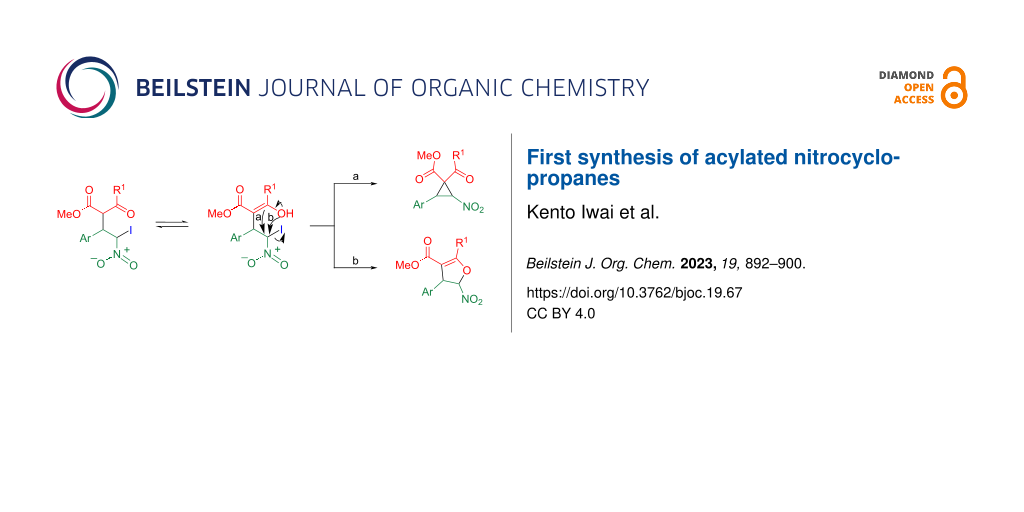
Abstract
Although nitrocyclopropanedicarboxylic acid esters are widely used in organic syntheses, nitrocyclopropanes with an acyl group have not yet been synthesized. When adducts of β-nitrostyrene and 1,3-dicarbonyl compounds are treated with (diacetoxyiodo)benzene and tetrabutylammonium iodide, iodination occurs at the α-position of the nitro group, and the subsequent O-attack of the enol moiety leads to 2,3-dihydrofuran. Cyclopropane was successfully synthesized through C-attack as the acyl group became bulkier. The obtained nitrocyclopropane was transformed into furan upon treatment with tin(II) chloride via a ring-opening/ring-closure process.

Graphical Abstract
Introduction
3-Arylated 2-nitrocyclopropane-1,1-dicarbonylic acid esters 1a have recently attracted considerable attention from synthetic organic chemists. In addition to their polyfunctionality, their ring strain and electron deficiency lead to a wide variety of reactivities. Based on their electron-deficient nature, these compounds have been used as substrates in the reaction with dinucleophiles such as 2-aminopyridines, which affords pyrido[1,2-a]pyrimidinones through ring opening (Scheme 1, reaction a) [1]. Chemical transformations that take advantage of polyfunctionality are also possible. A six-membered ring forms between the aryl group and ester functionality through an intramolecular aza-Wittig reaction, yielding cyclopropane-fused 2-quinolones [2]. A nitro group not only activates substrates and stabilizes the α-anion as an electron-withdrawing group but also acts as a nucleophile, electrophile, and leaving group, exhibiting diverse reactivities [3]. For example, when esters 1a are subjected to Lewis acid-induced denitration, highly electron-deficient enones (reaction b) [4] are obtained. The latter compounds are highly reactive and undergo reaction with, e.g., mercaptoacetaldehyde affording thiophenes (reaction c) [5] or with activated (hetero)aromatic compounds to give diarylated (oxoalkyl)malonates [6]. In the reaction using tin(II) chloride as the Lewis acid, the ring opening and nucleophilic attack of the nitro group occur, to produce functionalized isoxazolines (reaction d) [7]. In contrast, denitration under basic conditions generates highly reactive allenes (reaction e), which serve as synthetic intermediates for polyfunctionalized enynes [8]. The ring strain of the cyclopropane ring facilitates the cleavage of the C–C bond, and both cation and anion are stabilized by the adjacent phenyl group and ester functions, respectively (reaction f). These structural features enable the construction of a five-membered ring upon treatment with alkenes [9], diazo compounds (reaction g) [10], and nitriles [11].
![[1860-5397-19-67-i1]](/bjoc/content/inline/1860-5397-19-67-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Versatile reactivities of cyclopropanes 1a.
Scheme 1: Versatile reactivities of cyclopropanes 1a.
