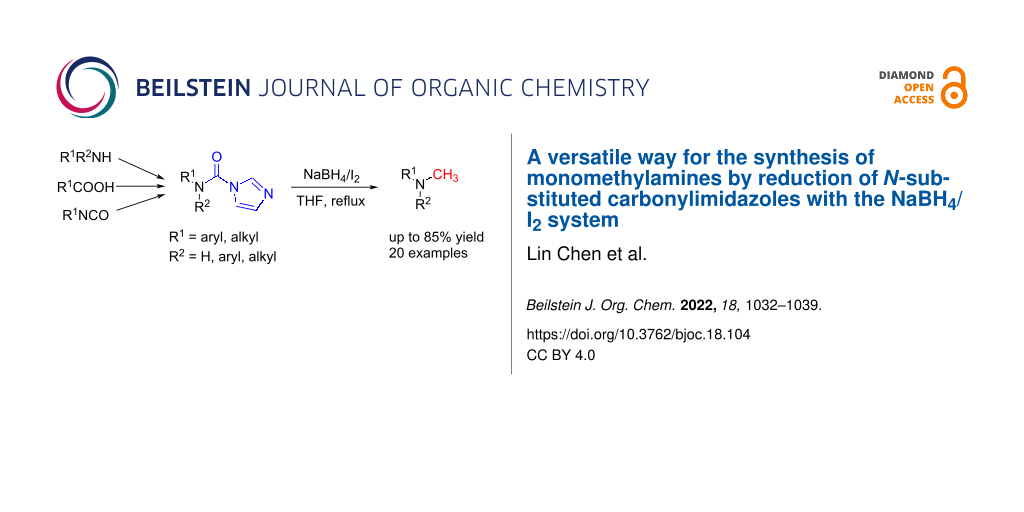
Abstract
An economical and versatile protocol for the one-pot synthesis of monomethylamines by reduction of N-substituted carbonylimidazoles with NaBH4/I2 in THF at reflux temperature is described. This method used no special catalyst and various monomethylamines can be easily obtained in moderate to good yields from a wide range of raw materials including amines (primary amines and secondary amines), carboxylic acids and isocyanates. Besides, an interesting reduction selectivity was observed. Exploration of the reaction process shows that it undergoes a two-step pathway via a formamide intermediate and the reduction of the formamide intermediate to monomethylamine as the rate-determining step. This work can contribute significantly expanding the applications of N-substituted carbonylimidazoles.

Graphical Abstract
Introduction
N-Methylamines are widely found in natural products, fine chemicals, agrochemicals, pharmaceuticals and dyes [1-4]. Traditional methods for the preparation of N-methylamines involve the direct methylation of amines by using methyl halides [5-7], dimethyl sulfate [8], diazomethane [9], methyl triflate [10,11] or dimethyl carbonate [12-15] as the methylation reagents and the reductive amination reactions by using formaldehyde or paraformaldehyde as the “indirect” alkylation reagents [16-19]. Recently, a variety of promising methylating agents or C1 sources such as formic acid [20,21], methanol [22-31] and carbon dioxide (CO2) [32-39] have been developed for the N-methylation of amines. However, these N-alkylation methods often require the employment of expensive catalysts, and the N-alkylation of primary amines generally does not stop with monomethylation as expected and inevitably provides a mixture of multiple methylated products because of the competing overalkylation reactions [14,16-43].
In order to obtain pure monomethylation product, the conventional method is to introduce alkyl formate, formacyl, methylene or their equivalents to amines, followed by reduction to give monomethylamines [44-51]. Protection/methylation/deprotection strategies have also been developed for the preparation of monomethylation objects, which are particularly suitable for peptide chemistry since protecting groups are often required in peptide synthesis [52,53]. These multistep reaction methods are conducive to avoid overmethylation products.
Although procedures for the synthesis of monomethylamines have been developed over the past years, the starting materials are mainly restricted regarding amines, in addition, expensive reagents or catalysts are usually required, which limit their applications to some extent. N-Substituted carbonylimidazoles, as important members of the carbonylimidazole family [54,55], are highly attractive intermediates with suitable stability for isolation or storage, and various good methods for their perparation have been developed by employing different starting materials such as amines [56-58], isocyanates [59-61] and carboxylic acids [62]. Since the carbonyl carbon atom of the carbonylimidazole moiety is easily attacked by a nucleophile the imidazole group readily dissociates. The N-substituted carbonylimidazoles have favorable reactivity and can be widely used in the synthesis of various valuable products such as ureas [63-70], carbamates [66,71-74], thiocarbamates [66], and cyanoformamides [75]. However, all of these works are primarily focused on the substitution reaction of N-substituted carbonylimidazoles. In our previous work, we conveniently prepared formamides by reducing N-substituted carbonylimidazoles with NaBH4 [62] (Scheme 1). The reaction mechanism shows that the H− ion acted as a nucleophile to attack the carbonyl carbon to cause the imidazolium ion to leave without reducing the carbonyl group. Although this work expands the application of N-substituted carbonylimidazoles, the reaction can still be regarded as a substitution reaction, which is attributed to the weak reducibility of NaBH4.
![[1860-5397-18-104-i1]](/bjoc/content/inline/1860-5397-18-104-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthesis of formamides and monomethylamines.
Scheme 1: The synthesis of formamides and monomethylamines.
In this work, our goal is to reduce the carbonyl group in N-substituted carbonylimidazoles. The inexpensive NaBH4/I2 system has great attraction because it is more reductive due to the generation of highly reactive BH3–THF by adding iodine to NaBH4 in THF [76-78] and the reaction conditions are not significantly changed compared to our previous preparation of formamide. With this reduction system, we achieved a one-step conversion from N-substituted carbonylimidazoles to methylamines. This interesting work will help to synthesize pure monomethylamines from a wide range of raw materials including amines, carboxylic acids and isocyanates under mild and safe reaction conditions.
Results and Discussion
Initially, N-phenethyl-1H-imidazole-1-carboxamide (1b) was chosen as a model substrate to react with 3.0 equiv of NaBH4 and 1.0 equiv of I2 in THF at reflux temperature, as expected, the carbonylimidazole moiety was successfully converted into a methyl group and the target monomethylamine (1c) was obtained in 70% yield after 6 h (Table 1, entry 1). When the amount of NaBH4 was increased from 3.0 equiv to 4.0 equiv and 5.0 equiv, the reaction time was shortened from 6 h to 4 h and 1 h, respectively. When further increasing the amount of NaBH4 to 6.0 equiv, only a slight decrease of the reaction time was observed. In addition, the yield of 1c showed few changes with the increase of the amount of NaBH4 from 3.0 equiv to 6.0 equiv. Obviously, 5.0 equiv of NaBH4 was optimal to perform the reaction. Since I2 was used to improve the reducibility of NaBH4, we next investigated the effect of the amount of I2 on the reaction. The use of 0.5 equiv of I2 in the presence of 5.0 equiv of NaBH4 afforded after 6 h only traces of methylamine but 62% of N-(phenethyl)formamide (Table 1, entry 5). Increasing the amount of I2 from 1.0 equiv to 1.5 equiv (Table 1, entry 6) did not significantly accelerated the reaction. All the above described results might indicate that an assembly of 1.0 equiv of I2 and 5.0 equiv of NaBH4 was sufficient to complete the reaction in one hour.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-104-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | NaBH4 (equiv) | I2 (equiv) | Time (h)b | Yield (%)c |
| 1 | 3.0 | 1.0 | 6 | 70 |
| 2 | 4.0 | 1.0 | 4 | 72 |
| 3 | 5.0 | 1.0 | 1 | 74 |
| 4 | 6.0 | 1.0 | 0.9 | 75 |
| 5 | 5.0 | 0.5 | 6 | traced |
| 6 | 5.0 | 1.5 | 1 | 73 |
aThe reactions were carried out with 1b (1.0 equiv, 2 mmol), NaBH4, I2, THF (25 mL) under reflux temperature. bThe reaction was monitored by TLC. cIsolated yield was based on 1b. dThe isolated yield of 1c and N-(phenethyl)formamide was 1% (trace) and 62%, respectively.
With the optimized reaction conditions in hand, we investigated the synthesis of other N-methylamines from various N-substituted carbonylimidazoles (Table 2). As a proof of the versatility and applicability of the proposed method, N-substituted carbonylimidazoles were prepared from amines (1b–14b) [56-58,62,64], isocyanates (15b–17b) [59-61], and carboxylic acids (18b–20b) [62], respectively (for detailed experimental procedures, see Supporting Information File 1). All types of these N-substituted carbonylimidazoles reacted smoothly with NaBH4/I2 to provide the corresponding N-methylamines in satisfactory yields.
