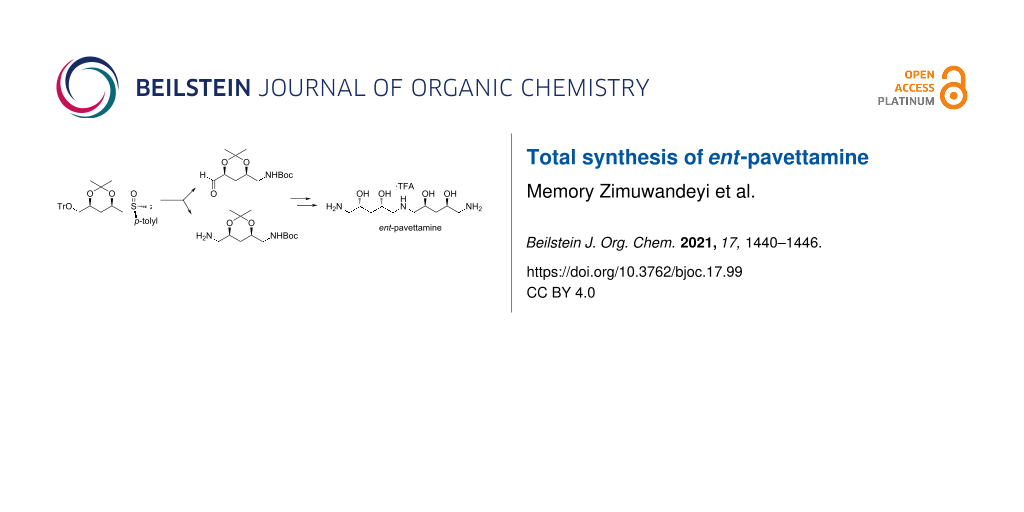
Abstract
Pavettamine, a plant toxin first isolated from Pavetta harborii in 1995, was previously identified as a polyamine with C2 symmetry and a 1,3-syn-diol moiety on a C10 carbon backbone – one of very few substituted polyamines to be isolated from nature. Its absolute configuration was later established by our first reported total synthesis in 2010. Herein we report the first total synthesis of the enantiomer of pavettamine, ent-pavettamine. The symmetrical structure of the molecule allows for the synthesis of a common C5 fragment that can be divergently transformed into two synthons for later convergent coupling to furnish the target carbon framework. Based on the success of the protocol we employed for the synthesis of the naturally occurring pavettamine, (S)-malic acid was again the starting material of choice for the synthesis of the two individual C5 fragments, with strategic differences in terminal-group manipulation allowing for the synthesis of ent-pavettamine rather than pavettamine. Chain extension and stereoselective ketone reduction were achieved using the (R)-methyl p-tolyl sulfoxide chiral auxiliary to give the desired 1,3-syn-diol C5 unit. A protecting-group strategy was also developed for the orthogonal protection of the alcohol and amine functional groups as they were unveiled. The functionalized C5 fragments were coupled via reductive amination revealing the C10 carbon backbone. Deprotection of the alcohol and amine functional groups successfully provided ent-pavettamine as a TFA salt.

Graphical Abstract
Introduction
The identification and first reported synthesis of pavettamine (1) heralded the arrival of a novel and uniquely hydroxylated polyamine (PA) (Figure 1) [1]. In general, polyamines are described as aliphatic organic compounds with two or more primary amine substituents, connected by one or more unsubstituted methylene linkages, within their structure [2]. The interest in polyamines arises from the diverse roles and functions they play in biological systems [3,4]; for example, as stabilizers of RNA and DNA, secondary messengers, nutrients, antioxidants, growth factors, and metabolic regulators [5]. In addition to this, some PAs are currently being used as therapeutic drugs, being incorporated as drug conjugates, or are under investigation for other applications [6-12].
![[1860-5397-17-99-1]](/bjoc/content/figures/1860-5397-17-99-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of pavettamine 1 and its enantiomer 2.
Figure 1: Structure of pavettamine 1 and its enantiomer 2.
