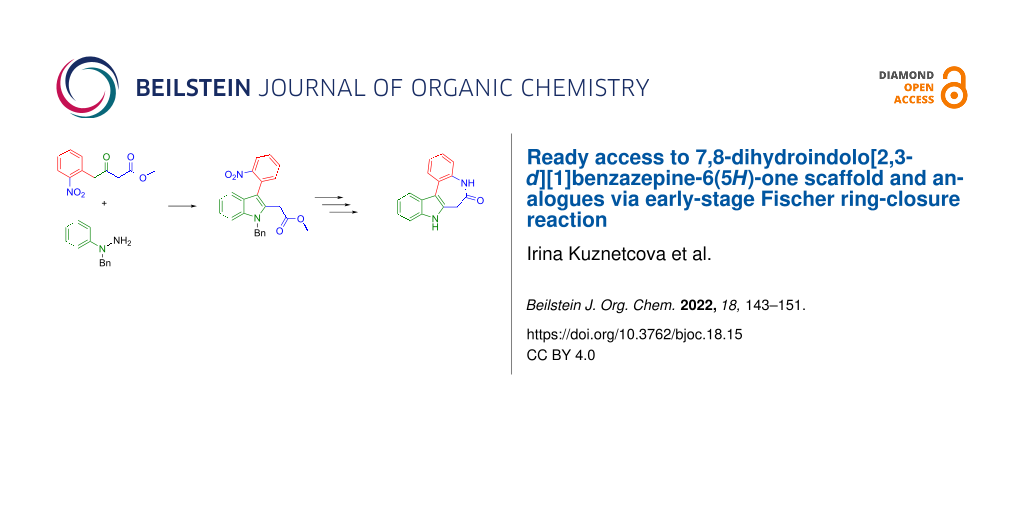
Abstract
Paullone isomers are known as inhibitors of tubulin polymerase and cyclin dependent kinases (Cdks), which are potential targets for cancer chemotherapy. Herein we report an efficient and clean pathway to the fourth isomer, which remained elusive so far, namely 7,8-dihydroindolo[2,3-d][1]benzazepin-6(5H)-one. Moreover, we demonstrate the generality of our pathway by synthesizing two closely related analogues, one containing a bromo substituent and the other one incorporating an 8-membered instead of a 7-membered ring. The key transformation in this four-step synthesis, with an overall yield of 29%, is the Fischer indole reaction of 2-nitrophenylacetyl acetoacetate with 1-benzyl-1-phenylhydrazine in acetic acid that delivers methyl 2-(1-benzyl-3-(2-nitrophenyl)-1H-indol-2-yl)acetate in 55% yield.

Graphical Abstract
Introduction
Indolobenzazepines are fused heterocyclic scaffolds with versatile medicinal properties, including anti-Alzheimer, anti-inflammatory, anticancer, antidiabetic, and antileishmanial activity [1]. Since the first synthesis of paullones (scaffold A in Figure 1) in 1992 [2], and disclosure of their Cdk inhibiting potential, several other analogues were designed and prepared [3], with the hope of developing more efficient anticancer drugs with either improved Cdk targeting or with a different mechanism of action [4,5]. The isomers B and D (Figure 1) are synthetic derivatives of paullones, in which either the lactam unit is shifted (B) or both the lactam unit is shifted and the indole ring is flipped (D). Isomer D has been shown to be a potent tubulin-polymerase inhibitor, while B was only mildly cytotoxic towards cancer cells at a concentration of 1 µM [5,6]. Backbone C, despite being very similar to the other three scaffolds, remained a synthetic challenge and practical synthesis is still missing in the literature. Its accessibility may enrich the arsenal of available tools for enzyme inhibitor design by increasing the number of hydrogen bonding donor and acceptor groups at the same side of the backbone, which may result in a tight binding with enzyme active sites and/or improved selectivity [7].
![[1860-5397-18-15-1]](/bjoc/content/figures/1860-5397-18-15-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Paullone related indolobenzazepinone isomers. 7,12-Dihydroindolo[3,2-d][1]benzazepin-6(5H)-one or paullone (A), 5,12-dihydroindolo[3,2-d][2]benzazepin-7(6H)-one (B), 7,8-dihydroindolo[2,3-d][1]benzazepin-6(5H)-one (C), and 5,8-dihydroindolo[2,3-d][2]benzazepin-7(6H)-one (D).
Figure 1: Paullone related indolobenzazepinone isomers. 7,12-Dihydroindolo[3,2-d][1]benzazepin-6(5H)-one or p...
One of the main drawbacks of paullones is their poor aqueous solubility. Therefore, in an attempt to overcome this shortcoming, the paullone backbone A was decorated with functional groups and coordinated to metal ions. Ruthenium(II), osmium(II), and copper(II) complexes prepared revealed an enhanced aqueous solubility and bioavailability indeed, along with very high cytotoxicity [8-21]. Moreover, the metal-free ligands and copper(II) complexes derived from backbone D revealed cytotoxicity in the nanomolar concentration range and selectivity for cancer cells over normal ones [22]. The synthesis of core structures A, B, and D is well-documented in the literature [2,6]. Herein we describe the first synthesis of 7,8-dihydroindolo[2,3-d][1]benzazepin-6(5H)-one (C) (Figure 1).