Table 2: Synthesis of monomethylamines from amines, carboxylic acids and isocyanates.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-104-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Carbamoylimidazole | Time (h)b | Product | Yield (%)c |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-104-i5.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-104-i6.svg?max-width=637&scale=1.0)
1b, 87% |
1 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-18-104-i7.svg?max-width=637&scale=1.0)
1c |
74 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-18-104-i8.svg?max-width=637&scale=1.0)
2a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-18-104-i9.svg?max-width=637&scale=1.0)
2b, 85% |
1 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-18-104-i10.svg?max-width=637&scale=1.0)
2c |
67 |
| 3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-18-104-i11.svg?max-width=637&scale=1.0)
3a |
![[Graphic 10]](/bjoc/content/inline/1860-5397-18-104-i12.svg?max-width=637&scale=1.0)
3b, 89% |
1 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-18-104-i13.svg?max-width=637&scale=1.0)
3c |
67 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-18-104-i14.svg?max-width=637&scale=1.0)
4a |
![[Graphic 13]](/bjoc/content/inline/1860-5397-18-104-i15.svg?max-width=637&scale=1.0)
4b, 77% |
1 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-18-104-i16.svg?max-width=637&scale=1.0)
4c |
65 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-18-104-i17.svg?max-width=637&scale=1.0)
5a |
![[Graphic 16]](/bjoc/content/inline/1860-5397-18-104-i18.svg?max-width=637&scale=1.0)
5b, 87% |
1 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-18-104-i19.svg?max-width=637&scale=1.0)
5c |
70 |
| 6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-18-104-i20.svg?max-width=637&scale=1.0)
1c |
![[Graphic 19]](/bjoc/content/inline/1860-5397-18-104-i21.svg?max-width=637&scale=1.0)
6b, 88% |
1 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-18-104-i22.svg?max-width=637&scale=1.0)
6c |
83 |
| 7 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-18-104-i23.svg?max-width=637&scale=1.0)
7a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-18-104-i24.svg?max-width=637&scale=1.0)
7b, 85% |
2 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-18-104-i25.svg?max-width=637&scale=1.0)
7c |
60 |
| 8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-18-104-i26.svg?max-width=637&scale=1.0)
8a |
![[Graphic 25]](/bjoc/content/inline/1860-5397-18-104-i27.svg?max-width=637&scale=1.0)
8b, 70% |
4 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-18-104-i28.svg?max-width=637&scale=1.0)
8c |
72 |
| 9 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-18-104-i29.svg?max-width=637&scale=1.0)
9a |
![[Graphic 28]](/bjoc/content/inline/1860-5397-18-104-i30.svg?max-width=637&scale=1.0)
9b, 78% |
4 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-18-104-i31.svg?max-width=637&scale=1.0)
9c |
85 |
| 10 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-18-104-i32.svg?max-width=637&scale=1.0)
10a |
![[Graphic 31]](/bjoc/content/inline/1860-5397-18-104-i33.svg?max-width=637&scale=1.0)
10b, 75% |
4 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-18-104-i34.svg?max-width=637&scale=1.0)
10c |
80 |
| 11 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-18-104-i35.svg?max-width=637&scale=1.0)
11a |
![[Graphic 34]](/bjoc/content/inline/1860-5397-18-104-i36.svg?max-width=637&scale=1.0)
11b, 55% |
4 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-18-104-i37.svg?max-width=637&scale=1.0)
11c |
70 |
| 12 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-18-104-i38.svg?max-width=637&scale=1.0)
8c |
![[Graphic 37]](/bjoc/content/inline/1860-5397-18-104-i39.svg?max-width=637&scale=1.0)
12b, 89% |
4 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-18-104-i40.svg?max-width=637&scale=1.0)
12c |
71 |
| 13 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-18-104-i41.svg?max-width=637&scale=1.0)
13a |
![[Graphic 40]](/bjoc/content/inline/1860-5397-18-104-i42.svg?max-width=637&scale=1.0)
13b, 57% |
6 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-18-104-i43.svg?max-width=637&scale=1.0)
13c |
67 |
| 14d |
![[Graphic 42]](/bjoc/content/inline/1860-5397-18-104-i44.svg?max-width=637&scale=1.0)
14a |
![[Graphic 43]](/bjoc/content/inline/1860-5397-18-104-i45.svg?max-width=637&scale=1.0)
14b, 83% |
10 |
![[Graphic 44]](/bjoc/content/inline/1860-5397-18-104-i46.svg?max-width=637&scale=1.0)
14c |
70 |
| 15 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-18-104-i47.svg?max-width=637&scale=1.0)
15a |
![[Graphic 46]](/bjoc/content/inline/1860-5397-18-104-i48.svg?max-width=637&scale=1.0)
15b, 95% |
1 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-18-104-i49.svg?max-width=637&scale=1.0)
15c |
74 |
| 16 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-18-104-i50.svg?max-width=637&scale=1.0)
16a |
![[Graphic 49]](/bjoc/content/inline/1860-5397-18-104-i51.svg?max-width=637&scale=1.0)
16b, 97% |
4 |
![[Graphic 50]](/bjoc/content/inline/1860-5397-18-104-i52.svg?max-width=637&scale=1.0)
16c |
74 |
| 17 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-18-104-i53.svg?max-width=637&scale=1.0)
17a |
![[Graphic 52]](/bjoc/content/inline/1860-5397-18-104-i54.svg?max-width=637&scale=1.0)
17b, 94% |
4 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-18-104-i55.svg?max-width=637&scale=1.0)
17c |
72 |
| 18 |
![[Graphic 54]](/bjoc/content/inline/1860-5397-18-104-i56.svg?max-width=637&scale=1.0)
18a |
![[Graphic 55]](/bjoc/content/inline/1860-5397-18-104-i57.svg?max-width=637&scale=1.0)
18b, 72% |
1 |
![[Graphic 56]](/bjoc/content/inline/1860-5397-18-104-i58.svg?max-width=637&scale=1.0)
18c |
77 |
| 19 |
![[Graphic 57]](/bjoc/content/inline/1860-5397-18-104-i59.svg?max-width=637&scale=1.0)
19a |
![[Graphic 58]](/bjoc/content/inline/1860-5397-18-104-i60.svg?max-width=637&scale=1.0)
19b, 91% |
1 |
![[Graphic 59]](/bjoc/content/inline/1860-5397-18-104-i61.svg?max-width=637&scale=1.0)
19c |
65 |
| 20 |
![[Graphic 60]](/bjoc/content/inline/1860-5397-18-104-i62.svg?max-width=637&scale=1.0)
20a |
![[Graphic 61]](/bjoc/content/inline/1860-5397-18-104-i63.svg?max-width=637&scale=1.0)
20b, 83% |
4 |
![[Graphic 62]](/bjoc/content/inline/1860-5397-18-104-i64.svg?max-width=637&scale=1.0)
20c |
68 |
aThe reactions were carried out with carbamoylimidazole (1.0 equiv, 2 mmol), NaBH4 (5.0 equiv, 10.0 mmol), I2 (1.0 equiv, 2 mmol) and THF (25 mL) under reflux. bMonitored by TLC. cIsolated yield was based on carbamoylimidazole. d10.0 equiv of NaBH4 and 2 equiv of I2 was used.
The impact of different substituents on the reaction were well investigated. As shown in Table 2, the alkyl substituents (R) in the N-alkyl carbonylimidazoles had a weak influence on the reaction (Table 2, entries 1–7, 18, and 19). Whether the N-alkyl carbonylimidazoles bear one substituent (e.g., 1b–5b, 15b, 18b, 19b) or two substituents (e.g., 6b and 7b), the reaction proceeded well, affording the desired product in 60–83% yields. Note that the reaction time of 7b (2 h) was obviously longer than that of 1b–6b (1 h), 18b (1 h) and 19b (1 h), possibly because the steric hindrance of the two benzyl groups on 7b slowed the reaction. Encouraged by the above mentioned results, we then tested N-aryl carbonylimidazoles in the reaction. To our delight, N-aryl carbonylimidazoles with either electron-donating (9b and 10b) or electron-withdrawing groups (11b, 16b and 17b) on the aryl rings were all transformed, affording the expected products in 70–85% yields. N-Aryl carbonylimidazoles with two substituents, such as 12b (R1 = methyl, R2 = phenyl) and 13b (R1 = phenyl, R2 = phenyl), were also amenable to this protocol, giving the corresponding products 12c and 13c in 71% and 67% yield, respectively. Furthermore, by using 2.0 equiv of I2 and 10.0 equiv of NaBH4, the substrate 14b with two N-substituted carbonylimidazole moieties could also undergo this reaction and provided the desired product 14c in moderate yield (70%). Additionally, our protocol was applicable to prepare N,N-dimethylamines by step-by-step methylation. Employing the mono-methylated products 1c and 8c, N,N-dimethylamine 6c and 12c can be obtained, respectively, via repeating our synthesis procedure.