The elucidation of the structure of pavettamine revealed a new class of polyamines with a substituted methylene linkage. Biological studies have shown that this toxin is responsible for “quick disease” (gousiekte) in ruminant animals, which causes inhibition of protein synthesis in the cardiovascular organs [13]. The unique structure coupled with the biological effects of this polyamine prompted the current study, which aimed to establish a method for the synthesis of ent-pavettamine (2) so as to contribute towards a comprehensive structure–activity relationship study of pavettamine. With the absolute stereochemistry of pavettamine having been established previously [1], this study focused on developing a route for the synthesis of enantiomer 2 by modification of the established protocol used for the synthesis of 1. The plan was that the alternative strategy would lead to an improvement in the overall yield whilst eliminating some of the troublesome steps.
Results and Discussion
The synthetic protocol we originally used for 1 was adapted for the synthesis of 2 owing to the excellent stereochemical control achieved using this route [1]. A modular approach was employed as it was envisioned that using this protocol would allow for the synthesis of the desired compound in a reasonable number of steps. As before, (S)-malic acid (3) was chosen as the starting material because its inherent chirality reduced the complexity of the synthetic process. Given the fact that the target molecule has C2 symmetry, it was prudent that a synthetic route starting from a common C5 subunit be chosen, with subsequent functionalization giving rise to two C5 units with compatible groups required for linking: in this case an amine and an aldehyde. In order for the synthesis to yield ent-pavettamine instead of pavettamine, the key was to functionalize the “opposite end” of each of the C5 units required for linking, when compared to our original pavettamine synthesis [1]. This opposite functionalization ensured that the enantiomer would result from the synthesis.
For purposes of comparison between the original route and the route described here, our efforts commenced with the synthesis of the common C5 unit 4, using our previously published methodology in an overall yield of 8% over 8 steps (Scheme 1) [1].
![[1860-5397-17-99-i1]](/bjoc/content/inline/1860-5397-17-99-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Established route for the synthesis of intermediate 4 [1].
Scheme 1: Established route for the synthesis of intermediate 4 [1].
An alternative strategy for the synthesis of 4 was developed so as to avoid a cumbersome continuous extraction step of a triol associated with the established method, in addition to improving the overall yield. The first step involved esterification of malic acid using methanol, followed by selective reduction of the resulting dimethyl ester using borane dimethyl sulfide (BMS) yielding diol 5 (Scheme 2) [1,14,15].
![[1860-5397-17-99-i2]](/bjoc/content/inline/1860-5397-17-99-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Alternative route. Reaction conditions: a) TrCl, pyridine, rt, overnight, 100%; b) DMAP, imidazole, TBS, DCM, rt, 12 h, 65%; c) LDA, (R)-methyl p-tolyl sulfoxide, THF, −78 °C to rt, 2.5 h, 55% yield of 8.
Scheme 2: Alternative route. Reaction conditions: a) TrCl, pyridine, rt, overnight, 100%; b) DMAP, imidazole,...
The step achieved chemo-differentiation of the carboxyl groups present in 3 whilst retaining the stereogenic center. Divergence from the original route to pavettamine occurred after recovery of diol 5; and a different protecting group strategy was applied to arrive at synthon 4. The primary alcohol of the diol 5 was regioselectively protected using the trityl group under basic conditions yielding 6 in quantitative yield. Our choice of the trityl protecting group was based on the fact that it could be selectively added in the presence of the secondary alcohol and selectively removed at a later stage in the presence of an orthogonally-protected secondary alcohol [16]. At this stage, the selective functionalization and protection of the primary and secondary hydroxy groups (and later amino groups) was critical for the successful synthesis of the target compound. Unfortunately, each protecting group incorporated increased the synthesis by two non-productive steps; nonetheless our aim to achieve an improved yield of intermediate 4 was met.