Results and Discussion
From a retrosynthetic point of view, we followed three main pathways, in order to accomplish the synthesis of scaffold C. The first retrosynthetic route (a) started with an alkyl halide precursor, which was expected to afford scaffold C after ring-closure reaction at position 2 of the indole ring [23]. The N-protected 3-(2-nitrophenyl)indole was considered an important intermediate in this synthetic route (Scheme 1). The second retrosynthetic pathway (b) involved cyclization at position 3 of the indole ring, with a halo-aryl precursor [24]. In this case N-protected indole-2-acetic acid was regarded as the key intermediate.
![[1860-5397-18-15-i1]](/bjoc/content/inline/1860-5397-18-15-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Investigated retrosynthetic pathways to scaffold C.
Scheme 1: Investigated retrosynthetic pathways to scaffold C.
The third pathway (c) was centered around a ring-closure reaction via lactam-bond formation from a precursor that contains a carboxylic ester in position 2 and an o-aniline moiety in position 3 of the indole ring by Fischer indole synthesis from methyl 4-(2-nitrophenyl)-3-oxobutanoate (Scheme 1).
To construct scaffold C by route (a) we first tried to synthesize 3-(2-nitrophenyl)indole by Heck reaction of indole and o-iodo-nitrobenzene [25]. However, in this case we obtained an isomeric mixture of 3- and 2-(2-nitrophenyl)indole in a 3:1 molar ratio, quite difficult to separate by column chromatography. Protection of the indole nitrogen by an ethoxymethyl group (Scheme 2) and the use of protected indole 4 in the subsequent Heck reaction resulted in a significantly lower conversion of 22% vs 64% for the unprotected indole. However, the protection of the indole nitrogen atom led exclusively to the desired isomer 5 with the nitrophenyl group in position 3, most likely due to the steric effect. Then, the nitro group in 5 was reduced by hydrogenation under 4 bar using Pd/C as catalyst to yield the desired amine 6 in 78% yield. Subsequently, the desired chloroacetyl derivative 7 was generated in 79% yield by treatment of amine 6 with chloroacetyl chloride. Next, the aminoacetyl group in 7 was benzyl-protected to give 8 in 88% yield. Halogen exchange reaction using excess sodium iodide in acetone gave the desired iodo-alkyl derivative 9 in 79% yield. Lastly, our attempt to accomplish organotin-mediated cyclization [23] of 9 led to a complex mixture of compounds, in which we have not identified the desired product. Therefore, we did not further pursue this route. Attempts to perform cyclization via intramolecular alkyl halide Heck reaction [26] also failed. This is most likely a result of the relatively high lability of the CH2 protons in the alkyl halide moiety of the starting molecule, which might lead to enolization instead of oxidative addition of palladium under strongly basic conditions needed in this type of reaction. Thus, we came to the conclusion that pathway (a) is not suitable for the synthesis of scaffold C. Nevertheless, a detailed description of the synthesis and spectral data for the intermediate species are given in Supporting Information File 1.
![[1860-5397-18-15-i2]](/bjoc/content/inline/1860-5397-18-15-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Attempted synthesis of scaffold C by route (a).
Scheme 2: Attempted synthesis of scaffold C by route (a).
Pathway (b) started with the synthesis of indole-2-acetic acid, which in turn was prepared in five steps as shown in Scheme 3. First, reduction of commercially available indole-2-carboxylate with lithium aluminum hydride in dry tetrahydrofuran gave (1H-indole-2-yl)methanol (10) in 89% yield. The obtained alcohol was exposed to benzoyl chloride and triethylamine to furnish benzoate 11, which was further reacted with potassium cyanide to yield nitrile 12 [27]. Saponification of the nitrile was performed by exploiting the Pinner reaction with saturated HCl gas in methanol. This led to methyl indol-2-ylacetate (13) in 83% yield [28]. The hydrolysis of ester 13 was performed in the presence of lithium hydroxide monohydrate [29] to give the desired indole-2-acetic acid (14) in 95% yield. The peptide coupling reaction [30] of indole-2-acetic acid (14) and 2-iodoaniline afforded 15 in 23% yield (Scheme 3). Subsequent protection of both the indole and the amide nitrogen with tert-butyloxycarbonyl groups in one step produced 16 in 86% yield. However, attempts to perform the ring-closure reaction of the Boc-protected amide 16 by an intramolecular Heck reaction failed and led to degradation of the starting material. Most likely, the electron-withdrawing tert-butyloxycarbonyl protecting group hampered this transformation [24]. Therefore, the same synthetic way was repeated with ethoxymethyl ether as the protecting group to give 17.
![[1860-5397-18-15-i3]](/bjoc/content/inline/1860-5397-18-15-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Attempted synthesis of C by route (b).
Scheme 3: Attempted synthesis of C by route (b).
The ESI mass spectrum provided evidence that cyclization occurred with formation of 18. However, protection with chloromethyl ethyl ether was achieved only in 18% yield. This is most likely due to the strongly basic conditions (NaH) needed for the attachment of the ethoxymethyl protecting group, which might lead to deprotonation at the CH2 group (C7) followed by the formation of undesired side products. Being disappointed by the inefficiency of this route with at least two low-yielding transformations we did not further pursue this pathway (Scheme 3).