In order to understand the reduction selectivity of the method, the substrates bearing acetamide groups (19b and 20b) were tested in the reaction. To our pleasure, both aliphatic and aromatic amides reacted smoothly and provided the expected products in satisfactory yields, with the acetyl groups being unaffected. This suggested that the N-acetyl groups in N-substituted carbonylimidazoles were well tolerated during the reduction, and our method showed interesting reduction selectivity.
To gain some preliminary insight into the reaction process, two representative intermediates for the synthesis of aliphatic methylamine 1c and aromatic methylamine 8c had been isolated and identified as corresponding formamides (see Supporting Information File 1). Furthermore, by follwoing the reaction with TLC, we found that the reaction time (hours) from the formamide intermediate to the corresponding methylamine product was much longer than the time (minutes) from the N-substituted carbonylimidazole to the formamide. This indicated that the reaction might undergo a two-step pathway via the formamide intermediate (Scheme 2).
![[1860-5397-18-104-i2]](/bjoc/content/inline/1860-5397-18-104-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The possible reaction mechanism. RDS = rate determining step.
Scheme 2: The possible reaction mechanism. RDS = rate determining step.
In the first step (step I), N-substituted carbonylimidazole was rapidly converted into the formamide intermediate by the attack of a hydrogen anion as we had reported before [62]. Subsequently, the carbonyl group of the formamide intermediate was reduced to furnish the desired N-methylamine in the second step (step II) [79-81]. Step II proceeded much slower than step I, so it could be treated as rate-determining step (RDS). The required longer reaction time for N-aryl carbonylimidazoles (over 4 h) than that for N-alkyl carbonylimidazoles (about 1 h) can be well explained by the two-step mechanism. In step I, the N-aryl carbonylimidazoles might react much faster than N-alkyl carbonylimidazoles, because the stronger conjugation system of the resulting N-aromatic formamides made them more stable and easier to generate. However, these more stable N-aryl formamide intermediates were less reactive and directly slowed the reaction in step II, which resulted in a longer reaction time of the N-aryl carbonylimidazoles in the whole reaction.
The substrate 13b, bearing two phenyl rings, which not only had a large steric hindrance like 7b, but also had a strong conjugation system, took much longer time (6 h) to complete the reaction.
As shown by the reaction mechanism, the methyl group was converted from the carbonylimidazole moiety by full reduction and therefore no competing overalkylation reactions occurred.
Although the N-methylamine could be prepared from carboxylic acid or amine by our method, the methyl source was remarkably different (Scheme S1, Supporting Information File 1). For amines, the carbon source of the methyl group is from the carbonyl group of the carbonyldiimidazole; while for carboxylic acids, the carbon source is the carboxyl group. When carboxylic acids were used, the carboxyl moiety was first converted to an isocyanate via Curtius rearrangement [82-85], then reacted with imidazole to form the carbonylimidazole, and eventually reduced to the methyl moiety. In the whole process, no extra carbon was introduced.
Conclusion
In conclusion, we have developed an economical and versatile protocol for the one-pot synthesis of monomethylamines by reduction of N-substituted carbonylimidazoles with the NaBH4/I2 system. This work further extends the application of N-substituted carbonylimidazoles. By employing inexpensive and commercially available reagents, a variety of aliphatic and aromatic monomethylamines were obtained in moderate to good yields from a broad substrate scope including not only amines (both primary amines and secondary amines) but also carboxylic acids or isocyanates. The acetamide group was well tolerated in our reduction, implying our method showed interesting reduction selectivity.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Paul, B.; Shee, S.; Chakrabarti, K.; Kundu, S. ChemSusChem 2017, 10, 2370–2374. doi:10.1002/cssc.201700503
Return to citation in text: [1] -
Davis, D. A.; Hamilton, A.; Yang, J.; Cremar, L. D.; Van Gough, D.; Potisek, S. L.; Ong, M. T.; Braun, P. V.; Martínez, T. J.; White, S. R.; Moore, J. S.; Sottos, N. R. Nature 2009, 459, 68–72. doi:10.1038/nature07970
Return to citation in text: [1] -
Seayad, A.; Ahmed, M.; Klein, H.; Jackstell, R.; Gross, T.; Beller, M. Science 2002, 297, 1676–1678. doi:10.1126/science.1074801
Return to citation in text: [1] -
Salvatore, R. N.; Yoon, C. H.; Jung, K. W. Tetrahedron 2001, 57, 7785–7811. doi:10.1016/s0040-4020(01)00722-0
Return to citation in text: [1] -
Olsen, R. K. J. Org. Chem. 1970, 35, 1912–1915. doi:10.1021/jo00831a042
Return to citation in text: [1] -
Schreiber, S. L.; Anthony, N. J.; Dorsey, B. D.; Hawley, R. C. Tetrahedron Lett. 1988, 29, 6577–6580. doi:10.1016/s0040-4039(00)82401-6
Return to citation in text: [1] -
Vedejs, E.; Kongkittingam, C. J. Org. Chem. 2001, 66, 7355–7364. doi:10.1021/jo0104882
Return to citation in text: [1] -
Prashad, M.; Har, D.; Hu, B.; Kim, H.-Y.; Repic, O.; Blacklock, T. J. Org. Lett. 2003, 5, 125–128. doi:10.1021/ol0268440
Return to citation in text: [1] -
Leggio, A.; Alò, D.; Belsito, E. L.; Di Gioia, M. L.; Romio, E.; Siciliano, C.; Liguori, A. J. Pept. Sci. 2015, 21, 644–650. doi:10.1002/psc.2777
Return to citation in text: [1] -
Lebleu, T.; Ma, X.; Maddaluno, J.; Legros, J. Chem. Commun. 2014, 50, 1836–1838. doi:10.1039/c3cc48997c