Several approaches are available for the synthesis of cyclopropanedicarboxylates 1a, which consist of three steps: 1) conjugate addition, 2) halogenation, and 3) ring closure (Scheme 2). β-Nitrostyrene 2 serves as an appropriate acceptor for conjugate addition by diethyl malonate (3a) to afford adduct 4a, in which the methine group flanked by two carbonyl groups is readily halogenated, and the subsequent intramolecular nucleophilic substitution by nitronic acid furnishes cyclopropane 1a (method a) [1,4,12,13]. Halogenated malonate 6a [7,14-17] and nitrostyrene 7 [18] can also be used as substrates in this protocol (methods b and c, respectively). In these methods, diesters are mostly used as 1,3-dicarbonyl compounds, with acetylacetone 3b used in only three cases [12,13,19], to the best of our knowledge. Extending beyond Scheme 2, only two syntheses of nitrocyclopropanes containing cyclic keto esters or diketones have been reported [20,21]. These findings indicate that the synthesis of nitrocyclopropanes with an acyl group is quite difficult, although they would be very useful in synthetic chemistry, if available. In this study, we investigated the synthesis of nitrocyclopropanes using keto esters 3c–f and diketones 3b and 3g instead of diester 3a as the starting 1,3-dicarbonyl compounds.
![[1860-5397-19-67-i2]](/bjoc/content/inline/1860-5397-19-67-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparative methods for cyclopropanedicarboxylates 1a.
Scheme 2: Preparative methods for cyclopropanedicarboxylates 1a.
Results and Discussion
We commenced our study using ethyl acetoacetate (3c) as starting 1,3-dicarbonyl compound according to method b (Scheme 2). After heating an acetonitrile solution of 3c, N-bromosuccinimide, and p-toluenesulfonic acid [7], α-bromoacetoacetate 6c was isolated with a 44% yield after purification of the reaction mixture by silica gel column chromatography (Scheme 3). In the subsequent reaction of the α-brominated product 6c with nitrostyrene 2a in the presence of triethylamine, the complete consumption of 2a was confirmed, however, the reaction mixture was complicated, and the desired cyclopropane 1c was not detected (Scheme 3).
![[1860-5397-19-67-i3]](/bjoc/content/inline/1860-5397-19-67-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Bromination of ethyl acetoacetate (3c) and reaction with nitrostyrene 2a.
Scheme 3: Bromination of ethyl acetoacetate (3c) and reaction with nitrostyrene 2a.
Next, method a (Scheme 2) was employed to synthesize nitrocyclopropanes 1 possessing an acyl group. Keto esters 3c–f and diketones 3b and 3g underwent conjugate addition to nitrostyrene 2a to afford the corresponding adducts 4b–g with moderate yields (Table 1). For unsymmetrical substrates, almost equal amounts of diastereomers were formed. Whereas 4b was isolated by recrystallization of the reaction mixture from ethanol, the other adducts 4c–g were easily isolated by column chromatography.
Table 1: Michael addition of 1,3-dicarbonyl compounds 3b–g to nitrostyrene 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-67-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R1 | R2 | Yield/% | dra | |
| 1 | Me | OEt | c | 74 | 58:42 |
| 2 | Et | OMe | d | 55 | 54:46 |
| 3 | iPr | OMe | e | 54 | 53:47 |
| 4 | Ph | OEt | f | 67 | 54:46 |
| 5 | Me | Me | b | 56 | — |
| 6 | Me | Ph | g | 66 | 57:43 |
aDiastereisomeric ratio determined by 1H NMR.
For comparison with previously reported results, adduct 4b, derived from acetylacetone (3b), was subjected to cyclopropanation according to a method described in the literature [13]. To a solution of adduct 4b in toluene, (diacetoxyiodo)benzene and tetrabutylammonium iodide were added, and the resulting mixture was stirred at room temperature for 14 h. Unexpectedly, from the reaction mixture, compound 8b was isolated after column chromatography with 21% yield instead of the desired cyclopropane 1b (Scheme 4).
![[1860-5397-19-67-i4]](/bjoc/content/inline/1860-5397-19-67-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of 4b with (diacetoxyiodo)benzene (top); structural determination of product 9 (bottom).
Scheme 4: Reaction of 4b with (diacetoxyiodo)benzene (top); structural determination of product 9 (bottom).
The NMR data for the ring protons of product 8b and compound 1b’ are listed in Table 2. Although the benzene ring in compound 8b is methyl-substituted, a 1H NMR spectrum similar to those in the literature was observed [12,13,19], indicating that both compounds have the same framework. Surprisingly, the coupling constants were considerably smaller than those of 1a (Ar = MeC6H4), and the same tendency was previously found in the literature [12,13]. However, a reasonable explanation was not given for the different coupling constants between diester 1a and diketone 1b’. In the 13C NMR spectrum of diester 1a, two separate signals of carbonyl groups were observed at 163.2 and 163.3 ppm, indicating that the two ester functionalities were not equivalent. Moreover, the spectrum of compound 8b revealed only a single signal of a carbonyl carbon at 193.2 ppm, and a signal of a quaternary carbon at 167.0 ppm, which could not be assigned. These spectral data prompted us to reconsider the structure of compound 8b.
Table 2: Comparison of the 1H NMR data of ring protons for compounds 1a, 1b’, and product 8b.