As an alternative approach to obtain indole-2-acetic acid with a protecting group at the indole nitrogen the application of conditions documented for Arndt–Eistert synthesis [31] was prompted from benzyl-protected indole-2-carboxylic acid 20 (Scheme 4) [32]. While the acid chloride of unsubstituted indole-2-carboxylic acid was reported to be highly unstable [33], we did not experience that issue with the benzyl-protected compound. Quantitative acid chloride formation using oxalyl chloride in dry dichloromethane was observed by TLC. However, the acid chloride reacted very sluggishly with (CH3)3SiCHN2, which is a safe alternative to the highly explosive diazomethane [34]. Another attempt to synthesize the open-chain precursor from indole and 2-bromo-N-(2-bromophenyl)acetamide by regioselective palladium-catalyzed norbornene-mediated C–H activation [35] failed and gave 1,4-bis(2-bromophenyl)piperazine-2,5-dione (22, Scheme 5) as the sole product in 61% yield, which to our knowledge is not documented in the literature yet.
![[1860-5397-18-15-i4]](/bjoc/content/inline/1860-5397-18-15-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Attempted synthesis of N-benzylated indole-2-acetic acid.
Scheme 4: Attempted synthesis of N-benzylated indole-2-acetic acid.
![[1860-5397-18-15-i5]](/bjoc/content/inline/1860-5397-18-15-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Attempt to obtain open-chain precursor N-(2-bromophenyl)-2-(1H-indol-2-yl)acetamide.
Scheme 5: Attempt to obtain open-chain precursor N-(2-bromophenyl)-2-(1H-indol-2-yl)acetamide.
Synthetic pathway (c) started from methyl 4-(2-nitrophenyl)-3-oxobutanoate prepared in two steps by following known protocols [36]. Fischer indole reaction of this precursor with 1-benzyl-1-phenylhydrazine in acetic acid gave 1a in 55% yield (Scheme 6). The protecting group on the hydrazine moiety is necessary in this case, otherwise amide bond formation would occur after the initial Schiff base reaction to give undesired 5-(2-nitrobenzyl)-2-phenyl-1,2-dihydro-3H-pyrazol-3-one [37]. Note that hypothetically two Fischer indole products could arise from this reaction, namely 1a and methyl 1-benzyl-2-(2-nitrobenzyl)-1H-indole-3-carboxylate, where the ester and nitroaryl moieties are interchanged. However, to our delight only the formation of the desired species 1a occurred, as evidenced by two-dimensional NMR measurements (for atom labeling scheme used for assignment of resonances see Figure S1 in Supporting Information File 1) and confirmed by SC-XRD (Figure 2), being in agreement with previous observations [38]. Reduction of the nitro group by palladium-catalyzed hydrogenation in dry methanol gave 2a in 92% yield. Interestingly compound 2a undergoes ring-closure reaction spontaneously at room temperature to give trace amounts of 3a (59 mg, 2%) after column chromatography, when a 3.1 g batch of 1a was reduced. It is likely that 2a can be cyclized by base catalysis, or by using common peptide coupling reagents (e.g., EDCI, HATU) upon saponification of the ester group. However, we opted for a trimethylaluminum-mediated amidation reaction [4,39] to give rise to 8-benzyl-7-hydroindolo[2,3-d][1]benzazepin-6(5H)-one (3a). This procedure delivered analytically pure 3a in 80% yield as also confirmed by SC-XRD (Figure 3). The final step to structure C required removal of the benzyl group. Debenzylation of amines is commonly performed by palladium-catalyzed hydrogenation [40]. However, this method is not viable for N-benzylated indoles, where reduction in liquid ammonia with sodium is usually used as a standard protocol instead, even though successful reports are rare [41]. Other methods involve strongly basic and oxidative conditions [42], which are not tolerated by the CH2 group in the lactam ring of 3a. Gratifyingly, we obtained scaffold C in 71% yield by refluxing 3a with aluminum chloride in benzene (Scheme 6) [43]. For comparison, debenzylation of paullone with sodium in liquid ammonia gave A in 40% yield [23].
![[1860-5397-18-15-i6]](/bjoc/content/inline/1860-5397-18-15-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of scaffold C and analogues by route (c).
Scheme 6: Synthesis of scaffold C and analogues by route (c).
![[1860-5397-18-15-2]](/bjoc/content/figures/1860-5397-18-15-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP view of 1a with thermal ellipsoids drawn at the 50% probability level.
Figure 2: ORTEP view of 1a with thermal ellipsoids drawn at the 50% probability level.
![[1860-5397-18-15-3]](/bjoc/content/figures/1860-5397-18-15-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP view of 3a with thermal ellipsoids drawn at the 50% probability level.
Figure 3: ORTEP view of 3a with thermal ellipsoids drawn at the 50% probability level.