Return to citation in text: [1] -
Kano, T.; Tanaka, Y.; Maruoka, K. Tetrahedron 2007, 63, 8658–8664. doi:10.1016/j.tet.2007.03.179
Return to citation in text: [1] -
Selva, M.; Tundo, P.; Perosa, A. J. Org. Chem. 2002, 67, 9238–9247. doi:10.1021/jo026057g
Return to citation in text: [1] -
Zheng, J.; Darcel, C.; Sortais, J.-B. Chem. Commun. 2014, 50, 14229–14232. doi:10.1039/c4cc05517a
Return to citation in text: [1] -
Cabrero-Antonino, J. R.; Adam, R.; Junge, K.; Beller, M. Catal. Sci. Technol. 2016, 6, 7956–7966. doi:10.1039/c6cy01401a
Return to citation in text: [1] [2] -
Seo, H.; Bédard, A.-C.; Chen, W. P.; Hicklin, R. W.; Alabugin, A.; Jamison, T. F. Tetrahedron 2018, 74, 3124–3128. doi:10.1016/j.tet.2017.11.068
Return to citation in text: [1] -
Borch, R. F.; Hassid, A. I. J. Org. Chem. 1972, 37, 1673–1674. doi:10.1021/jo00975a049
Return to citation in text: [1] [2] -
Alinezhad, H.; Tajbakhsh, M.; Salehian, F.; Fazli, K. Synth. Commun. 2010, 40, 2415–2420. doi:10.1080/00397910903249606
Return to citation in text: [1] [2] -
Ge, X.; Luo, C.; Qian, C.; Yu, Z.; Chen, X. RSC Adv. 2014, 4, 43195–43203. doi:10.1039/c4ra04414b
Return to citation in text: [1] [2] -
Petricci, E.; Santillo, N.; Castagnolo, D.; Cini, E.; Taddei, M. Adv. Synth. Catal. 2018, 360, 2560–2565. doi:10.1002/adsc.201701619
Return to citation in text: [1] [2] -
Savourey, S.; Lefèvre, G.; Berthet, J.-C.; Cantat, T. Chem. Commun. 2014, 50, 14033–14036. doi:10.1039/c4cc05908e
Return to citation in text: [1] [2] -
Sorribes, I.; Junge, K.; Beller, M. Chem. – Eur. J. 2014, 20, 7878–7883. doi:10.1002/chem.201402124
Return to citation in text: [1] [2] -
Zhang, L.; Zhang, Y.; Deng, Y.; Shi, F. RSC Adv. 2015, 5, 14514–14521. doi:10.1039/c4ra13848a
Return to citation in text: [1] [2] -
Bruneau-Voisine, A.; Wang, D.; Dorcet, V.; Roisnel, T.; Darcel, C.; Sortais, J.-B. J. Catal. 2017, 347, 57–62. doi:10.1016/j.jcat.2017.01.004
Return to citation in text: [1] [2] -
Neumann, J.; Elangovan, S.; Spannenberg, A.; Junge, K.; Beller, M. Chem. – Eur. J. 2017, 23, 5410–5413. doi:10.1002/chem.201605218
Return to citation in text: [1] [2] -
Dang, T. T.; Ramalingam, B.; Seayad, A. M. ACS Catal. 2015, 5, 4082–4088. doi:10.1021/acscatal.5b00606
Return to citation in text: [1] [2] -
Liang, R.; Li, S.; Wang, R.; Lu, L.; Li, F. Org. Lett. 2017, 19, 5790–5793. doi:10.1021/acs.orglett.7b02723
Return to citation in text: [1] [2] -
Choi, G.; Hong, S. H. Angew. Chem., Int. Ed. 2018, 57, 6166–6170. doi:10.1002/anie.201801524
Return to citation in text: [1] [2] -
Maji, M.; Chakrabarti, K.; Paul, B.; Roy, B. C.; Kundu, S. Adv. Synth. Catal. 2018, 360, 722–729. doi:10.1002/adsc.201701117
Return to citation in text: [1] [2] -
Jiang, L.; Zhang, X.; Wang, Y.; Guo, F.; Hou, Z. Asian J. Org. Chem. 2021, 10, 2165–2169. doi:10.1002/ajoc.202100339
Return to citation in text: [1] [2] -
Wang, J.; Qiang, W.; Ye, S.; Zhu, L.; Liu, X.; Loh, T.-P. Catal. Sci. Technol. 2021, 11, 3364–3375. doi:10.1039/d0cy02442b
Return to citation in text: [1] [2] -
Paul, B.; Maji, M.; Panja, D.; Kundu, S. Asian J. Org. Chem. 2022, 11, e202100678. doi:10.1002/ajoc.202100678
Return to citation in text: [1] [2] -
Jacquet, O.; Frogneux, X.; Das Neves Gomes, C.; Cantat, T. Chem. Sci. 2013, 4, 2127–2131. doi:10.1039/c3sc22240c
Return to citation in text: [1] [2] -
Li, Y.; Fang, X.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 9568–9571. doi:10.1002/anie.201301349
Return to citation in text: [1] [2] -
Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Ji, G.; Ma, Z.; Gao, X.; Liu, Z. Green Chem. 2015, 17, 4189–4193. doi:10.1039/c5gc01386k
Return to citation in text: [1] [2] -
Niu, H.; Lu, L.; Shi, R.; Chiang, C.-W.; Lei, A. Chem. Commun. 2017, 53, 1148–1151. doi:10.1039/c6cc09072a
Return to citation in text: [1] [2] -
Cui, X.; Dai, X.; Zhang, Y.; Deng, Y.; Shi, F. Chem. Sci. 2014, 5, 649–655. doi:10.1039/c3sc52676c
Return to citation in text: [1] [2] -
Tamura, M.; Miura, A.; Gu, Y.; Nakagawa, Y.; Tomishige, K. Chem. Lett. 2017, 46, 1243–1246. doi:10.1246/cl.170419
Return to citation in text: [1] [2] -
Toyao, T.; Siddiki, S. M. A. H.; Morita, Y.; Kamachi, T.; Touchy, A. S.; Onodera, W.; Kon, K.; Furukawa, S.; Ariga, H.; Asakura, K.; Yoshizawa, K.; Shimizu, K.-i. Chem. – Eur. J. 2017, 23, 14848–14859. doi:10.1002/chem.201702801
Return to citation in text: [1] [2] -
Lin, S.; Liu, J.; Ma, L. J. CO2 Util. 2021, 54, 101759. doi:10.1016/j.jcou.2021.101759
Return to citation in text: [1] [2] -
Du, X.-L.; Tang, G.; Bao, H.-L.; Jiang, Z.; Zhong, X.-H.; Su, D. S.; Wang, J.-Q. ChemSusChem 2015, 8, 3489–3496. doi:10.1002/cssc.201500486
Return to citation in text: [1] -
Santoro, O.; Lazreg, F.; Minenkov, Y.; Cavallo, L.; Cazin, C. S. J. Dalton Trans. 2015, 44, 18138–18144. doi:10.1039/c5dt03506f
Return to citation in text: [1] -
Jiang, X.; Wang, C.; Wei, Y.; Xue, D.; Liu, Z.; Xiao, J. Chem. – Eur. J. 2014, 20, 58–63. doi:10.1002/chem.201303802
Return to citation in text: [1] -
Goyal, V.; Naik, G.; Narani, A.; Natte, K.; Jagadeesh, R. V. Tetrahedron 2021, 98, 132414. doi:10.1016/j.tet.2021.132414
Return to citation in text: [1] -
Paquette, L. A.; Tae, J. J. Org. Chem. 1998, 63, 2022–2030. doi:10.1021/jo9722921
Return to citation in text: [1] -
Benoit, W. L.; Parvez, M.; Keay, B. A. Tetrahedron: Asymmetry 2009, 20, 69–77. doi:10.1016/j.tetasy.2008.12.027
Return to citation in text: [1] -
Fuentes de Arriba, Á. L.; Seisdedos, D. G.; Simón, L.; Alcázar, V.; Raposo, C.; Morán, J. R. J. Org. Chem. 2010, 75, 8303–8306. doi:10.1021/jo101723v
Return to citation in text: [1] -
Sun, N.; Wang, S.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. Tetrahedron 2010, 66, 7142–7148. doi:10.1016/j.tet.2010.06.091