Subsequent TBS protection of the secondary alcohol 6 gave rise to compound 7 in a moderate yield of 65%. Once the ester precursor 7 had been successfully synthesized, the next step involved conversion of the ester into its β-keto sulfoxide via substitution with the anion of (R)-methyl p-tolyl sulfoxide according to published procedures [1,17]. The desired product 8 was recovered in a yield of 55% and a significant amount of side product 9 was recovered in a yield of 24%. The identity of the side product was not apparent at first because both products were light yellow solids with the same melting point of 115–116 °C and both had the same measured mass when analyzed by HRMS. The differences were that the specific optical rotation was measured to be +67.0° for compound 8 and +91.4° for compound 9 and their 1H NMR spectra differed for the signals close to the OTBS center.
At this point, it was crucial to conclusively characterize the unexpected product 9. Fortuitously, we were able to obtain crystals for this compound after several recrystallization attempts. The crystal structure for 9 (Figure 2) very surprisingly showed the OTBS group on the carbon atom α to the carbonyl group, instead of β to the carbonyl carbon for the desired compound 8. These results were puzzling, and how this product could have been formed from reaction of compound 7 with (R)-methyl p-tolyl sulfoxide was not obvious.
![[1860-5397-17-99-2]](/bjoc/content/figures/1860-5397-17-99-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of compound 9.
Figure 2: Crystal structure of compound 9.
After investigating several possibilities coupled with careful examination of the preceding steps it became clear that compound 9 was not formed in the reaction of 7 with the sulfoxide auxiliary. Unbeknownst to us at the time, the formation of diol 5 was not as highly selective as expected and this reaction produced two inseparable diols, the desired product 5 and unwanted side product 11 (Scheme 3) which were not distinguishable by 1H NMR spectroscopy. Carrying side product 11 through the subsequent synthetic steps resulted in compounds 12 and 13 and ultimately product 9 (Scheme 2), the first point in the sequence where the isomeric products were separable and clearly distinguishable by 1H NMR spectroscopy.
![[1860-5397-17-99-i3]](/bjoc/content/inline/1860-5397-17-99-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Sequence showing the source of compound 9.
Scheme 3: Sequence showing the source of compound 9.
The formation of 11 was not apparent when using an acetonide protection strategy (Scheme 1) because the desired product 14 was easily separated by column chromatography from the undesired compound 15, emanating from diol 11 (Scheme 3). However, compound 12 was not separable from 6 by column chromatography and TLC visualization only showed a single spot. The presence of the unwanted side product 12 only became evident retrospectively when a close examination of the 1H NMR spectrum revealed a small extra methoxy signal, showing a ratio of between 21:1 and 9:1 of the desired product 6 to the unwanted material 12. The product ratios changed with each subsequent synthetic step, which suggests different reaction rates occurred for each of the two isomers. Ultimately, a ratio of 55:24 was observed for compounds 8 and 9. We attempted to eliminate or minimize the formation of diol 11 by performing the reduction step at temperatures lower than 15 °C but this significantly reduced the yield of the desired product, even with longer reaction times. Therefore, we had to be satisfied with separation of desired product 8 from unwanted 9 after the addition of the chiral auxiliary.
Our attention then turned to the stereoselective reduction of 8. Chelation-controlled reduction of 8 using ZnCl2 and DIBALH allowed for the successful formation of 16 as a single diastereomer (Scheme 4), as evidenced by 1H NMR spectroscopy [18].
![[1860-5397-17-99-i4]](/bjoc/content/inline/1860-5397-17-99-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Stereoselective reduction of intermediate 8 as key step towards intermediate 4. Reaction conditions: a) i) ZnCl2, rt, 2 h; ii) DIBALH, THF, −78 °C to rt, 2 h, 87%; b) TBAF, THF, 0 °C, 4 h, 87%; c) 5% p-TsOH, 2,2-dimethoxypropane, acetone, rt, 1 h, 86%.
Scheme 4: Stereoselective reduction of intermediate 8 as key step towards intermediate 4. Reaction conditions...
