| 88. | Aydogan, F.; Yolacan, C. J. Chem. 2013, 2013, 976724. doi:10.1155/2013/976724 |
| 18. | Battilocchio, C.; Poce, G.; Alfonso, S.; Porretta, G. C.; Consalvi, S.; Sautebin, L.; Pace, S.; Rossi, A.; Ghelardini, C.; Di Cesare Mannelli, L.; Schenone, S.; Giordani, A.; Di Francesco, L.; Patrignani, P.; Biava, M. Bioorg. Med. Chem. 2013, 21, 3695–3701. doi:10.1016/j.bmc.2013.04.031 |
| 19. | Said Fatahala, S.; Hasabelnaby, S.; Goudah, A.; Mahmoud, G. I.; Helmy Abd-El Hameed, R. Molecules 2017, 22, 461. doi:10.3390/molecules22030461 |
| 85. | Mahmoudi, H.; Jafari, A. A. ChemCatChem 2013, 5, 3743–3749. doi:10.1002/cctc.201300623 |
| 16. | Palmer, M. J.; Deng, X.; Watts, S.; Krilov, G.; Gerasyuto, A.; Kokkonda, S.; El Mazouni, F.; White, J.; White, K. L.; Striepen, J.; Bath, J.; Schindler, K. A.; Yeo, T.; Shackleford, D. M.; Mok, S.; Deni, I.; Lawong, A.; Huang, A.; Chen, G.; Wang, W.; Jayaseelan, J.; Katneni, K.; Patil, R.; Saunders, J.; Shahi, S. P.; Chittimalla, R.; Angulo-Barturen, I.; Jiménez-Díaz, M. B.; Wittlin, S.; Tumwebaze, P. K.; Rosenthal, P. J.; Cooper, R. A.; Aguiar, A. C. C.; Guido, R. V. C.; Pereira, D. B.; Mittal, N.; Winzeler, E. A.; Tomchick, D. R.; Laleu, B.; Burrows, J. N.; Rathod, P. K.; Fidock, D. A.; Charman, S. A.; Phillips, M. A. J. Med. Chem. 2021, 64, 6085–6136. doi:10.1021/acs.jmedchem.1c00173 |
| 17. | Kancharla, P.; Li, Y.; Yeluguri, M.; Dodean, R. A.; Reynolds, K. A.; Kelly, J. X. J. Med. Chem. 2021, 64, 8739–8754. doi:10.1021/acs.jmedchem.1c00748 |
| 25. | McArthur, K. A.; Mitchell, S. S.; Tsueng, G.; Rheingold, A.; White, D. J.; Grodberg, J.; Lam, K. S.; Potts, B. C. M. J. Nat. Prod. 2008, 71, 1732–1737. doi:10.1021/np800286d |
| 26. | Ding, X.-B.; Furkert, D. P.; Brimble, M. A. Chem. Commun. 2016, 52, 12638–12641. doi:10.1039/c6cc07532k |
| 86. | Rohit, K. R.; Meera, G.; Anilkumar, G. J. Heterocycl. Chem. 2022, 59, 194–200. doi:10.1002/jhet.4372 |
| 47. | Plasencia, C.; Grande, F.; Oshima, T.; Cao, X.; Yamada, R.; Sanchez, T.; Aiello, F.; Garofalo, A.; Neamati, N. Cancer Biol. Ther. 2009, 8, 458–465. doi:10.4161/cbt.8.5.7741 |
| 48. | Butini, S.; Brindisi, M.; Gemma, S.; Minetti, P.; Cabri, W.; Gallo, G.; Vincenti, S.; Talamonti, E.; Borsini, F.; Caprioli, A.; Stasi, M. A.; Di Serio, S.; Ros, S.; Borrelli, G.; Maramai, S.; Fezza, F.; Campiani, G.; Maccarrone, M. J. Med. Chem. 2012, 55, 6898–6915. doi:10.1021/jm300689c |
| 42. | Barton, D. H. R.; Zard, S. Z. J. Chem. Soc., Chem. Commun. 1985, 1098–1100. doi:10.1039/c39850001098 |
| 43. | He, X.-L.; Zhao, H.-R.; Song, X.; Jiang, B.; Du, W.; Chen, Y.-C. ACS Catal. 2019, 9, 4374–4381. doi:10.1021/acscatal.9b00767 |
| 44. | Ono, N. Heterocycles 2008, 75, 243. doi:10.3987/rev-07-622 |
| 45. | Yoshimitsu, T.; Ino, T.; Tanaka, T. Org. Lett. 2008, 10, 5457–5460. doi:10.1021/ol802225g |
| 46. | Ma, K.; Yin, X.; Dai, M. Angew. Chem. 2018, 130, 15429–15432. doi:10.1002/ange.201809114 |
| 92. | Gullapelli, K.; Brahmeshwari, G.; Ravichander, M. Bull. Chem. Soc. Ethiop. 2019, 33, 143–148. doi:10.4314/bcse.v33i1.14 |
| 57. | Kumar, S.; Krishnakanth, S.; Mathew, J.; Pomerantz, Z.; Lellouche, J.-P.; Ghosh, S. J. Phys. Chem. C 2014, 118, 2570–2579. doi:10.1021/jp411098y |
| 58. | Fang, Y.; Leysen, D.; Ottenheijm, H. C. J. Synth. Commun. 1995, 25, 1857–1861. doi:10.1080/00397919508015431 |
| 55. | Wang, Z. Clauson-Kaas Reaction. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, 2010; pp 665–668. doi:10.1002/9780470638859.conrr146 |
| 56. | Sonnet, P.; Dallemagne, P.; Guillon, J.; Enguehard, C.; Stiebing, S.; Tanguy, J.; Bureau, R.; Rault, S.; Auvray, P.; Moslemi, S.; Sourdaine, P.; Séralini, G.-E. Bioorg. Med. Chem. 2000, 8, 945–955. doi:10.1016/s0968-0896(00)00024-9 |
| 54. | Wynn, T. A. Pyrroles and Pyrrolidines. In Name Reactions in Heterocyclic Chemistry II; Li, J. J.; Corey, E. J., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp 41–82. doi:10.1002/9781118092828.ch2 |
| 37. | Clauson-Kaas, N.; Tyle, Z. Acta Chem. Scand. 1952, 6, 667–670. doi:10.3891/acta.chem.scand.06-0667 |
| 49. | Kiskan, B.; Gacal, B.; Asan, M.; Gunaydin, E. C.; Yilmaz, I.; Yagci, Y. Polym. Bull. 2011, 67, 609–621. doi:10.1007/s00289-010-0412-9 |
| 50. | Gursoy, S. S.; Uygun (GOK), A.; Tilki, T. J. Macromol. Sci., Part A: Pure Appl. Chem. 2010, 47, 681–688. doi:10.1080/10601325.2010.483376 |
| 51. | Singh, D. K.; Nath, M. Beilstein J. Org. Chem. 2014, 10, 808–813. doi:10.3762/bjoc.10.76 |
| 52. | Singh, D. K.; Nath, M. Org. Biomol. Chem. 2015, 13, 1836–1845. doi:10.1039/c4ob02370f |
| 53. | Berionni Berna, B.; Nardis, S.; Galloni, P.; Savoldelli, A.; Stefanelli, M.; Fronczek, F. R.; Smith, K. M.; Paolesse, R. Org. Lett. 2016, 18, 3318–3321. doi:10.1021/acs.orglett.6b01314 |
© 2023 Singh and Kumar; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.