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-67-i8.svg?max-width=637&scale=1.0)
|
|||||||||
| Ref. | Chemical shift/ppm | Coupling constant | Ref. | Chemical shift/ppm | Coupling constant | ||||
| Ha | Hb | J [Hz] | Ha | Hb | J [Hz] | ||||
| 8b | 4.61 (br s) | 5.71 (br s) | – | ||||||
| [12] | 1b’ | 4.66 (d) | 5.77 (d) | 2.0 | [12] | 1a | 4.17 (d) | 5.39 (d) | 6.0 |
| [13] | 1b’ | 4.66 (d) | 5.74 (d) | 1.8 | [13] | 1a | 4.15 (d) | 5.38 (d) | 6.0 |
| [19] | 1b’ | 4.59 (s) | 5.67 (s) | – | [7] | 1a | 4.15 (d) | 5.37 (d) | 5.9 |
The reaction of chlorinated nitrostyrene 2b with acetylacetone (3b) was in the same way, and the obtained product 9 was converted to the 2,4-dinitrophenylhydrazone 10 to facilitate the crystallization for X-ray crystallography, which showed that a 2,3-dihydrofuran framework had formed (Scheme 4, bottom). Hence, we clarified that product 8b was not the desired cyclopropane, but a dihydrofuran, so the cyclopropane 1b’ reported in the literature [12,13,19] is presumably incorrect [22]. In the cases of donor–acceptor cyclopropanes possessing an electron-donating group such as an alkoxy or amino group, ring expansion caused by an intramolecular attack of nitro oxygen occurs, leading to five-membered cyclic nitronates [23]. To the contrary, such reaction was not observed at all, which is presumably due to the lower electron-donating ability of the benzene ring.
The other adducts 4c–g were subjected to the cyclization under the same conditions (Table 3). When the adduct of ethyl acetoacetate (4c) was used, dihydrofuran 8c was obtained as the main product, with small amounts of cyclopropane 1c (Table 3, entry 1).
Table 3: Cyclization of other adducts 4.
![[Graphic 3]](/bjoc/content/inline/1860-5397-19-67-i9.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | R1 | R2 | Yield [%] | Recovery of 4/% | |||
| 1 | 8 | 11 | |||||
| 1 | Me | OEt | c | 8 | 39 | 0 | 0 |
| 2 | Et | OMe | d | 12 | 44 | trace | 0 |
| 3 | iPr | OMe | e | 24 | 36 | 9 | 0 |
| 4 | Ph | OEt | f | 56 | 9 | 17 | 0 |
| 5 | Me | Me | b | 0 | 21 | 0 | 17 |
| 6 | Me | Ph | g | 5 | 30a | 0 | 7 |
aTotal yield of two isomers 8g (14%) and 8g’ (R1 = Ph, R2 = Me, 16%).
With a bulkier acyl group the yield of 1 was higher, and the yield increased up to 56% in the reaction using adduct 4f, which was derived from ethyl benzoylacetate (3f) (Table 3, entries 1–4). The yield of aromatized furans 8 also increased with an increase in the bulkiness of the acyl group. A similar tendency was observed when adducts 4b and 4g derived from diketones 3b and 3g were reacted in the same way (Table 3, entries 5 and 6).
To obtain insight into the two cyclization modes, the reaction of 4e was monitored by 1H NMR in 5 min intervals (Figure 1). In Figure 1, the red triangles are the total yields of furans 8e and 11e. The yields of cyclopropane 1e and furans 8e and 11e increased with increasing reaction time, without disturbing the shape of the graph. In addition, we confirmed that isolated cyclopropane 1e did not change upon treatment with (diacetoxyiodo)benzene and tetrabutylammonium iodide. These findings indicate that no equilibrium existed between cyclopropane 1e and dihydrofuran 8e, and that these products were competitively formed.
![[1860-5397-19-67-1]](/bjoc/content/figures/1860-5397-19-67-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Monitoring the cyclization reaction using 4e by 1H NMR.
Figure 1: Monitoring the cyclization reaction using 4e by 1H NMR.
A plausible mechanism explaining the experimental results is illustrated in Scheme 5. In this reaction, acetoxyiodine serves as the active species [13,24]. Nitronic acid, one of the tautomers of 4, attacks the acetoxyiodine to afford α-iododerivative 12. After the carbonyl moiety tautomerized to the enol form, a C-attack (path a) furnishes cyclopropane 1, and an O-attack (path b) furnishes dihydrofuran 8. When the R1 group becomes bulkier, the hydroxy group may be far from the reaction site because of the steric repulsion in the stable conformation. Another possibility is that the bulky substituent may prevent the attack of other reagents and suppress the decomposition of the nitrocyclopropane framework.
![[1860-5397-19-67-i5]](/bjoc/content/inline/1860-5397-19-67-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A plausible mechanism for formation of cyclopropane 1 and dihydrofuran 8.
Scheme 5: A plausible mechanism for formation of cyclopropane 1 and dihydrofuran 8.
The chemical transformation of cyclopropane 1e was investigated. When a solution of 1e and tin(II) chloride in benzene was heated at 100 °C for 14 h, successive ring-opening/ring-closure proceeded, to produce furan 13 with a 46% yield (Scheme 6). The coordination of two carbonyl groups to the tin species facilitated the ring opening of the cyclopropane ring to afford betaine [7], then the oxygen atom of the enolate attacked the benzyl cation to construct a five-membered ring. The subsequent elimination of nitrous acid, accompanied by aromatization, yielded furan 13. In addition to the stepwise mechanism, a concerted ring-expansion can be also acceptable [25].