TBS deprotection furnished 17, followed by acetonide protection of the 1,3-diol functionality to yield the previously synthesized intermediate 4. The acetonide protecting group was chosen because of its stability under the future reaction conditions in addition to it being a convenient monitor of the relative configuration of the 1,3-diol motif [19]. The group is also associated with ease of deprotection, which is well documented in carbohydrate and sugar chemistry [20]. Comparison of our new route involving trityl and TBS protections with our previously published acetonide protection route for the synthesis of 4, showed that the new route doubled the overall yield to 16% for a similar number of steps. The specific optical rotation for 4 of +28.1° in acetone was comparable to that obtained previously [1]. Single crystal X-ray analysis of compound 4 (Figure 3), further confirmed the identity and stereochemistry of the compound.
![[1860-5397-17-99-3]](/bjoc/content/figures/1860-5397-17-99-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Single crystal X-ray structure of compound 4.
Figure 3: Single crystal X-ray structure of compound 4.
Once the crucial 1,3-syn-diol moiety had been obtained, attention shifted to functionalization and differentiation of the terminal carbon atoms for the synthesis of the two C5 fragments. Intermediate 4 was subjected to the Pummerer rearrangement by treatment with TFAA in collidine at 0 °C, followed by hydrolysis with solid K2CO3 and water for 30 min (Scheme 5). The desired aldehyde 18 was recovered in an excellent yield of 99%, and product epimerization was not detected based on 13C NMR spectroscopic analysis. This was confirmed by analysis of the 13C chemical shifts of the acetonide group, whose values differ for the syn-1,3 diol when compared to the anti-1,3-diol as described after an extensive study by Rychnovsky et al. [21].
![[1860-5397-17-99-i5]](/bjoc/content/inline/1860-5397-17-99-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of the C5 fragments from intermediate 4. Reaction conditions: a) i) TFAA, collidine, 0 °C, 30 min; ii) K2CO3, rt, 30 min, 99%; b) benzylamine, DCE, THF, sodium triacetoxyborohydride, N2, rt, 24 h, 60%; c) ethanol, 10% Pd/C, H2, rt, 24 h, 84%; d) Boc anhydride, DMAP, THF, rt, overnight, 100%; e) Na, NH3, THF, -60 °C, 1 h, 81%.
Scheme 5: Synthesis of the C5 fragments from intermediate 4. Reaction conditions: a) i) TFAA, collidine, 0 °C...
The reductive amination of the aldehyde with benzylamine allowed for the introduction of a protected terminal amine group to the C5 fragment, yielding 19 in a modest yield of 60%. The next step involved the selective removal of the trityl group so as to facilitate the synthesis of the two separate C5 fragments which would be coupled to yield the desired product. Unfortunately, the anticipated selective removal of the trityl group in the presence of an N-benzyl group did not yield the desired product [22-26]. Several selective trityl deprotection attempts gave different results: either the reaction did not proceed at all yielding starting material, or both the trityl and benzyl group were removed or all three protecting groups were removed. Based on these results, it was evident that the selective removal of the trityl group in the presence of the benzyl group is substrate specific. Eventually, hydrogenation under neutral conditions at atmospheric pressure for 24 h allowed for the selective removal of the benzyl group, affording amine 20 in a yield of 84%. The Boc group was chosen for the re-protection of the amine, furnishing 21 in quantitative yield. Due to the significant difference between the trityl and the Boc groups, selective trityl removal was now possible under Birch reduction conditions. The primary alcohol 22 was recovered in an excellent yield of 81%.
The alcohol was apportioned into two parts to synthesize the two required C5 fragments separately, by converting the alcohol to an amine for one fragment and an aldehyde for the second fragment. The conversion of the alcohol to the aldehyde was first attempted using MnO2 but only the starting material was recovered. Use of PCC resulted in product epimerization and so the method was abandoned. Success was achieved by use of IBX in DMSO, overnight, resulting in the recovery of a quantitative yield of 23 (Scheme 6). The amine was synthesized by first converting the primary alcohol to a tosylate under basic conditions affording 24 in a yield of 83%. The displacement of the tosyl group with an azide whilst heating the reaction at 80 °C allowed for the isolation of azide 25 in a good yield of 75%. Heating at higher temperatures resulted in product decomposition. Hydrogenation of the azide 25 at 4 atm whilst incorporating Pd/C as a catalyst furnished the desired amine 26 in quantitative yield. Performing the reaction at a lower pressure resulted in the recovery of starting material only.