In addition, the generality of synthetic pathway (c) (Scheme 1) was confirmed by the successful preparation of two closely related analogues, namely 3b, bearing a bromo substituent at position 11 of the backbone C (Scheme 6), and 3c, with an 8-membered azocinone ring instead of the 7-membered one. The synthesis of 3b started with a Fischer indole synthesis from methyl 4-(2-nitrophenyl)-3-oxobutanoate [36] and 1-benzyl-1-(4-bromophenyl)hydrazine in the presence of sodium acetate and sulfuric acid in glacial acetic acid affording 1b in 55% yield. It is of note that the treatment of 1b with hydrogen in the presence of Pd/C as catalyst led not only to reduction of the nitro group but also to cyclization and the reductive elimination of bromine to afford 3a. The synthesis of 3b could be realized in 58% yield by using iron powder under acidic conditions. Reaction of methyl 5-(2-nitrophenyl)-4-oxopentanoate [44] and 1-benzyl-1-phenylhydrazine [36] hydrochloride in glacial acetic acid at 100 °C gave 1c in 53% yield. Compound 2c was obtained by palladium-catalyzed reduction of 1c with hydrogen in an excellent yield and its structure was confirmed by SC-XRD (Figure S4 in Supporting Information File 1). The following ring-closure reaction of 2c with trimethylaluminum delivered 3c in 53% yield (Scheme 6).
In an alternative approach we tried to synthesize non-benzylated species 1a from methyl indol-2-ylacetate. Iodination at position 3 of the indole backbone in the presence of N-iodosuccinimide [45] followed by Ullmann cross-coupling with o-bromo-nitrobenzene [46] was expected to give non-benzylated 1a. However, iodination of methyl indol-2-ylacetate led to polymerization reactions involving the CH2 protons. In order to investigate the general viability of this synthetic way, we performed iodination at position 3 of ethyl indole-2-carboxylate, which does not contain labile CH2 protons. This afforded the iodinated compound 23 in 80% yield. However, Ullmann cross-coupling with o-bromo-nitrobenzene gave only trace amounts of the desired product, which we could not separate by chromatographic methods from the major homo-coupling species 24 of this reaction (Scheme 7).
![[1860-5397-18-15-i7]](/bjoc/content/inline/1860-5397-18-15-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Attempted Ullmann cross-coupling of 23 with o-bromo-nitrobenzene.
Scheme 7: Attempted Ullmann cross-coupling of 23 with o-bromo-nitrobenzene.
Conclusion
In summary, a concise, simple, clean and efficient synthesis of paullone isomer C was achieved in 4 steps with a crucial transformation, the Fischer indole ring-closure reaction, at the early stage of the synthesis (Scheme 6). The synthetic utility of the elaborated route was validated by gram-scale synthesis with only one step requiring chromatographic purification and inexpensive starting materials. In addition, two failed routes (a) and (b) are described as well, providing useful information for organic chemists and enlarging the scope of this work (Scheme 1). We believe that accessibility to isomer C and a number of closely related derivatives with modified electronic and steric properties, e.g., bromo-substituted species, backbone with 8-membered azocinone ring, will trigger the design and synthesis of new exciting anticancer drug candidates, including metal-based, by targeting particular enzymes. Preliminary results indicate that scaffold C shows good affinity to proto-oncogene tyrosine-protein kinase Src, a well-known anticancer target [47], with IC50 = 0.26 µM and is a suitable candidate for further structural optimization.
Supporting Information
CCDC 2072772−2072777 and 2103781 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
| Supporting Information File 1: Experimental procedures, characterization data and 1H and 13C NMR spectra of compounds, NMR numbering schemes and X-ray crystallography data. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Kadagathur, M.; Patra, S.; Sigalapalli, D. K.; Shankaraiah, N.; Tangellamudi, N. D. Org. Biomol. Chem. 2021, 19, 738–764. doi:10.1039/d0ob02181d
Return to citation in text: [1] -
Kunick, C. Arch. Pharm. (Weinheim, Ger.) 1992, 325, 297–299. doi:10.1002/ardp.19923250509
Return to citation in text: [1] [2] -
Singh, A. K.; Raj, V.; Saha, S. Eur. J. Med. Chem. 2017, 142, 244–265. doi:10.1016/j.ejmech.2017.07.042
Return to citation in text: [1] -
Joucla, L.; Popowycz, F.; Lozach, O.; Meijer, L.; Joseph, B. Helv. Chim. Acta 2007, 90, 753–763. doi:10.1002/hlca.200790076
Return to citation in text: [1] [2] -
Putey, A.; Popowycz, F.; Do, Q.-T.; Bernard, P.; Talapatra, S. K.; Kozielski, F.; Galmarini, C. M.; Joseph, B. J. Med. Chem. 2009, 52, 5916–5925. doi:10.1021/jm900476c
Return to citation in text: [1] [2] -
Putey, A.; Joucla, L.; Picot, L.; Besson, T.; Joseph, B. Tetrahedron 2007, 63, 867–879. doi:10.1016/j.tet.2006.11.042
Return to citation in text: [1] [2] -