![[1860-5397-19-67-i6]](/bjoc/content/inline/1860-5397-19-67-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Tin(II)-mediated ring expansion of nitrocyclopropane 1e.
Scheme 6: Tin(II)-mediated ring expansion of nitrocyclopropane 1e.
Conclusion
Although nitrocyclopropanedicarboxylic acid esters 1a have been used in organic syntheses, nitrocyclopropanes possessing an acyl group are unknown, except for a derivative of cyclic diketone [20,21]. Although diacetyl derivative 13 has been reported in some studies [12,13,19], we corrected its structure to dihydrofuran 8. This is the first report of the synthesis of acylated nitrocyclopropanes 1. After the conjugate addition of the 1,3-dicarbonyl compound 3 to nitrostyrene 2 and the α-iodination of the adduct 4, two cyclization modes became possible owing to the ambident property of enol 12. Dihydrofuran 8 was formed in the case of an O-attack, and nitrocyclopropane 1 was formed in the case of a C-attack. Furthermore, the latter cyclization predominantly proceeded as the acyl group became bulkier. The polyfunctionality of products 1 and 8 facilitate further chemical conversion [22]. Nitrocyclopropane 1e was converted into furan 13 by treatment with tin(II) chloride. The findings obtained herein will be useful for researchers studying organic syntheses using functionalized cyclopropanes.
Experimental
General
All reagents were purchased from commercial sources and used without further purification. 1H and13C NMR spectra were recorded on Bruker DPX-400 and JEOL JMN-ECZ400S spectrometers (400 MHz and 100 MHz, respectively) in CDCl3 using TMS as an internal standard. The assignments of the 13C NMR signals were performed by DEPT experiments. IR spectra were recorded on a JASCO FT/IR-4200 spectrometer equipped with an ATR detector. High-resolution mass spectra were obtained on AB SCIEX Triplet TOF 4600 and Bruker Compact mass spectrometers. All measurements were made on a Rigaku AFC7R diffractometer with graphite monochromatized Mo Kα radiation. Melting points were recorded on an SRS-Optimelt automated melting point system and were uncorrected.
Preparation of nitrostyrenes 2
Nitrostyrenes 2 were prepared according to the literature [26]. To a solution of ammonium acetate (2.63 g, 36 mmol) in acetic acid (10 mL), were added 4-methylbenzaldehyde (2 mL, 15 mmol) and nitromethane (5.3 mL, 98 mmol), and the resultant mixture was heated at 100 °C for 6 h. After adjusting the pH value to 7 by 2 M sodium hydroxide (40 mL, 80 mmol), the mixture was extracted with ethyl acetate (30 mL × 3), and the organic layer was washed with brine (30 mL × 1), dried over magnesium sulfate, and concentrated to afford nitrostyrene 2a (17.4 g, 71%, mp 56–58 °C) as a yellow solid. Nitrostyrene 2b (mp 112–114 °C) was prepared in the same way. The structure was confirmed by comparison with those reported in the literature [26].
Conjugate addition of 1,3-dicarbonyl compound 3 to nitrostyrene 2
Adduct 4 was prepared according to the literature [27]. To a solution of nitrostyrene 2a (978 mg, 6 mmol) in dichloromethane (8 mL), were added ethyl benzoylacetate (3f, 1.04 mL, 6 mmol) and triethylamine (84 μL, 0.6 mmol), and the resultant solution was stirred at room temperature for 14 h. After removal of the solvent under reduced pressure, the residual pale-yellow solid was extracted with hot hexane (20 mL × 4). The hexane was concentrated, and the residual yellow oil was subjected to column chromatography on silica gel to afford ethyl 2-benzoyl-3-(4-methylphenyl)-4-nitrobutanoate (4f) [28] (eluted with hexane/ethyl acetate 95:5, 1.43 g, 4.02 mmol, 67%) as a pale-yellow oil. Major isomer 1H NMR (400 MHz, CDCl3) δ 0.92 (t, J = 7.2 Hz, 3H), 4.17 (q, J = 1.8, 7.2 Hz, 2H), 4.48–4.37 (m, 1H), 4.81–4.72 (m, 1H), 4.97–4.88 (m, 2H), 7.09 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.48 (dd, J = 7.6, 7.2 Hz, 2H), 7.61 (t, J = 7.6 Hz, 1H), 8.05 (d, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.6 (CH3), 21.1 (CH3), 42.8 (CH), 57.1 (CH), 61.9 (CH2), 78.1 (CH2), 128.1 (CH), 128.6 (CH), 128.9 (CH), 128.9 (CH), 133.8 (C), 134.2 (CH), 135.9 (C), 138.0 (C), 167.8 (C), 192.9 (C). Minor isomer 1H NMR (400 MHz, CDCl3) δ 1.17 (dd, J = 7.2, 7.2 Hz, 3H), 3.88 (dq, J = 7.2 Hz, 1H), 3.88 (dq, J = 7.2 Hz, 1H), 4.48–4.37 (m, 1H), 4.81–4.72 (m, 1H), 4.97–4.88 (m, 2H), 7.02 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 7.41 (dd, J = 7.6, 7.2 Hz, 2H), 7.55 (t, J = 7.6 Hz, 1H), 7.86 (d, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 21.0 (CH3), 42.8 (CH), 56.5 (CH), 62.2 (CH2), 78.1 (CH2), 127.8 (CH), 128.7 (CH), 129.6 (CH), 129.6 (CH), 133.2 (C), 133.8 (CH), 135.1 (C), 137.8 (C), 167.0 (C), 192.7 (C).