![[1860-5397-17-99-i6]](/bjoc/content/inline/1860-5397-17-99-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of ent-pavettamine as the TFA salt 28. Reaction conditions: a) IBX, DMSO, rt, overnight, quantitative yield; b) TsCl, DMAP, DCM, rt, 24 h, 83%; c) NaN3, DMF, 80 °C, 3 h, 75% yield; d) 10% Pd/C, H2, ethanol, rt, 12 h, quantitative yield; e) sodium triacetoxyborohydride, 1,2-DCE, THF, rt, 24 h, 95%; f) 95% TFA, 2.5% H2O, 2.5% triisopropylamine, rt, overnight, quantitative yield.
Scheme 6: Synthesis of ent-pavettamine as the TFA salt 28. Reaction conditions: a) IBX, DMSO, rt, overnight, ...
With the two C5 fragments in hand, convergent coupling of aldehyde 23 and amine 26 was achieved via reductive amination employing sodium triacetoxyborohydride as the reducing agent. The desired product precursor 27 was successfully recovered as a yellow oil in a yield of 95%. HRMS showed the desired mass whilst NMR spectroscopy showing a single set of signals for each half of the structure, confirmed the desired C2 symmetry and stereochemical integrity. The ultimate deprotection step proved to be more difficult than expected. Ordinarily, there are a number of methods that can be employed to simultaneously deprotect the Boc and acetonide groups [27-29], but they could not effect the desired transformation in this case. After several failed attempts, a procedure typically used for Boc deprotection in peptide synthesis utilizing TFA and triisopropylsilane was successfully used. Product purification furnished the desired enantiomer of pavettamine as a TFA salt (28). The presence of TFA was evident in the 13C NMR spectrum due to the observed distorted quartet at δ 163.7 for the C=O group displaying 2JCF coupling and a quartet at δ 120.3 with a 1JCF coupling constant of 291 Hz. 19F NMR and HRMS further confirmed the presence of the recovered TFA salt of ent-pavettamine (28). However, attempts to neutralize the TFA salt to obtain the free amine were unsuccessful, most often resulting in product decomposition. Also the attempted product purification by recrystallization from methanol did not improve the purity of the product. Spectroscopic data of the synthesized ent-pavettamine and the pavettamine originally synthesized could unfortunately not be directly compared because the enantiomer was recovered as a TFA salt, whilst pavettamine was recovered as a neutral polyamine.
Conclusion
ent-Pavettamine, isolated as the TFA salt, was successfully synthesized for the first time. Key steps in the route were the synthesis of the C5 intermediate 4 in twice the yield previously achieved, and the effective use of reductive amination for coupling of the two C5 units. The successful strategy employed was to functionalize the two C5 units at the opposite end to that previously described for the synthesis of pavettamine, in order to obtain the enantiomer. The final deprotection step proved particularly efficient, with two protecting groups being removed simultaneously to unveil the desired target.
Supporting Information
Detailed experimental methods and NMR spectra for all compounds prepared and crystal structure data for compounds 4 and 9 is available as supporting information.