Return to citation in text: [1] -
Barluenga, J.; Bayón, A. M.; Campos, P.; Asensio, G.; Gonzalez-Nuñez, E.; Molina, Y. J. Chem. Soc., Perkin Trans. 1 1988, 1631–1636. doi:10.1039/p19880001631
Return to citation in text: [1] -
Crochet, R. A., Jr.; Blanton, C. D., Jr. Synthesis 1974, 55–56. doi:10.1055/s-1974-23242
Return to citation in text: [1] -
Zhang, J.; Chang, H.-M.; Kane, R. R. Synlett 2001, 643–645. doi:10.1055/s-2001-13378
Return to citation in text: [1] -
Blackburn, C.; LaMarche, M. J.; Brown, J.; Che, J. L.; Cullis, C. A.; Lai, S.; Maguire, M.; Marsilje, T.; Geddes, B.; Govek, E.; Kadambi, V.; Doherty, C.; Dayton, B.; Brodjian, S.; Marsh, K. C.; Collins, C. A.; Kym, P. R. Bioorg. Med. Chem. Lett. 2006, 16, 2621–2627. doi:10.1016/j.bmcl.2006.02.044
Return to citation in text: [1] -
Das, D.; Khan, H. P. A.; Shivahare, R.; Gupta, S.; Sarkar, J.; Siddiqui, M. I.; Ampapathi, R. S.; Chakraborty, T. K. Org. Biomol. Chem. 2017, 15, 3337–3352. doi:10.1039/c6ob02610a
Return to citation in text: [1] -
Avula, K.; Mohapatra, D. K. Tetrahedron Lett. 2016, 57, 1715–1717. doi:10.1016/j.tetlet.2016.03.017
Return to citation in text: [1] -
Heller, S. T.; Fu, T.; Sarpong, R. Org. Lett. 2012, 14, 1970–1973. doi:10.1021/ol300339q
Return to citation in text: [1] -
Meng, G.; Szostak, R.; Szostak, M. Org. Lett. 2017, 19, 3596–3599. doi:10.1021/acs.orglett.7b01575
Return to citation in text: [1] -
Rawling, T.; McDonagh, A. M.; Tattam, B.; Murray, M. Tetrahedron 2012, 68, 6065–6070. doi:10.1016/j.tet.2012.05.002
Return to citation in text: [1] [2] -
Velavan, A.; Sumathi, S.; Balasubramanian, K. K. Org. Biomol. Chem. 2012, 10, 6420–6431. doi:10.1039/c2ob25412c
Return to citation in text: [1] [2] -
McBurney, R. T.; Walton, J. C. J. Am. Chem. Soc. 2013, 135, 7349–7354. doi:10.1021/ja402833w
Return to citation in text: [1] [2] -
Jöst, C.; Nitsche, C.; Scholz, T.; Roux, L.; Klein, C. D. J. Med. Chem. 2014, 57, 7590–7599. doi:10.1021/jm5006918
Return to citation in text: [1] [2] -
Ren, Y.; Rousseaux, S. A. L. J. Org. Chem. 2018, 83, 913–920. doi:10.1021/acs.joc.7b02905
Return to citation in text: [1] [2] -
Meng, H.; Sun, K.; Xu, Z.; Tian, L.; Wang, Y. Eur. J. Org. Chem. 2021, 1768–1772. doi:10.1002/ejoc.202100113
Return to citation in text: [1] [2] -
Chen, Z.; Cao, Y.; Tian, Z.; Zhou, X.; Xu, W.; Yang, J.; Teng, H. Tetrahedron Lett. 2017, 58, 2166–2170. doi:10.1016/j.tetlet.2017.04.072
Return to citation in text: [1] [2] [3] [4] [5] -
Peng, Z.; Wong, J. W.; Hansen, E. C.; Puchlopek-Dermenci, A. L. A.; Clarke, H. J. Org. Lett. 2014, 16, 860–863. doi:10.1021/ol403630g
Return to citation in text: [1] -
Padiya, K. J.; Gavade, S.; Kardile, B.; Tiwari, M.; Bajare, S.; Mane, M.; Gaware, V.; Varghese, S.; Harel, D.; Kurhade, S. Org. Lett. 2012, 14, 2814–2817. doi:10.1021/ol301009d
Return to citation in text: [1] [2] -
de Greef, T. F. A.; Nieuwenhuizen, M. M. L.; Sijbesma, R. P.; Meijer, E. W. J. Org. Chem. 2010, 75, 598–610. doi:10.1021/jo902053t
Return to citation in text: [1] -
Duspara, P. A.; Islam, M. S.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2012, 77, 10362–10368. doi:10.1021/jo302084a
Return to citation in text: [1] [2] [3] -
Wong, E. C. N.; Reekie, T. A.; Werry, E. L.; O'Brien-Brown, J.; Bowyer, S. L.; Kassiou, M. Bioorg. Med. Chem. Lett. 2017, 27, 2439–2442. doi:10.1016/j.bmcl.2017.04.005
Return to citation in text: [1] -
Venter, J.; Perez, C.; van Otterlo, W. A. L.; Martínez, A.; Blackie, M. A. L. Bioorg. Med. Chem. Lett. 2019, 29, 1597–1600. doi:10.1016/j.bmcl.2019.04.049
Return to citation in text: [1] -
Jones, C. D.; Simmons, H. T. D.; Horner, K. E.; Liu, K.; Thompson, R. L.; Steed, J. W. Nat. Chem. 2019, 11, 375–381. doi:10.1038/s41557-019-0222-0
Return to citation in text: [1] -
Kondoh, A.; Ishikawa, S.; Terada, M. J. Am. Chem. Soc. 2020, 142, 3724–3728. doi:10.1021/jacs.9b13922
Return to citation in text: [1] -
Rannard, S. P.; Davis, N. J.; Herbert, I. Macromolecules 2004, 37, 9418–9430. doi:10.1021/ma0489218
Return to citation in text: [1] -
Griffin, R. J.; Evers, E.; Davison, R.; Gibson, A. E.; Layton, D.; Irwin, W. J. J. Chem. Soc., Perkin Trans. 1 1996, 1205–1211. doi:10.1039/p19960001205
Return to citation in text: [1] -
Alsarraf, J.; Petitpoisson, L.; Pichette, A. Org. Lett. 2021, 23, 6052–6056. doi:10.1021/acs.orglett.1c02116
Return to citation in text: [1] -
Zhao, C.; Ye, Z.; Ma, Z.-x.; Wildman, S. A.; Blaszczyk, S. A.; Hu, L.; Guizei, I. A.; Tang, W. Nat. Commun. 2019, 10, 4015. doi:10.1038/s41467-019-11976-2
Return to citation in text: [1] -
Nugent, J.; Campbell, S. G.; Vo, Y.; Schwartz, B. D. Eur. J. Org. Chem. 2017, 5110–5118. doi:10.1002/ejoc.201700974
Return to citation in text: [1] -
Prasad, A. S. B.; Kanth, J. V. B.; Periasamy, M. Tetrahedron 1992, 48, 4623–4628. doi:10.1016/s0040-4020(01)81236-9
Return to citation in text: [1] -
Rodrigues, A.; Olivato, P. R.; Rittner, R. Synthesis 2005, 2578–2582. doi:10.1055/s-2005-872101
Return to citation in text: [1] -
Harish, V.; Periasamy, M. Tetrahedron: Asymmetry 2017, 28, 175–180. doi:10.1016/j.tetasy.2016.12.002
Return to citation in text: [1] -
Abinet, E.; Martin, D.; Standfuss, S.; Kulinna, H.; Spaniol, T. P.; Okuda, J. Chem. – Eur. J. 2011, 17, 15014–15026. doi:10.1002/chem.201102145
Return to citation in text: [1] -
Garnier-Amblard, E. C.; Mays, S. G.; Arrendale, R. F.; Baillie, M. T.; Bushnev, A. S.; Culver, D. G.; Evers, T. J.; Holt, J. J.; Howard, R. B.; Liebeskind, L. S.; Menaldino, D. S.; Natchus, M. G.; Petros, J. A.; Ramaraju, H.; Reddy, G. P.; Liotta, D. C. ACS Med. Chem. Lett. 2011, 2, 438–443. doi:10.1021/ml2000164
Return to citation in text: [1] -