Mulcahy, S. P.; Meggers, E. Organometallics as Structural Scaffolds for Enzyme Inhibitor Design. In Medicinal Organometallic Chemistry; Jaouen, G.; Metzler-Nolte, N., Eds.; Topics in Organometallic Chemistry, Vol. 32; Springer: Berlin, Heidelberg, 2010; pp 141–153. doi:10.1007/978-3-642-13185-1_6
Return to citation in text: [1] -
Primik, M. F.; Filak, L. K.; Arion, V. B. Metal-Based Indolobenzazepines and Indoloquinolines: From Moderate CDK Inhibitors to Potential Antitumor Drugs. In Advances in Organometallic Chemistry and Catalysis; Pombeiro, A. J. L., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp 605–617. doi:10.1002/9781118742952.ch45
Return to citation in text: [1] -
Filak, L. K.; Kalinowski, D. S.; Bauer, T. J.; Richardson, D. R.; Arion, V. B. Inorg. Chem. 2014, 53, 6934–6943. doi:10.1021/ic500825j
Return to citation in text: [1] -
Dobrov, A.; Göschl, S.; Jakupec, M. A.; Popović-Bijelić, A.; Gräslund, A.; Rapta, P.; Arion, V. B. Chem. Commun. 2013, 49, 10007–10009. doi:10.1039/c3cc45743e
Return to citation in text: [1] -
Primik, M. F.; Göschl, S.; Meier, S. M.; Eberherr, N.; Jakupec, M. A.; Enyedy, É. A.; Novitchi, G.; Arion, V. B. Inorg. Chem. 2013, 52, 10137–10146. doi:10.1021/ic401573d
Return to citation in text: [1] -
Filak, L. K.; Göschl, S.; Heffeter, P.; Ghannadzadeh Samper, K.; Egger, A. E.; Jakupec, M. A.; Keppler, B. K.; Berger, W.; Arion, V. B. Organometallics 2013, 32, 903–914. doi:10.1021/om3012272
Return to citation in text: [1] -
Arion, V. B.; Dobrov, A.; Göschl, S.; Jakupec, M. A.; Keppler, B. K.; Rapta, P. Chem. Commun. 2012, 48, 8559–8561. doi:10.1039/c2cc33786j
Return to citation in text: [1] -
Filak, L. K.; Göschl, S.; Hackl, S.; Jakupec, M. A.; Arion, V. B. Inorg. Chim. Acta 2012, 393, 252–260. doi:10.1016/j.ica.2012.06.004
Return to citation in text: [1] -
Mühlgassner, G.; Bartel, C.; Schmid, W. F.; Jakupec, M. A.; Arion, V. B.; Keppler, B. K. J. Inorg. Biochem. 2012, 116, 180–187. doi:10.1016/j.jinorgbio.2012.06.003
Return to citation in text: [1] -
Primik, M. F.; Göschl, S.; Jakupec, M. A.; Roller, A.; Keppler, B. K.; Arion, V. B. Inorg. Chem. 2010, 49, 11084–11095. doi:10.1021/ic101633z
Return to citation in text: [1] -
Filak, L. K.; Mühlgassner, G.; Jakupec, M. A.; Heffeter, P.; Berger, W.; Arion, V. B.; Keppler, B. K. J. Biol. Inorg. Chem. 2010, 15, 903–918. doi:10.1007/s00775-010-0653-y
Return to citation in text: [1] -
Primik, M. F.; Mühlgassner, G.; Jakupec, M. A.; Zava, O.; Dyson, P. J.; Arion, V. B.; Keppler, B. K. Inorg. Chem. 2010, 49, 302–311. doi:10.1021/ic902042a
Return to citation in text: [1] -
Schmid, W. F.; John, R. O.; Arion, V. B.; Jakupec, M. A.; Keppler, B. K. Organometallics 2007, 26, 6643–6652. doi:10.1021/om700813c
Return to citation in text: [1] -
Schmid, W. F.; John, R. O.; Mühlgassner, G.; Heffeter, P.; Jakupec, M. A.; Galanski, M.; Berger, W.; Arion, V. B.; Keppler, B. K. J. Med. Chem. 2007, 50, 6343–6355. doi:10.1021/jm701042w
Return to citation in text: [1] -
Schmid, W. F.; Zorbas-Seifried, S.; John, R. O.; Arion, V. B.; Jakupec, M. A.; Roller, A.; Galanski, M.; Chiorescu, I.; Zorbas, H.; Keppler, B. K. Inorg. Chem. 2007, 46, 3645–3656. doi:10.1021/ic070098j
Return to citation in text: [1] -
Bacher, F.; Wittmann, C.; Nové, M.; Spengler, G.; Marć, M. A.; Enyedy, E. A.; Darvasiová, D.; Rapta, P.; Reiner, T.; Arion, V. B. Dalton Trans. 2019, 48, 10464–10478. doi:10.1039/c9dt01238a
Return to citation in text: [1] -
Bremner, J. B.; Sengpracha, W. Tetrahedron 2005, 61, 5489–5498. doi:10.1016/j.tet.2005.03.133
Return to citation in text: [1] [2] [3] -
Joucla, L.; Putey, A.; Joseph, B. Tetrahedron Lett. 2005, 46, 8177–8179. doi:10.1016/j.tetlet.2005.09.122
Return to citation in text: [1] [2] -
Agarwal, P. K.; Sawant, D.; Sharma, S.; Kundu, B. Eur. J. Org. Chem. 2009, 292–303. doi:10.1002/ejoc.200800929
Return to citation in text: [1] -
Firmansjah, L.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 11340–11341. doi:10.1021/ja075245r
Return to citation in text: [1] -
Mendoza-Figueroa, H. L.; Serrano-Alva, M. T.; Aparicio-Ozores, G.; Martínez-Gudiño, G.; Suárez-Castillo, O. R.; Pérez-Rojas, N. A.; Morales-Ríos, M. S. Med. Chem. Res. 2018, 27, 1624–1633. doi:10.1007/s00044-018-2177-x
Return to citation in text: [1] -
Adams, R.; Thal, A. F. Org. Synth. 1922, 2, 27. doi:10.15227/orgsyn.002.0027
Return to citation in text: [1] -
Sharpe, R. J.; Johnson, J. S. J. Org. Chem. 2015, 80, 9740–9766. doi:10.1021/acs.joc.5b01844
Return to citation in text: [1] -
Lepri, S.; Ceccarelli, M.; Milani, N.; Tortorella, S.; Cucco, A.; Valeri, A.; Goracci, L.; Brink, A.; Cruciani, G. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E3178–E3187. doi:10.1073/pnas.1618881114
Return to citation in text: [1] -
Podlech, J. J. Prakt. Chem./Chem.-Ztg. 1998, 340, 679–682. doi:10.1002/prac.19983400714
Return to citation in text: [1] -
Parker, A. N.; Martin, M. C.; Shenje, R.; France, S. Org. Lett. 2019, 21, 7268–7273. doi:10.1021/acs.orglett.9b02498
Return to citation in text: [1] -
Matell, M. Ark. Kemi 1956, 10, 179–181.