| Supporting Information File 1: NMR data of all compounds. | ||
| Format: PDF | Size: 7.0 MB | Download |
| Supporting Information File 2: Crystal structure data for compound 4. | ||
| Format: CIF | Size: 975.1 KB | Download |
| Supporting Information File 3: Crystal structure data for compound 9. | ||
| Format: CIF | Size: 743.7 KB | Download |
References
-
Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Bienz, S.; Bisegger, P.; Guggisberg, A.; Hesse, M. Nat. Prod. Rep. 2005, 22, 647–658. doi:10.1039/b413742f
Return to citation in text: [1] -
Casero, R. A., Jr.; Marton, L. J. Nat. Rev. Drug Discovery 2007, 6, 373–390. doi:10.1038/nrd2243
Return to citation in text: [1] -
Wallace, H. M. Eur. J. Clin. Invest. 2000, 30, 1–3. doi:10.1046/j.1365-2362.2000.00585.x
Return to citation in text: [1] -
Manku, S.; Laplante, C.; Kopac, D.; Chan, T.; Hall, D. G. J. Org. Chem. 2001, 66, 874–885. doi:10.1021/jo005647g
Return to citation in text: [1] -
Seiler, N.; Raul, F. J. Cell. Mol. Med. 2005, 9, 623–642. doi:10.1111/j.1582-4934.2005.tb00493.x
Return to citation in text: [1] -
Bergeron, R. J.; Wiegand, J.; Fannin, T. L. Dig. Dis. Sci. 2001, 46, 2615–2623. doi:10.1023/a:1012750723644
Return to citation in text: [1] -
Liew, L. P. P.; Kaiser, M.; Copp, B. R. Bioorg. Med. Chem. Lett. 2013, 23, 452–454. doi:10.1016/j.bmcl.2012.11.072
Return to citation in text: [1] -
Pearce, A. N.; Chia, E. W.; Berridge, M. V.; Maas, E. W.; Page, M. J.; Harper, J. L.; Webb, V. L.; Copp, B. R. Tetrahedron 2008, 64, 5748–5755. doi:10.1016/j.tet.2008.04.012
Return to citation in text: [1] -
Lizzi, F.; Veronesi, G.; Belluti, F.; Bergamini, C.; López-Sánchez, A.; Kaiser, M.; Brun, R.; Krauth-Siegel, R. L.; Hall, D. G.; Rivas, L.; Bolognesi, M. L. J. Med. Chem. 2012, 55, 10490–10500. doi:10.1021/jm301112z
Return to citation in text: [1] -
Bolognesi, M. L.; Banzi, R.; Bartolini, M.; Cavalli, A.; Tarozzi, A.; Andrisano, V.; Minarini, A.; Rosini, M.; Tumiatti, V.; Bergamini, C.; Fato, R.; Lenaz, G.; Hrelia, P.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. J. Med. Chem. 2007, 50, 4882–4897. doi:10.1021/jm070559a
Return to citation in text: [1] -
Bitonti, A. J.; McCann, P. P.; Sjoerdsma, A. The Effects of Polyamine Analogues on Malaria Parasites In Vitro and In Vivo. In Progress in Polyamine Research: Novel Biochemical, Pharmacological, and Clinical Aspects; Zappia, V.; Pegg, A. E., Eds.; Springer US: Boston, MA, USA, 1988; pp 717–726. doi:10.1007/978-1-4684-5637-0_63
Return to citation in text: [1] -
Hay, L.; Schultz, R. A.; Schutte, P. J. Onderstepoort J. Vet. Res. 2008, 75, 249–253. doi:10.4102/ojvr.v75i3.100
Return to citation in text: [1] -
Yadav, J. S.; Reddy, P. A. N.; Reddy, Y. J.; Meraj, S.; Prasad, A. R. Eur. J. Org. Chem. 2013, 6317–6324. doi:10.1002/ejoc.201300623
Return to citation in text: [1] -
Dardonville, C.; Gilbert, I. H. Org. Biomol. Chem. 2003, 1, 552–559. doi:10.1039/b210606j
Return to citation in text: [1] -
Wuts, P. G. M.; Greene, T. W. Greene's Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons: New York, NY, USA, 2006. doi:10.1002/0470053488