Nakagawa, Y.; Irie, K.; Yanagita, R. C.; Ohigashi, H.; Tsuda, K.-i.; Kashiwagi, K.; Saito, N. J. Med. Chem. 2006, 49, 2681–2688. doi:10.1021/jm050857c
Return to citation in text: [1] -
Teng, H.; He, Y.; Wu, L.; Su, J.; Feng, X.; Qiu, G.; Liang, S.; Hu, X. Synlett 2006, 877–880. doi:10.1055/s-2006-939044
Return to citation in text: [1] -
Teng, H.; Zhang, Z.; Zhou, Y.; Chen, Z.; Chen, Q.; Liu, Y.; Xu, W. RSC Adv. 2015, 5, 71868–71872. doi:10.1039/c5ra10622b
Return to citation in text: [1] -
Balci, M. Synthesis 2018, 50, 1373–1401. doi:10.1055/s-0036-1589527
Return to citation in text: [1] -
Wang, L.; Qin, R.-Q.; Yan, H.-Y.; Ding, M.-U. Synthesis 2015, 3522–3528. doi:10.1055/s-0034-1378874
Return to citation in text: [1]
| 79. | Abinet, E.; Martin, D.; Standfuss, S.; Kulinna, H.; Spaniol, T. P.; Okuda, J. Chem. – Eur. J. 2011, 17, 15014–15026. doi:10.1002/chem.201102145 |
| 80. | Garnier-Amblard, E. C.; Mays, S. G.; Arrendale, R. F.; Baillie, M. T.; Bushnev, A. S.; Culver, D. G.; Evers, T. J.; Holt, J. J.; Howard, R. B.; Liebeskind, L. S.; Menaldino, D. S.; Natchus, M. G.; Petros, J. A.; Ramaraju, H.; Reddy, G. P.; Liotta, D. C. ACS Med. Chem. Lett. 2011, 2, 438–443. doi:10.1021/ml2000164 |
| 81. | Nakagawa, Y.; Irie, K.; Yanagita, R. C.; Ohigashi, H.; Tsuda, K.-i.; Kashiwagi, K.; Saito, N. J. Med. Chem. 2006, 49, 2681–2688. doi:10.1021/jm050857c |
| 82. | Teng, H.; He, Y.; Wu, L.; Su, J.; Feng, X.; Qiu, G.; Liang, S.; Hu, X. Synlett 2006, 877–880. doi:10.1055/s-2006-939044 |
| 83. | Teng, H.; Zhang, Z.; Zhou, Y.; Chen, Z.; Chen, Q.; Liu, Y.; Xu, W. RSC Adv. 2015, 5, 71868–71872. doi:10.1039/c5ra10622b |
| 84. | Balci, M. Synthesis 2018, 50, 1373–1401. doi:10.1055/s-0036-1589527 |
| 85. | Wang, L.; Qin, R.-Q.; Yan, H.-Y.; Ding, M.-U. Synthesis 2015, 3522–3528. doi:10.1055/s-0034-1378874 |
| 1. | Paul, B.; Shee, S.; Chakrabarti, K.; Kundu, S. ChemSusChem 2017, 10, 2370–2374. doi:10.1002/cssc.201700503 |
| 2. | Davis, D. A.; Hamilton, A.; Yang, J.; Cremar, L. D.; Van Gough, D.; Potisek, S. L.; Ong, M. T.; Braun, P. V.; Martínez, T. J.; White, S. R.; Moore, J. S.; Sottos, N. R. Nature 2009, 459, 68–72. doi:10.1038/nature07970 |
| 3. | Seayad, A.; Ahmed, M.; Klein, H.; Jackstell, R.; Gross, T.; Beller, M. Science 2002, 297, 1676–1678. doi:10.1126/science.1074801 |
| 4. | Salvatore, R. N.; Yoon, C. H.; Jung, K. W. Tetrahedron 2001, 57, 7785–7811. doi:10.1016/s0040-4020(01)00722-0 |
| 10. | Lebleu, T.; Ma, X.; Maddaluno, J.; Legros, J. Chem. Commun. 2014, 50, 1836–1838. doi:10.1039/c3cc48997c |
| 11. | Kano, T.; Tanaka, Y.; Maruoka, K. Tetrahedron 2007, 63, 8658–8664. doi:10.1016/j.tet.2007.03.179 |
| 56. | Rawling, T.; McDonagh, A. M.; Tattam, B.; Murray, M. Tetrahedron 2012, 68, 6065–6070. doi:10.1016/j.tet.2012.05.002 |
| 57. | Velavan, A.; Sumathi, S.; Balasubramanian, K. K. Org. Biomol. Chem. 2012, 10, 6420–6431. doi:10.1039/c2ob25412c |
| 58. | McBurney, R. T.; Walton, J. C. J. Am. Chem. Soc. 2013, 135, 7349–7354. doi:10.1021/ja402833w |
| 9. | Leggio, A.; Alò, D.; Belsito, E. L.; Di Gioia, M. L.; Romio, E.; Siciliano, C.; Liguori, A. J. Pept. Sci. 2015, 21, 644–650. doi:10.1002/psc.2777 |
| 59. | Jöst, C.; Nitsche, C.; Scholz, T.; Roux, L.; Klein, C. D. J. Med. Chem. 2014, 57, 7590–7599. doi:10.1021/jm5006918 |
| 60. | Ren, Y.; Rousseaux, S. A. L. J. Org. Chem. 2018, 83, 913–920. doi:10.1021/acs.joc.7b02905 |
| 61. | Meng, H.; Sun, K.; Xu, Z.; Tian, L.; Wang, Y. Eur. J. Org. Chem. 2021, 1768–1772. doi:10.1002/ejoc.202100113 |
| 8. | Prashad, M.; Har, D.; Hu, B.; Kim, H.-Y.; Repic, O.; Blacklock, T. J. Org. Lett. 2003, 5, 125–128. doi:10.1021/ol0268440 |
| 52. | Das, D.; Khan, H. P. A.; Shivahare, R.; Gupta, S.; Sarkar, J.; Siddiqui, M. I.; Ampapathi, R. S.; Chakraborty, T. K. Org. Biomol. Chem. 2017, 15, 3337–3352. doi:10.1039/c6ob02610a |
| 53. | Avula, K.; Mohapatra, D. K. Tetrahedron Lett. 2016, 57, 1715–1717. doi:10.1016/j.tetlet.2016.03.017 |
| 5. | Olsen, R. K. J. Org. Chem. 1970, 35, 1912–1915. doi:10.1021/jo00831a042 |
| 6. | Schreiber, S. L.; Anthony, N. J.; Dorsey, B. D.; Hawley, R. C. Tetrahedron Lett. 1988, 29, 6577–6580. doi:10.1016/s0040-4039(00)82401-6 |
| 7. | Vedejs, E.; Kongkittingam, C. J. Org. Chem. 2001, 66, 7355–7364. doi:10.1021/jo0104882 |
| 54. | Heller, S. T.; Fu, T.; Sarpong, R. Org. Lett. 2012, 14, 1970–1973. doi:10.1021/ol300339q |
| 55. | Meng, G.; Szostak, R.; Szostak, M. Org. Lett. 2017, 19, 3596–3599. doi:10.1021/acs.orglett.7b01575 |
| 22. | Zhang, L.; Zhang, Y.; Deng, Y.; Shi, F. RSC Adv. 2015, 5, 14514–14521. doi:10.1039/c4ra13848a |
| 23. | Bruneau-Voisine, A.; Wang, D.; Dorcet, V.; Roisnel, T.; Darcel, C.; Sortais, J.-B. J. Catal. 2017, 347, 57–62. doi:10.1016/j.jcat.2017.01.004 |
| 24. | Neumann, J.; Elangovan, S.; Spannenberg, A.; Junge, K.; Beller, M. Chem. – Eur. J. 2017, 23, 5410–5413. doi:10.1002/chem.201605218 |
| 25. | Dang, T. T.; Ramalingam, B.; Seayad, A. M. ACS Catal. 2015, 5, 4082–4088. doi:10.1021/acscatal.5b00606 |
| 26. | Liang, R.; Li, S.; Wang, R.; Lu, L.; Li, F. Org. Lett. 2017, 19, 5790–5793. doi:10.1021/acs.orglett.7b02723 |
| 27. | Choi, G.; Hong, S. H. Angew. Chem., Int. Ed. 2018, 57, 6166–6170. doi:10.1002/anie.201801524 |
| 28. | Maji, M.; Chakrabarti, K.; Paul, B.; Roy, B. C.; Kundu, S. Adv. Synth. Catal. 2018, 360, 722–729. doi:10.1002/adsc.201701117 |
| 29. | Jiang, L.; Zhang, X.; Wang, Y.; Guo, F.; Hou, Z. Asian J. Org. Chem. 2021, 10, 2165–2169. doi:10.1002/ajoc.202100339 |
| 30. | Wang, J.; Qiang, W.; Ye, S.; Zhu, L.; Liu, X.; Loh, T.-P. Catal. Sci. Technol. 2021, 11, 3364–3375. doi:10.1039/d0cy02442b |
| 31. | Paul, B.; Maji, M.; Panja, D.; Kundu, S. Asian J. Org. Chem. 2022, 11, e202100678. doi:10.1002/ajoc.202100678 |