Return to citation in text: [1] -
Aoyama, T.; Shioiri, T. Chem. Pharm. Bull. 1981, 29, 3249–3255. doi:10.1248/cpb.29.3249
Return to citation in text: [1] -
Jiao, L.; Bach, T. J. Am. Chem. Soc. 2011, 133, 12990–12993. doi:10.1021/ja2055066
Return to citation in text: [1] -
Peng, L.; Zhang, T.; Li, Y.; Li, Y. Synth. Commun. 2002, 32, 785–791. doi:10.1081/scc-120002520
Return to citation in text: [1] [2] [3] -
Liu, C.; Zhu, X.; Zhang, P.; Yang, H.; Zhu, C.; Fu, H. iScience 2018, 10, 11–22. doi:10.1016/j.isci.2018.11.018
Return to citation in text: [1] -
Teotino, U. M. Gazz. Chim. Ital. 1959, 89, 1853–1862.
Return to citation in text: [1] -
Drissi-Amraoui, S. Synlett 2015, 26, 1424–1425. doi:10.1055/s-0034-1380539
Return to citation in text: [1] -
Zhou, Z.-L.; Keana, J. F. W. J. Org. Chem. 1999, 64, 3763–3766. doi:10.1021/jo9824697
Return to citation in text: [1] -
Srinivasa Rao, T.; Pandey, P. S. Synth. Commun. 2004, 34, 3121–3127. doi:10.1081/scc-200028570
Return to citation in text: [1] -
Haddach, A. A.; Kelleman, A.; Deaton-Rewolinski, M. V. Tetrahedron Lett. 2002, 43, 399–402. doi:10.1016/s0040-4039(01)02192-x
Return to citation in text: [1] -
Watanabe, T.; Kobayashi, A.; Nishiura, M.; Takahashi, H.; Usui, T.; Kamiyama, I.; Mochizuki, N.; Noritake, K.; Yokoyama, Y.; Murakami, Y. Chem. Pharm. Bull. 1991, 39, 1152–1156. doi:10.1248/cpb.39.1152
Return to citation in text: [1] -
Bunce, R. A.; Nammalwar, B. J. Heterocycl. Chem. 2009, 46, 172–177. doi:10.1002/jhet.22
Return to citation in text: [1] -
Jana, G. K.; Sinha, S. Tetrahedron 2012, 68, 7155–7165. doi:10.1016/j.tet.2012.06.027
Return to citation in text: [1] -
Banwell, M. G.; Lupton, D. W.; Ma, X.; Renner, J.; Sydnes, M. O. Org. Lett. 2004, 6, 2741–2744. doi:10.1021/ol0490375
Return to citation in text: [1] -
Roskoski, R., Jr. Pharmacol. Res. 2015, 94, 9–25. doi:10.1016/j.phrs.2015.01.003
Return to citation in text: [1]
| 4. | Joucla, L.; Popowycz, F.; Lozach, O.; Meijer, L.; Joseph, B. Helv. Chim. Acta 2007, 90, 753–763. doi:10.1002/hlca.200790076 |
| 39. | Drissi-Amraoui, S. Synlett 2015, 26, 1424–1425. doi:10.1055/s-0034-1380539 |
| 40. | Zhou, Z.-L.; Keana, J. F. W. J. Org. Chem. 1999, 64, 3763–3766. doi:10.1021/jo9824697 |
| 41. | Srinivasa Rao, T.; Pandey, P. S. Synth. Commun. 2004, 34, 3121–3127. doi:10.1081/scc-200028570 |
| 1. | Kadagathur, M.; Patra, S.; Sigalapalli, D. K.; Shankaraiah, N.; Tangellamudi, N. D. Org. Biomol. Chem. 2021, 19, 738–764. doi:10.1039/d0ob02181d |
| 5. | Putey, A.; Popowycz, F.; Do, Q.-T.; Bernard, P.; Talapatra, S. K.; Kozielski, F.; Galmarini, C. M.; Joseph, B. J. Med. Chem. 2009, 52, 5916–5925. doi:10.1021/jm900476c |
| 6. | Putey, A.; Joucla, L.; Picot, L.; Besson, T.; Joseph, B. Tetrahedron 2007, 63, 867–879. doi:10.1016/j.tet.2006.11.042 |
| 27. | Mendoza-Figueroa, H. L.; Serrano-Alva, M. T.; Aparicio-Ozores, G.; Martínez-Gudiño, G.; Suárez-Castillo, O. R.; Pérez-Rojas, N. A.; Morales-Ríos, M. S. Med. Chem. Res. 2018, 27, 1624–1633. doi:10.1007/s00044-018-2177-x |
| 45. | Jana, G. K.; Sinha, S. Tetrahedron 2012, 68, 7155–7165. doi:10.1016/j.tet.2012.06.027 |
| 4. | Joucla, L.; Popowycz, F.; Lozach, O.; Meijer, L.; Joseph, B. Helv. Chim. Acta 2007, 90, 753–763. doi:10.1002/hlca.200790076 |