Return to citation in text: [1] -
Solladié, G.; Hutt, J.; Girardin, A. Synthesis 1987, 173. doi:10.1055/s-1987-27877
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2006, 62, 5559–5601. doi:10.1016/j.tet.2006.03.093
Return to citation in text: [1] -
Rychnovsky, S. D. J. Org. Chem. 1989, 54, 4982–4984. doi:10.1021/jo00282a005
Return to citation in text: [1] -
Coppola, G. M.; Schuster, H. F. α-Hydroxy Acids in Enantioselective Syntheses; Wiley-VCH: Weinheim, Germany, 1997. doi:10.1002/352760085x
Return to citation in text: [1] -
Rychnovsky, S. D.; Skalitzky, D. J. Tetrahedron Lett. 1990, 31, 945–948. doi:10.1016/s0040-4039(00)94399-5
Return to citation in text: [1] -
Das, B.; Mahender, G.; Sunil Kumar, V.; Chowdhury, N. Tetrahedron Lett. 2004, 45, 6709–6711. doi:10.1016/j.tetlet.2004.07.075
Return to citation in text: [1] -
Hwu, J. R.; Jain, M. L.; Tsai, F.-Y.; Tsay, S.-C.; Balakumar, A.; Hakimelahi, G. H. J. Org. Chem. 2000, 65, 5077–5088. doi:10.1021/jo000024o
Return to citation in text: [1] -
Kohli, V.; Blöcker, H.; Köster, H. Tetrahedron Lett. 1980, 21, 2683–2686. doi:10.1016/s0040-4039(00)78579-0
Return to citation in text: [1] -
Behloul, C.; Chouti, A.; Chabour, I.; Bey, H. B.; Guijarro, D.; Foubelo, F.; Nájera, C.; Yus, M. Tetrahedron Lett. 2016, 57, 3526–3528. doi:10.1016/j.tetlet.2016.06.110
Return to citation in text: [1] -
Bessodes, M.; Komiotis, D.; Antonakis, K. Tetrahedron Lett. 1986, 27, 579–580. doi:10.1016/s0040-4039(00)84045-9
Return to citation in text: [1] -
Dandepally, S. R.; Williams, A. L. Tetrahedron Lett. 2009, 50, 1071–1074. doi:10.1016/j.tetlet.2008.12.074
Return to citation in text: [1] -
Jih Ru Hwu; Jain, M. L.; Shwu-Chen, T.; Hakimelahi, G. H. Tetrahedron Lett. 1996, 37, 2035–2038. doi:10.1016/0040-4039(96)00211-0
Return to citation in text: [1] -
Jana, S.; Roy, S. C. Indian J. Chem. 2007, 46B, 707–709.
Return to citation in text: [1]
| 21. | Rychnovsky, S. D.; Skalitzky, D. J. Tetrahedron Lett. 1990, 31, 945–948. doi:10.1016/s0040-4039(00)94399-5 |
| 20. | Coppola, G. M.; Schuster, H. F. α-Hydroxy Acids in Enantioselective Syntheses; Wiley-VCH: Weinheim, Germany, 1997. doi:10.1002/352760085x |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 6. | Seiler, N.; Raul, F. J. Cell. Mol. Med. 2005, 9, 623–642. doi:10.1111/j.1582-4934.2005.tb00493.x |
| 7. | Bergeron, R. J.; Wiegand, J.; Fannin, T. L. Dig. Dis. Sci. 2001, 46, 2615–2623. doi:10.1023/a:1012750723644 |
| 8. | Liew, L. P. P.; Kaiser, M.; Copp, B. R. Bioorg. Med. Chem. Lett. 2013, 23, 452–454. doi:10.1016/j.bmcl.2012.11.072 |
| 9. | Pearce, A. N.; Chia, E. W.; Berridge, M. V.; Maas, E. W.; Page, M. J.; Harper, J. L.; Webb, V. L.; Copp, B. R. Tetrahedron 2008, 64, 5748–5755. doi:10.1016/j.tet.2008.04.012 |
| 10. | Lizzi, F.; Veronesi, G.; Belluti, F.; Bergamini, C.; López-Sánchez, A.; Kaiser, M.; Brun, R.; Krauth-Siegel, R. L.; Hall, D. G.; Rivas, L.; Bolognesi, M. L. J. Med. Chem. 2012, 55, 10490–10500. doi:10.1021/jm301112z |