Although the adduct was obtained as a mixture of diastereomeric isomers (54:46), these isomers were subjected to subsequent cyclization without separation. When other 1,3-dicarbonyl compounds 3 or nitrostyrene 2b were used, the reaction was conducted in the same way.
Cyclization of adduct 4
Cyclization was conducted according to the literature [13]. To a solution of adduct 4f (593 mg, 1.67 mmol) in toluene (7 mL), were added (diacetoxyiodo)benzene (539 mg, 2.5 mmol) and tetrabutylammonium iodide (618 mg, 2.5 mmol), and the resultant mixture was stirred at room temperature for 14 h. The solution was subjected to column chromatography on silica gel to afford ethyl 4,5-dihydro-4-(4-methylphenyl)-5-nitro-2-phenylfuran-3-carboxylate (8f) (eluted with hexane/ethyl acetate 95:5, 53 mg, 0.15 mmol, 9%) as a pale-yellow oil, ethyl 1-benzoyl-2-(4-methylphenyl)-3-nitrocyclopropanecarboxylate (1f) (eluted with hexane/ethyl acetate 90:10, 330 mg, 0.94 mmol, 56%) as a yellow oil, and ethyl 4-(4-methylphenyl)-5-nitro-2-phenylfuran-3-carboxylate (11f) (eluted with hexane/ethyl acetate 90:10, 100 mg, 0.28 mmol, 17%) as a yellow oil, respectively.
Ethyl 1-benzoyl-2-(4-methylphenyl)-3-nitrocyclopropanecarboxylate (1f): Major isomer: 1H NMR (400 MHz, CDCl3) δ 0.82 (t, J = 7.1 Hz, 3H), 2.34 (s, 3H), 3.90 (q, J = 7.1 Hz, 2H), 4.34 (d, J = 5.8 Hz, 1H), 5.73 (d, J = 5.8 Hz, 1H), 7.16 (d, J = 7.9 Hz, 2H), 7.24 (d, J = 7.9 Hz, 2H), 7.48 (dd, J = 7.5, 7.8 Hz, 2H), 7.59 (t, J = 7.5 Hz, 1H), 7.96 (d, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5 (CH3), 21.1 (CH3), 36.5 (CH), 49.8 (C), 62.7 (CH2), 67.8 (CH), 127.5 (C), 128.2 (CH), 128.3 (CH), 129.1 (CH), 129.5 (CH), 134.1 (CH), 135.1 (C), 138.3 (C), 164.3 (C), 186.6 (C); IR (ATR): 1362, 1557, 1682, 1695 cm−1; HRESIMS–TOF (m/z): [M + H]+ calcd for C20H19NO5, 354.1336; found, 354.1328. Minor isomer: 1H NMR (400 MHz, CDCl3) δ 0.99 (t, J = 7.1 Hz, 3H), 2.20 (s, 3H), 4.11 (q, J = 7.1 Hz, 2H), 4.44 (d, J = 6.2 Hz, 1H), 5.73–5.72 (overlapped, 1H), 7.00 (d, J = 8.0 Hz, 2H), 7.07 (d, J = 8.0 Hz, 2H), 7.37 (dd, J = 7.7, 7.8 Hz, 2H), 7.79 (d, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5 (CH3), 21.0 (CH3), 38.8 (CH), 52.4 (C), 63.1 (CH2), 66.2 (CH), 126.9 (C), 127.6 (CH), 128.6 (CH), 128.6 (CH), 129.5 (CH), 133.8 (CH), 135.6 (C), 138.4 (C), 164.8 (C), 187.8 (C).
Ethyl 4,5-dihydro-4-(4-methylphenyl)-5-nitro-2-phenylfuran-3-carboxylate (8f): 1H NMR (400 MHz, CDCl3) δ 1.08 (dd, J = 7.2, 7.2 Hz, 3H), 4.02 (dq, J = 7.2, 10.8 Hz, 1H), 4.06 (dq, J = 7.2, 10.8 Hz, 1H), 4.80 (d, J = 1.7 Hz, 1H), 5.86 (d, J = 1.7 Hz, 1H), 7.21–7.20 (br s, 4H), 7.54–7.46 (m, 3H), 8.09–8.07 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 13.9 (CH3), 21.1 (CH3), 57.0 (CH), 60.6 (CH2), 107.7 (C), 108.9 (CH), 127.0 (CH), 127.7 (C), 128.0 (CH), 129.9 (CH), 130.0 (CH), 134.9 (C), 138.3 (C), 162.7 (C), 163.5 (C); IR (ATR): 1371, 1572, 1697 cm−1; HRESIMS–TOF (m/z): [M + H]+ calcd for C14H15NO4, 354.1336; found, 354.1324.