| 5. | Putey, A.; Popowycz, F.; Do, Q.-T.; Bernard, P.; Talapatra, S. K.; Kozielski, F.; Galmarini, C. M.; Joseph, B. J. Med. Chem. 2009, 52, 5916–5925. doi:10.1021/jm900476c |
| 46. | Banwell, M. G.; Lupton, D. W.; Ma, X.; Renner, J.; Sydnes, M. O. Org. Lett. 2004, 6, 2741–2744. doi:10.1021/ol0490375 |
| 3. | Singh, A. K.; Raj, V.; Saha, S. Eur. J. Med. Chem. 2017, 142, 244–265. doi:10.1016/j.ejmech.2017.07.042 |
| 23. | Bremner, J. B.; Sengpracha, W. Tetrahedron 2005, 61, 5489–5498. doi:10.1016/j.tet.2005.03.133 |
| 44. | Bunce, R. A.; Nammalwar, B. J. Heterocycl. Chem. 2009, 46, 172–177. doi:10.1002/jhet.22 |
| 2. | Kunick, C. Arch. Pharm. (Weinheim, Ger.) 1992, 325, 297–299. doi:10.1002/ardp.19923250509 |
| 26. | Firmansjah, L.; Fu, G. C. J. Am. Chem. Soc. 2007, 129, 11340–11341. doi:10.1021/ja075245r |
| 36. | Peng, L.; Zhang, T.; Li, Y.; Li, Y. Synth. Commun. 2002, 32, 785–791. doi:10.1081/scc-120002520 |
| 2. | Kunick, C. Arch. Pharm. (Weinheim, Ger.) 1992, 325, 297–299. doi:10.1002/ardp.19923250509 |
| 6. | Putey, A.; Joucla, L.; Picot, L.; Besson, T.; Joseph, B. Tetrahedron 2007, 63, 867–879. doi:10.1016/j.tet.2006.11.042 |
| 24. | Joucla, L.; Putey, A.; Joseph, B. Tetrahedron Lett. 2005, 46, 8177–8179. doi:10.1016/j.tetlet.2005.09.122 |
| 23. | Bremner, J. B.; Sengpracha, W. Tetrahedron 2005, 61, 5489–5498. doi:10.1016/j.tet.2005.03.133 |
| 22. | Bacher, F.; Wittmann, C.; Nové, M.; Spengler, G.; Marć, M. A.; Enyedy, E. A.; Darvasiová, D.; Rapta, P.; Reiner, T.; Arion, V. B. Dalton Trans. 2019, 48, 10464–10478. doi:10.1039/c9dt01238a |
| 25. | Agarwal, P. K.; Sawant, D.; Sharma, S.; Kundu, B. Eur. J. Org. Chem. 2009, 292–303. doi:10.1002/ejoc.200800929 |
| 36. | Peng, L.; Zhang, T.; Li, Y.; Li, Y. Synth. Commun. 2002, 32, 785–791. doi:10.1081/scc-120002520 |
| 8. | Primik, M. F.; Filak, L. K.; Arion, V. B. Metal-Based Indolobenzazepines and Indoloquinolines: From Moderate CDK Inhibitors to Potential Antitumor Drugs. In Advances in Organometallic Chemistry and Catalysis; Pombeiro, A. J. L., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp 605–617. doi:10.1002/9781118742952.ch45 |
| 9. | Filak, L. K.; Kalinowski, D. S.; Bauer, T. J.; Richardson, D. R.; Arion, V. B. Inorg. Chem. 2014, 53, 6934–6943. doi:10.1021/ic500825j |
| 10. | Dobrov, A.; Göschl, S.; Jakupec, M. A.; Popović-Bijelić, A.; Gräslund, A.; Rapta, P.; Arion, V. B. Chem. Commun. 2013, 49, 10007–10009. doi:10.1039/c3cc45743e |
| 11. | Primik, M. F.; Göschl, S.; Meier, S. M.; Eberherr, N.; Jakupec, M. A.; Enyedy, É. A.; Novitchi, G.; Arion, V. B. Inorg. Chem. 2013, 52, 10137–10146. doi:10.1021/ic401573d |
| 12. | Filak, L. K.; Göschl, S.; Heffeter, P.; Ghannadzadeh Samper, K.; Egger, A. E.; Jakupec, M. A.; Keppler, B. K.; Berger, W.; Arion, V. B. Organometallics 2013, 32, 903–914. doi:10.1021/om3012272 |
| 13. | Arion, V. B.; Dobrov, A.; Göschl, S.; Jakupec, M. A.; Keppler, B. K.; Rapta, P. Chem. Commun. 2012, 48, 8559–8561. doi:10.1039/c2cc33786j |
| 14. | Filak, L. K.; Göschl, S.; Hackl, S.; Jakupec, M. A.; Arion, V. B. Inorg. Chim. Acta 2012, 393, 252–260. doi:10.1016/j.ica.2012.06.004 |