| 11. | Bolognesi, M. L.; Banzi, R.; Bartolini, M.; Cavalli, A.; Tarozzi, A.; Andrisano, V.; Minarini, A.; Rosini, M.; Tumiatti, V.; Bergamini, C.; Fato, R.; Lenaz, G.; Hrelia, P.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. J. Med. Chem. 2007, 50, 4882–4897. doi:10.1021/jm070559a |
| 12. | Bitonti, A. J.; McCann, P. P.; Sjoerdsma, A. The Effects of Polyamine Analogues on Malaria Parasites In Vitro and In Vivo. In Progress in Polyamine Research: Novel Biochemical, Pharmacological, and Clinical Aspects; Zappia, V.; Pegg, A. E., Eds.; Springer US: Boston, MA, USA, 1988; pp 717–726. doi:10.1007/978-1-4684-5637-0_63 |
| 18. | Pellissier, H. Tetrahedron 2006, 62, 5559–5601. doi:10.1016/j.tet.2006.03.093 |
| 5. | Manku, S.; Laplante, C.; Kopac, D.; Chan, T.; Hall, D. G. J. Org. Chem. 2001, 66, 874–885. doi:10.1021/jo005647g |
| 3. | Casero, R. A., Jr.; Marton, L. J. Nat. Rev. Drug Discovery 2007, 6, 373–390. doi:10.1038/nrd2243 |
| 4. | Wallace, H. M. Eur. J. Clin. Invest. 2000, 30, 1–3. doi:10.1046/j.1365-2362.2000.00585.x |
| 16. | Wuts, P. G. M.; Greene, T. W. Greene's Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons: New York, NY, USA, 2006. doi:10.1002/0470053488 |
| 2. | Bienz, S.; Bisegger, P.; Guggisberg, A.; Hesse, M. Nat. Prod. Rep. 2005, 22, 647–658. doi:10.1039/b413742f |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 17. | Solladié, G.; Hutt, J.; Girardin, A. Synthesis 1987, 173. doi:10.1055/s-1987-27877 |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 14. | Yadav, J. S.; Reddy, P. A. N.; Reddy, Y. J.; Meraj, S.; Prasad, A. R. Eur. J. Org. Chem. 2013, 6317–6324. doi:10.1002/ejoc.201300623 |
| 15. | Dardonville, C.; Gilbert, I. H. Org. Biomol. Chem. 2003, 1, 552–559. doi:10.1039/b210606j |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 22. | Das, B.; Mahender, G.; Sunil Kumar, V.; Chowdhury, N. Tetrahedron Lett. 2004, 45, 6709–6711. doi:10.1016/j.tetlet.2004.07.075 |
| 23. | Hwu, J. R.; Jain, M. L.; Tsai, F.-Y.; Tsay, S.-C.; Balakumar, A.; Hakimelahi, G. H. J. Org. Chem. 2000, 65, 5077–5088. doi:10.1021/jo000024o |
| 24. | Kohli, V.; Blöcker, H.; Köster, H. Tetrahedron Lett. 1980, 21, 2683–2686. doi:10.1016/s0040-4039(00)78579-0 |
| 25. | Behloul, C.; Chouti, A.; Chabour, I.; Bey, H. B.; Guijarro, D.; Foubelo, F.; Nájera, C.; Yus, M. Tetrahedron Lett. 2016, 57, 3526–3528. doi:10.1016/j.tetlet.2016.06.110 |
| 26. | Bessodes, M.; Komiotis, D.; Antonakis, K. Tetrahedron Lett. 1986, 27, 579–580. doi:10.1016/s0040-4039(00)84045-9 |
| 13. | Hay, L.; Schultz, R. A.; Schutte, P. J. Onderstepoort J. Vet. Res. 2008, 75, 249–253. doi:10.4102/ojvr.v75i3.100 |
| 1. | Bode, M. L.; Gates, P. J.; Gebretnsae, S. Y.; Vleggaar, R. Tetrahedron 2010, 66, 2026–2036. doi:10.1016/j.tet.2010.01.043 |
| 27. | Dandepally, S. R.; Williams, A. L. Tetrahedron Lett. 2009, 50, 1071–1074. doi:10.1016/j.tetlet.2008.12.074 |
| 28. | Jih Ru Hwu; Jain, M. L.; Shwu-Chen, T.; Hakimelahi, G. H. Tetrahedron Lett. 1996, 37, 2035–2038. doi:10.1016/0040-4039(96)00211-0 |
| 29. | Jana, S.; Roy, S. C. Indian J. Chem. 2007, 46B, 707–709. |
© 2021 Zimuwandeyi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)