| 14. | Cabrero-Antonino, J. R.; Adam, R.; Junge, K.; Beller, M. Catal. Sci. Technol. 2016, 6, 7956–7966. doi:10.1039/c6cy01401a |
| 16. | Borch, R. F.; Hassid, A. I. J. Org. Chem. 1972, 37, 1673–1674. doi:10.1021/jo00975a049 |
| 17. | Alinezhad, H.; Tajbakhsh, M.; Salehian, F.; Fazli, K. Synth. Commun. 2010, 40, 2415–2420. doi:10.1080/00397910903249606 |
| 18. | Ge, X.; Luo, C.; Qian, C.; Yu, Z.; Chen, X. RSC Adv. 2014, 4, 43195–43203. doi:10.1039/c4ra04414b |
| 19. | Petricci, E.; Santillo, N.; Castagnolo, D.; Cini, E.; Taddei, M. Adv. Synth. Catal. 2018, 360, 2560–2565. doi:10.1002/adsc.201701619 |
| 20. | Savourey, S.; Lefèvre, G.; Berthet, J.-C.; Cantat, T. Chem. Commun. 2014, 50, 14033–14036. doi:10.1039/c4cc05908e |
| 21. | Sorribes, I.; Junge, K.; Beller, M. Chem. – Eur. J. 2014, 20, 7878–7883. doi:10.1002/chem.201402124 |
| 22. | Zhang, L.; Zhang, Y.; Deng, Y.; Shi, F. RSC Adv. 2015, 5, 14514–14521. doi:10.1039/c4ra13848a |
| 23. | Bruneau-Voisine, A.; Wang, D.; Dorcet, V.; Roisnel, T.; Darcel, C.; Sortais, J.-B. J. Catal. 2017, 347, 57–62. doi:10.1016/j.jcat.2017.01.004 |
| 24. | Neumann, J.; Elangovan, S.; Spannenberg, A.; Junge, K.; Beller, M. Chem. – Eur. J. 2017, 23, 5410–5413. doi:10.1002/chem.201605218 |
| 25. | Dang, T. T.; Ramalingam, B.; Seayad, A. M. ACS Catal. 2015, 5, 4082–4088. doi:10.1021/acscatal.5b00606 |
| 26. | Liang, R.; Li, S.; Wang, R.; Lu, L.; Li, F. Org. Lett. 2017, 19, 5790–5793. doi:10.1021/acs.orglett.7b02723 |
| 27. | Choi, G.; Hong, S. H. Angew. Chem., Int. Ed. 2018, 57, 6166–6170. doi:10.1002/anie.201801524 |
| 28. | Maji, M.; Chakrabarti, K.; Paul, B.; Roy, B. C.; Kundu, S. Adv. Synth. Catal. 2018, 360, 722–729. doi:10.1002/adsc.201701117 |
| 29. | Jiang, L.; Zhang, X.; Wang, Y.; Guo, F.; Hou, Z. Asian J. Org. Chem. 2021, 10, 2165–2169. doi:10.1002/ajoc.202100339 |
| 30. | Wang, J.; Qiang, W.; Ye, S.; Zhu, L.; Liu, X.; Loh, T.-P. Catal. Sci. Technol. 2021, 11, 3364–3375. doi:10.1039/d0cy02442b |
| 31. | Paul, B.; Maji, M.; Panja, D.; Kundu, S. Asian J. Org. Chem. 2022, 11, e202100678. doi:10.1002/ajoc.202100678 |
| 32. | Jacquet, O.; Frogneux, X.; Das Neves Gomes, C.; Cantat, T. Chem. Sci. 2013, 4, 2127–2131. doi:10.1039/c3sc22240c |
| 33. | Li, Y.; Fang, X.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 9568–9571. doi:10.1002/anie.201301349 |
| 34. | Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Ji, G.; Ma, Z.; Gao, X.; Liu, Z. Green Chem. 2015, 17, 4189–4193. doi:10.1039/c5gc01386k |
| 35. | Niu, H.; Lu, L.; Shi, R.; Chiang, C.-W.; Lei, A. Chem. Commun. 2017, 53, 1148–1151. doi:10.1039/c6cc09072a |
| 36. | Cui, X.; Dai, X.; Zhang, Y.; Deng, Y.; Shi, F. Chem. Sci. 2014, 5, 649–655. doi:10.1039/c3sc52676c |
| 37. | Tamura, M.; Miura, A.; Gu, Y.; Nakagawa, Y.; Tomishige, K. Chem. Lett. 2017, 46, 1243–1246. doi:10.1246/cl.170419 |
| 38. | Toyao, T.; Siddiki, S. M. A. H.; Morita, Y.; Kamachi, T.; Touchy, A. S.; Onodera, W.; Kon, K.; Furukawa, S.; Ariga, H.; Asakura, K.; Yoshizawa, K.; Shimizu, K.-i. Chem. – Eur. J. 2017, 23, 14848–14859. doi:10.1002/chem.201702801 |
| 39. | Lin, S.; Liu, J.; Ma, L. J. CO2 Util. 2021, 54, 101759. doi:10.1016/j.jcou.2021.101759 |
| 40. | Du, X.-L.; Tang, G.; Bao, H.-L.; Jiang, Z.; Zhong, X.-H.; Su, D. S.; Wang, J.-Q. ChemSusChem 2015, 8, 3489–3496. doi:10.1002/cssc.201500486 |
| 41. | Santoro, O.; Lazreg, F.; Minenkov, Y.; Cavallo, L.; Cazin, C. S. J. Dalton Trans. 2015, 44, 18138–18144. doi:10.1039/c5dt03506f |
| 42. | Jiang, X.; Wang, C.; Wei, Y.; Xue, D.; Liu, Z.; Xiao, J. Chem. – Eur. J. 2014, 20, 58–63. doi:10.1002/chem.201303802 |
| 43. | Goyal, V.; Naik, G.; Narani, A.; Natte, K.; Jagadeesh, R. V. Tetrahedron 2021, 98, 132414. doi:10.1016/j.tet.2021.132414 |
| 20. | Savourey, S.; Lefèvre, G.; Berthet, J.-C.; Cantat, T. Chem. Commun. 2014, 50, 14033–14036. doi:10.1039/c4cc05908e |
| 21. | Sorribes, I.; Junge, K.; Beller, M. Chem. – Eur. J. 2014, 20, 7878–7883. doi:10.1002/chem.201402124 |
| 44. | Paquette, L. A.; Tae, J. J. Org. Chem. 1998, 63, 2022–2030. doi:10.1021/jo9722921 |
| 45. | Benoit, W. L.; Parvez, M.; Keay, B. A. Tetrahedron: Asymmetry 2009, 20, 69–77. doi:10.1016/j.tetasy.2008.12.027 |
| 46. | Fuentes de Arriba, Á. L.; Seisdedos, D. G.; Simón, L.; Alcázar, V.; Raposo, C.; Morán, J. R. J. Org. Chem. 2010, 75, 8303–8306. doi:10.1021/jo101723v |
| 47. | Sun, N.; Wang, S.; Mo, W.; Hu, B.; Shen, Z.; Hu, X. Tetrahedron 2010, 66, 7142–7148. doi:10.1016/j.tet.2010.06.091 |
| 48. | Barluenga, J.; Bayón, A. M.; Campos, P.; Asensio, G.; Gonzalez-Nuñez, E.; Molina, Y. J. Chem. Soc., Perkin Trans. 1 1988, 1631–1636. doi:10.1039/p19880001631 |
| 49. | Crochet, R. A., Jr.; Blanton, C. D., Jr. Synthesis 1974, 55–56. doi:10.1055/s-1974-23242 |
| 50. | Zhang, J.; Chang, H.-M.; Kane, R. R. Synlett 2001, 643–645. doi:10.1055/s-2001-13378 |
| 51. | Blackburn, C.; LaMarche, M. J.; Brown, J.; Che, J. L.; Cullis, C. A.; Lai, S.; Maguire, M.; Marsilje, T.; Geddes, B.; Govek, E.; Kadambi, V.; Doherty, C.; Dayton, B.; Brodjian, S.; Marsh, K. C.; Collins, C. A.; Kym, P. R. Bioorg. Med. Chem. Lett. 2006, 16, 2621–2627. doi:10.1016/j.bmcl.2006.02.044 |
| 16. | Borch, R. F.; Hassid, A. I. J. Org. Chem. 1972, 37, 1673–1674. doi:10.1021/jo00975a049 |
| 17. | Alinezhad, H.; Tajbakhsh, M.; Salehian, F.; Fazli, K. Synth. Commun. 2010, 40, 2415–2420. doi:10.1080/00397910903249606 |
| 18. | Ge, X.; Luo, C.; Qian, C.; Yu, Z.; Chen, X. RSC Adv. 2014, 4, 43195–43203. doi:10.1039/c4ra04414b |
| 19. | Petricci, E.; Santillo, N.; Castagnolo, D.; Cini, E.; Taddei, M. Adv. Synth. Catal. 2018, 360, 2560–2565. doi:10.1002/adsc.201701619 |
| 12. | Selva, M.; Tundo, P.; Perosa, A. J. Org. Chem. 2002, 67, 9238–9247. doi:10.1021/jo026057g |