Ethyl 4-(4-methylphenyl)-5-nitro-2-phenylfuran-3-carboxylate (11f): 1H NMR (400 MHz, CDCl3) δ 0.99 (t, J = 7.2 Hz, 3H), 2.42 (s, 3H), 4.12 (q, J = 7.2 Hz, 2H), 7.26 (d, J = 6.8 Hz, 2H), 7.32 (d, J = 6.8 Hz, 2H), 7.52–7.47 (m, 3H), 7.93 (dd, J = 7.2, 1.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.5 (CH3), 21.4 (CH3), 61.6 (CH2), 118.2 (C), 125.6 (C), 127.3 (C), 128.4 (CH), 128.7 (CH), 128.9 (CH), 129.2 (CH), 131.2 (CH), 139.3 (C), 154.9 (C), 162.5 (C), Two signals were lacked presumably due to overlapping. IR (ATR): 1359, 1593, 1730 cm−1; HRESIMS–TOF (m/z): [M + Na]+ calcd for C19H19NO6, 374.0999; found, 374.1000.
Supporting Information
| Supporting Information File 1: Spectral data for 1, 4, 8, 10, 13 and NMR charts (1H and 13C NMR), and information of X-ray analysis for 10. | ||
| Format: PDF | Size: 3.6 MB | Download |
| Supporting Information File 2: Crystallographic information file for compound 10. | ||
| Format: CIF | Size: 180.9 KB | Download |
| Supporting Information File 3: CheckCIF/PLATON report for compound 10. | ||
| Format: PDF | Size: 138.3 KB | Download |
References
-
Selvi, S.; Srinivasan, K. Eur. J. Org. Chem. 2017, 5644–5648. doi:10.1002/ejoc.201700765
Return to citation in text: [1] [2] -
Diaz, J.; Rodenas, D.; Ballester, F.-J.; Alajarin, M.; Orenes, R.-A.; Sanchez-Andrada, P.; Vidal, A. Arabian J. Chem. 2020, 13, 2702–2714. doi:10.1016/j.arabjc.2018.07.002
Return to citation in text: [1] -
Nishiwaki, N. Molecules 2020, 25, 3680. doi:10.3390/molecules25163680
Return to citation in text: [1] -
Selvi, T.; Srinivasan, K. J. Org. Chem. 2014, 79, 3653–3658. doi:10.1021/jo402848v
Return to citation in text: [1] [2] -
Selvi, T.; Vanmathi, G.; Srinivasan, K. RSC Adv. 2015, 5, 49326–49329. doi:10.1039/c5ra09111j
Return to citation in text: [1] -
Selvi, T.; Srinivasan, K. Adv. Synth. Catal. 2015, 357, 2111–2118. doi:10.1002/adsc.201500143
Return to citation in text: [1] -
Asahara, H.; Kamidate, R.; Nishiwaki, N. Heterocycles 2021, 103, 379–391. doi:10.3987/com-20-s(k)21
Return to citation in text: [1] [2] [3] [4] [5] -
Zhou, Z.-Y.; Xu, Z.-Y.; Shen, Q.-Y.; Huang, L.-S.; Zhang, X.; Xia, A.-B.; Xu, D.-Q.; Xu, Z.-Y. Chem. Commun. 2021, 57, 6424–6427. doi:10.1039/d1cc01573g
Return to citation in text: [1] -
Wang, C.; Ren, X.; Xie, H.; Lu, Z. Chem. – Eur. J. 2015, 21, 9676–9680. doi:10.1002/chem.201500873
Return to citation in text: [1] -
Yang, C.; Liu, W.; He, Z.; He, Z. Org. Lett. 2016, 18, 4936–4939. doi:10.1021/acs.orglett.6b02415
Return to citation in text: [1] -
Selvi, T.; Srinivasan, K. Chem. Commun. 2014, 50, 10845–10848. doi:10.1039/c4cc04764h
Return to citation in text: [1] -
Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Motornov, V. A.; Tabolin, A. A.; Novikov, R. A.; Nelyubina, Y. V.; Nenajdenko, V. G.; Ioffe, S. L. Org. Chem. Front. 2018, 5, 2588–2594. doi:10.1039/c8qo00623g
Return to citation in text: [1] -
Rapi, Z.; Démuth, B.; Keglevich, G.; Grün, A.; Drahos, L.; Sóti, P. L.; Bakó, P. Tetrahedron: Asymmetry 2014, 25, 141–147. doi:10.1016/j.tetasy.2013.12.007
Return to citation in text: [1] -
Cai, S.; Zhang, S.; Zhao, Y.; Wang, D. Z. Org. Lett. 2013, 15, 2660–2663. doi:10.1021/ol4009443
Return to citation in text: [1] -
Lee, H. J.; Kim, S. M.; Kim, D. Y. Tetrahedron Lett. 2012, 53, 3437–3439. doi:10.1016/j.tetlet.2012.04.072
Return to citation in text: [1] -