| 15. | Mühlgassner, G.; Bartel, C.; Schmid, W. F.; Jakupec, M. A.; Arion, V. B.; Keppler, B. K. J. Inorg. Biochem. 2012, 116, 180–187. doi:10.1016/j.jinorgbio.2012.06.003 |
| 16. | Primik, M. F.; Göschl, S.; Jakupec, M. A.; Roller, A.; Keppler, B. K.; Arion, V. B. Inorg. Chem. 2010, 49, 11084–11095. doi:10.1021/ic101633z |
| 17. | Filak, L. K.; Mühlgassner, G.; Jakupec, M. A.; Heffeter, P.; Berger, W.; Arion, V. B.; Keppler, B. K. J. Biol. Inorg. Chem. 2010, 15, 903–918. doi:10.1007/s00775-010-0653-y |
| 18. | Primik, M. F.; Mühlgassner, G.; Jakupec, M. A.; Zava, O.; Dyson, P. J.; Arion, V. B.; Keppler, B. K. Inorg. Chem. 2010, 49, 302–311. doi:10.1021/ic902042a |
| 19. | Schmid, W. F.; John, R. O.; Arion, V. B.; Jakupec, M. A.; Keppler, B. K. Organometallics 2007, 26, 6643–6652. doi:10.1021/om700813c |
| 20. | Schmid, W. F.; John, R. O.; Mühlgassner, G.; Heffeter, P.; Jakupec, M. A.; Galanski, M.; Berger, W.; Arion, V. B.; Keppler, B. K. J. Med. Chem. 2007, 50, 6343–6355. doi:10.1021/jm701042w |
| 21. | Schmid, W. F.; Zorbas-Seifried, S.; John, R. O.; Arion, V. B.; Jakupec, M. A.; Roller, A.; Galanski, M.; Chiorescu, I.; Zorbas, H.; Keppler, B. K. Inorg. Chem. 2007, 46, 3645–3656. doi:10.1021/ic070098j |
| 42. | Haddach, A. A.; Kelleman, A.; Deaton-Rewolinski, M. V. Tetrahedron Lett. 2002, 43, 399–402. doi:10.1016/s0040-4039(01)02192-x |
| 7. | Mulcahy, S. P.; Meggers, E. Organometallics as Structural Scaffolds for Enzyme Inhibitor Design. In Medicinal Organometallic Chemistry; Jaouen, G.; Metzler-Nolte, N., Eds.; Topics in Organometallic Chemistry, Vol. 32; Springer: Berlin, Heidelberg, 2010; pp 141–153. doi:10.1007/978-3-642-13185-1_6 |
| 23. | Bremner, J. B.; Sengpracha, W. Tetrahedron 2005, 61, 5489–5498. doi:10.1016/j.tet.2005.03.133 |
| 43. | Watanabe, T.; Kobayashi, A.; Nishiura, M.; Takahashi, H.; Usui, T.; Kamiyama, I.; Mochizuki, N.; Noritake, K.; Yokoyama, Y.; Murakami, Y. Chem. Pharm. Bull. 1991, 39, 1152–1156. doi:10.1248/cpb.39.1152 |
| 24. | Joucla, L.; Putey, A.; Joseph, B. Tetrahedron Lett. 2005, 46, 8177–8179. doi:10.1016/j.tetlet.2005.09.122 |
| 29. | Sharpe, R. J.; Johnson, J. S. J. Org. Chem. 2015, 80, 9740–9766. doi:10.1021/acs.joc.5b01844 |
| 47. | Roskoski, R., Jr. Pharmacol. Res. 2015, 94, 9–25. doi:10.1016/j.phrs.2015.01.003 |
| 30. | Lepri, S.; Ceccarelli, M.; Milani, N.; Tortorella, S.; Cucco, A.; Valeri, A.; Goracci, L.; Brink, A.; Cruciani, G. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E3178–E3187. doi:10.1073/pnas.1618881114 |
| 37. | Liu, C.; Zhu, X.; Zhang, P.; Yang, H.; Zhu, C.; Fu, H. iScience 2018, 10, 11–22. doi:10.1016/j.isci.2018.11.018 |
| 35. | Jiao, L.; Bach, T. J. Am. Chem. Soc. 2011, 133, 12990–12993. doi:10.1021/ja2055066 |
| 36. | Peng, L.; Zhang, T.; Li, Y.; Li, Y. Synth. Commun. 2002, 32, 785–791. doi:10.1081/scc-120002520 |
| 34. | Aoyama, T.; Shioiri, T. Chem. Pharm. Bull. 1981, 29, 3249–3255. doi:10.1248/cpb.29.3249 |
| 31. | Podlech, J. J. Prakt. Chem./Chem.-Ztg. 1998, 340, 679–682. doi:10.1002/prac.19983400714 |
| 32. | Parker, A. N.; Martin, M. C.; Shenje, R.; France, S. Org. Lett. 2019, 21, 7268–7273. doi:10.1021/acs.orglett.9b02498 |
© 2022 Kuznetcova et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.