| 13. | Zheng, J.; Darcel, C.; Sortais, J.-B. Chem. Commun. 2014, 50, 14229–14232. doi:10.1039/c4cc05517a |
| 14. | Cabrero-Antonino, J. R.; Adam, R.; Junge, K.; Beller, M. Catal. Sci. Technol. 2016, 6, 7956–7966. doi:10.1039/c6cy01401a |
| 15. | Seo, H.; Bédard, A.-C.; Chen, W. P.; Hicklin, R. W.; Alabugin, A.; Jamison, T. F. Tetrahedron 2018, 74, 3124–3128. doi:10.1016/j.tet.2017.11.068 |
| 32. | Jacquet, O.; Frogneux, X.; Das Neves Gomes, C.; Cantat, T. Chem. Sci. 2013, 4, 2127–2131. doi:10.1039/c3sc22240c |
| 33. | Li, Y.; Fang, X.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 9568–9571. doi:10.1002/anie.201301349 |
| 34. | Yang, Z.; Yu, B.; Zhang, H.; Zhao, Y.; Ji, G.; Ma, Z.; Gao, X.; Liu, Z. Green Chem. 2015, 17, 4189–4193. doi:10.1039/c5gc01386k |
| 35. | Niu, H.; Lu, L.; Shi, R.; Chiang, C.-W.; Lei, A. Chem. Commun. 2017, 53, 1148–1151. doi:10.1039/c6cc09072a |
| 36. | Cui, X.; Dai, X.; Zhang, Y.; Deng, Y.; Shi, F. Chem. Sci. 2014, 5, 649–655. doi:10.1039/c3sc52676c |
| 37. | Tamura, M.; Miura, A.; Gu, Y.; Nakagawa, Y.; Tomishige, K. Chem. Lett. 2017, 46, 1243–1246. doi:10.1246/cl.170419 |
| 38. | Toyao, T.; Siddiki, S. M. A. H.; Morita, Y.; Kamachi, T.; Touchy, A. S.; Onodera, W.; Kon, K.; Furukawa, S.; Ariga, H.; Asakura, K.; Yoshizawa, K.; Shimizu, K.-i. Chem. – Eur. J. 2017, 23, 14848–14859. doi:10.1002/chem.201702801 |
| 39. | Lin, S.; Liu, J.; Ma, L. J. CO2 Util. 2021, 54, 101759. doi:10.1016/j.jcou.2021.101759 |
| 66. | Duspara, P. A.; Islam, M. S.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2012, 77, 10362–10368. doi:10.1021/jo302084a |
| 71. | Rannard, S. P.; Davis, N. J.; Herbert, I. Macromolecules 2004, 37, 9418–9430. doi:10.1021/ma0489218 |
| 72. | Griffin, R. J.; Evers, E.; Davison, R.; Gibson, A. E.; Layton, D.; Irwin, W. J. J. Chem. Soc., Perkin Trans. 1 1996, 1205–1211. doi:10.1039/p19960001205 |
| 73. | Alsarraf, J.; Petitpoisson, L.; Pichette, A. Org. Lett. 2021, 23, 6052–6056. doi:10.1021/acs.orglett.1c02116 |
| 74. | Zhao, C.; Ye, Z.; Ma, Z.-x.; Wildman, S. A.; Blaszczyk, S. A.; Hu, L.; Guizei, I. A.; Tang, W. Nat. Commun. 2019, 10, 4015. doi:10.1038/s41467-019-11976-2 |
| 62. | Chen, Z.; Cao, Y.; Tian, Z.; Zhou, X.; Xu, W.; Yang, J.; Teng, H. Tetrahedron Lett. 2017, 58, 2166–2170. doi:10.1016/j.tetlet.2017.04.072 |
| 63. | Peng, Z.; Wong, J. W.; Hansen, E. C.; Puchlopek-Dermenci, A. L. A.; Clarke, H. J. Org. Lett. 2014, 16, 860–863. doi:10.1021/ol403630g |
| 64. | Padiya, K. J.; Gavade, S.; Kardile, B.; Tiwari, M.; Bajare, S.; Mane, M.; Gaware, V.; Varghese, S.; Harel, D.; Kurhade, S. Org. Lett. 2012, 14, 2814–2817. doi:10.1021/ol301009d |
| 65. | de Greef, T. F. A.; Nieuwenhuizen, M. M. L.; Sijbesma, R. P.; Meijer, E. W. J. Org. Chem. 2010, 75, 598–610. doi:10.1021/jo902053t |
| 66. | Duspara, P. A.; Islam, M. S.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2012, 77, 10362–10368. doi:10.1021/jo302084a |
| 67. | Wong, E. C. N.; Reekie, T. A.; Werry, E. L.; O'Brien-Brown, J.; Bowyer, S. L.; Kassiou, M. Bioorg. Med. Chem. Lett. 2017, 27, 2439–2442. doi:10.1016/j.bmcl.2017.04.005 |
| 68. | Venter, J.; Perez, C.; van Otterlo, W. A. L.; Martínez, A.; Blackie, M. A. L. Bioorg. Med. Chem. Lett. 2019, 29, 1597–1600. doi:10.1016/j.bmcl.2019.04.049 |
| 69. | Jones, C. D.; Simmons, H. T. D.; Horner, K. E.; Liu, K.; Thompson, R. L.; Steed, J. W. Nat. Chem. 2019, 11, 375–381. doi:10.1038/s41557-019-0222-0 |
| 70. | Kondoh, A.; Ishikawa, S.; Terada, M. J. Am. Chem. Soc. 2020, 142, 3724–3728. doi:10.1021/jacs.9b13922 |
| 62. | Chen, Z.; Cao, Y.; Tian, Z.; Zhou, X.; Xu, W.; Yang, J.; Teng, H. Tetrahedron Lett. 2017, 58, 2166–2170. doi:10.1016/j.tetlet.2017.04.072 |
| 62. | Chen, Z.; Cao, Y.; Tian, Z.; Zhou, X.; Xu, W.; Yang, J.; Teng, H. Tetrahedron Lett. 2017, 58, 2166–2170. doi:10.1016/j.tetlet.2017.04.072 |
| 56. | Rawling, T.; McDonagh, A. M.; Tattam, B.; Murray, M. Tetrahedron 2012, 68, 6065–6070. doi:10.1016/j.tet.2012.05.002 |
| 57. | Velavan, A.; Sumathi, S.; Balasubramanian, K. K. Org. Biomol. Chem. 2012, 10, 6420–6431. doi:10.1039/c2ob25412c |
| 58. | McBurney, R. T.; Walton, J. C. J. Am. Chem. Soc. 2013, 135, 7349–7354. doi:10.1021/ja402833w |
| 62. | Chen, Z.; Cao, Y.; Tian, Z.; Zhou, X.; Xu, W.; Yang, J.; Teng, H. Tetrahedron Lett. 2017, 58, 2166–2170. doi:10.1016/j.tetlet.2017.04.072 |
| 64. | Padiya, K. J.; Gavade, S.; Kardile, B.; Tiwari, M.; Bajare, S.; Mane, M.; Gaware, V.; Varghese, S.; Harel, D.; Kurhade, S. Org. Lett. 2012, 14, 2814–2817. doi:10.1021/ol301009d |
| 59. | Jöst, C.; Nitsche, C.; Scholz, T.; Roux, L.; Klein, C. D. J. Med. Chem. 2014, 57, 7590–7599. doi:10.1021/jm5006918 |
| 60. | Ren, Y.; Rousseaux, S. A. L. J. Org. Chem. 2018, 83, 913–920. doi:10.1021/acs.joc.7b02905 |
| 61. | Meng, H.; Sun, K.; Xu, Z.; Tian, L.; Wang, Y. Eur. J. Org. Chem. 2021, 1768–1772. doi:10.1002/ejoc.202100113 |
| 62. | Chen, Z.; Cao, Y.; Tian, Z.; Zhou, X.; Xu, W.; Yang, J.; Teng, H. Tetrahedron Lett. 2017, 58, 2166–2170. doi:10.1016/j.tetlet.2017.04.072 |
| 76. | Prasad, A. S. B.; Kanth, J. V. B.; Periasamy, M. Tetrahedron 1992, 48, 4623–4628. doi:10.1016/s0040-4020(01)81236-9 |
| 77. | Rodrigues, A.; Olivato, P. R.; Rittner, R. Synthesis 2005, 2578–2582. doi:10.1055/s-2005-872101 |
| 78. | Harish, V.; Periasamy, M. Tetrahedron: Asymmetry 2017, 28, 175–180. doi:10.1016/j.tetasy.2016.12.002 |
| 66. | Duspara, P. A.; Islam, M. S.; Lough, A. J.; Batey, R. A. J. Org. Chem. 2012, 77, 10362–10368. doi:10.1021/jo302084a |
| 75. | Nugent, J.; Campbell, S. G.; Vo, Y.; Schwartz, B. D. Eur. J. Org. Chem. 2017, 5110–5118. doi:10.1002/ejoc.201700974 |
© 2022 Chen et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.