McCooey, S. H.; McCabe, T.; Connon, S. J. J. Org. Chem. 2006, 71, 7494–7497. doi:10.1021/jo0613838
Return to citation in text: [1] -
Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016
Return to citation in text: [1] [2] [3] [4] [5] -
Das, U.; Tsai, Y.-L.; Lin, W. Org. Biomol. Chem. 2013, 11, 44–47. doi:10.1039/c2ob26943k
Return to citation in text: [1] [2] -
Piovesana, S.; Scarpino Schietroma, D. M.; Tulli, L. G.; Monaco, M. R.; Bella, M. Chem. Commun. 2010, 46, 5160–5162. doi:10.1039/c003296d
Return to citation in text: [1] [2] -
Nikerov, D. S.; Ashatkina, M. A.; Shiryaev, V. A.; Tkachenko, I. M.; Rybakov, V. B.; Reznikov, A. N.; Klimochkin, Y. N. Tetrahedron 2021, 84, 132029. doi:10.1016/j.tet.2021.132029
Return to citation in text: [1] [2] -
Schmidt, C. D.; Kaschel, J.; Schneider, T. F.; Kratzert, D.; Stalke, D.; Werz, D. B. Org. Lett. 2013, 15, 6098–6101. doi:10.1021/ol402990j
Return to citation in text: [1] -
Kandrnálová, M.; Kokan, Z.; Havel, V.; Nečas, M.; Šindelář, V. Angew. Chem., Int. Ed. 2019, 58, 18182–18185. doi:10.1002/anie.201908953
Return to citation in text: [1] -
Schneider, T. F.; Kaschel, J.; Awan, S. I.; Dittrich, B.; Werz, D. B. Chem. – Eur. J. 2010, 16, 11276–11288. doi:10.1002/chem.201000468
Return to citation in text: [1] -
Lopchuk, J. M.; Hughes, R. P.; Gribble, G. W. Org. Lett. 2013, 15, 5218–5221. doi:10.1021/ol402385v
Return to citation in text: [1] [2] -
Zenz, I.; Mayr, H. J. Org. Chem. 2011, 76, 9370–9378. doi:10.1021/jo201678u
Return to citation in text: [1] -
Jiang, X.; Zhang, Y.; Liu, X.; Zhang, G.; Lai, L.; Wu, L.; Zhang, J.; Wang, R. J. Org. Chem. 2009, 74, 5562–5567. doi:10.1021/jo9009276
Return to citation in text: [1]
| 22. | Nikerov, D. S.; Ashatkina, M. A.; Shiryaev, V. A.; Tkachenko, I. M.; Rybakov, V. B.; Reznikov, A. N.; Klimochkin, Y. N. Tetrahedron 2021, 84, 132029. doi:10.1016/j.tet.2021.132029 |
| 23. | Schmidt, C. D.; Kaschel, J.; Schneider, T. F.; Kratzert, D.; Stalke, D.; Werz, D. B. Org. Lett. 2013, 15, 6098–6101. doi:10.1021/ol402990j |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 24. | Kandrnálová, M.; Kokan, Z.; Havel, V.; Nečas, M.; Šindelář, V. Angew. Chem., Int. Ed. 2019, 58, 18182–18185. doi:10.1002/anie.201908953 |
| 1. | Selvi, S.; Srinivasan, K. Eur. J. Org. Chem. 2017, 5644–5648. doi:10.1002/ejoc.201700765 |
| 5. | Selvi, T.; Vanmathi, G.; Srinivasan, K. RSC Adv. 2015, 5, 49326–49329. doi:10.1039/c5ra09111j |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 19. | Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016 |
| 26. | Lopchuk, J. M.; Hughes, R. P.; Gribble, G. W. Org. Lett. 2013, 15, 5218–5221. doi:10.1021/ol402385v |
| 4. | Selvi, T.; Srinivasan, K. J. Org. Chem. 2014, 79, 3653–3658. doi:10.1021/jo402848v |
| 20. | Das, U.; Tsai, Y.-L.; Lin, W. Org. Biomol. Chem. 2013, 11, 44–47. doi:10.1039/c2ob26943k |
| 21. | Piovesana, S.; Scarpino Schietroma, D. M.; Tulli, L. G.; Monaco, M. R.; Bella, M. Chem. Commun. 2010, 46, 5160–5162. doi:10.1039/c003296d |
| 7. | Asahara, H.; Kamidate, R.; Nishiwaki, N. Heterocycles 2021, 103, 379–391. doi:10.3987/com-20-s(k)21 |
| 14. | Motornov, V. A.; Tabolin, A. A.; Novikov, R. A.; Nelyubina, Y. V.; Nenajdenko, V. G.; Ioffe, S. L. Org. Chem. Front. 2018, 5, 2588–2594. doi:10.1039/c8qo00623g |
| 15. | Rapi, Z.; Démuth, B.; Keglevich, G.; Grün, A.; Drahos, L.; Sóti, P. L.; Bakó, P. Tetrahedron: Asymmetry 2014, 25, 141–147. doi:10.1016/j.tetasy.2013.12.007 |
| 16. | Cai, S.; Zhang, S.; Zhao, Y.; Wang, D. Z. Org. Lett. 2013, 15, 2660–2663. doi:10.1021/ol4009443 |
| 17. | Lee, H. J.; Kim, S. M.; Kim, D. Y. Tetrahedron Lett. 2012, 53, 3437–3439. doi:10.1016/j.tetlet.2012.04.072 |
| 22. | Nikerov, D. S.; Ashatkina, M. A.; Shiryaev, V. A.; Tkachenko, I. M.; Rybakov, V. B.; Reznikov, A. N.; Klimochkin, Y. N. Tetrahedron 2021, 84, 132029. doi:10.1016/j.tet.2021.132029 |
| 2. | Diaz, J.; Rodenas, D.; Ballester, F.-J.; Alajarin, M.; Orenes, R.-A.; Sanchez-Andrada, P.; Vidal, A. Arabian J. Chem. 2020, 13, 2702–2714. doi:10.1016/j.arabjc.2018.07.002 |
| 18. | McCooey, S. H.; McCabe, T.; Connon, S. J. J. Org. Chem. 2006, 71, 7494–7497. doi:10.1021/jo0613838 |
| 26. | Lopchuk, J. M.; Hughes, R. P.; Gribble, G. W. Org. Lett. 2013, 15, 5218–5221. doi:10.1021/ol402385v |
| 9. | Wang, C.; Ren, X.; Xie, H.; Lu, Z. Chem. – Eur. J. 2015, 21, 9676–9680. doi:10.1002/chem.201500873 |
| 11. | Selvi, T.; Srinivasan, K. Chem. Commun. 2014, 50, 10845–10848. doi:10.1039/c4cc04764h |
| 20. | Das, U.; Tsai, Y.-L.; Lin, W. Org. Biomol. Chem. 2013, 11, 44–47. doi:10.1039/c2ob26943k |
| 21. | Piovesana, S.; Scarpino Schietroma, D. M.; Tulli, L. G.; Monaco, M. R.; Bella, M. Chem. Commun. 2010, 46, 5160–5162. doi:10.1039/c003296d |
| 8. | Zhou, Z.-Y.; Xu, Z.-Y.; Shen, Q.-Y.; Huang, L.-S.; Zhang, X.; Xia, A.-B.; Xu, D.-Q.; Xu, Z.-Y. Chem. Commun. 2021, 57, 6424–6427. doi:10.1039/d1cc01573g |
| 1. | Selvi, S.; Srinivasan, K. Eur. J. Org. Chem. 2017, 5644–5648. doi:10.1002/ejoc.201700765 |
| 4. | Selvi, T.; Srinivasan, K. J. Org. Chem. 2014, 79, 3653–3658. doi:10.1021/jo402848v |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 19. | Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016 |
| 7. | Asahara, H.; Kamidate, R.; Nishiwaki, N. Heterocycles 2021, 103, 379–391. doi:10.3987/com-20-s(k)21 |
| 7. | Asahara, H.; Kamidate, R.; Nishiwaki, N. Heterocycles 2021, 103, 379–391. doi:10.3987/com-20-s(k)21 |
| 6. | Selvi, T.; Srinivasan, K. Adv. Synth. Catal. 2015, 357, 2111–2118. doi:10.1002/adsc.201500143 |
| 10. | Yang, C.; Liu, W.; He, Z.; He, Z. Org. Lett. 2016, 18, 4936–4939. doi:10.1021/acs.orglett.6b02415 |
| 25. | Schneider, T. F.; Kaschel, J.; Awan, S. I.; Dittrich, B.; Werz, D. B. Chem. – Eur. J. 2010, 16, 11276–11288. doi:10.1002/chem.201000468 |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 19. | Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016 |
| 7. | Asahara, H.; Kamidate, R.; Nishiwaki, N. Heterocycles 2021, 103, 379–391. doi:10.3987/com-20-s(k)21 |
| 28. | Jiang, X.; Zhang, Y.; Liu, X.; Zhang, G.; Lai, L.; Wu, L.; Zhang, J.; Wang, R. J. Org. Chem. 2009, 74, 5562–5567. doi:10.1021/jo9009276 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 7. | Asahara, H.; Kamidate, R.; Nishiwaki, N. Heterocycles 2021, 103, 379–391. doi:10.3987/com-20-s(k)21 |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 19. | Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 19. | Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016 |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
| 13. | Fan, R.; Ye, Y.; Li, W.; Wang, L. Adv. Synth. Catal. 2008, 350, 2488–2492. doi:10.1002/adsc.200800452 |
| 12. | Li, Y.; Guo, H.; Fan, R. Synthesis 2020, 52, 928–932. doi:10.1055/s-0039-1690809 |
© 2023 Iwai et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.