Abstract
A series of P-stereogenic chiral phosphorus acids (CPAs) were synthesized to determine the requirements for efficient asymmetric organocatalysis. In order to eliminate the need for C2-symmetry in common CPAs, various scaffolds containing C1-symmetrical thiophosphorus acids were chosen. These new compounds were synthesized and evaluated in the asymmetric transfer hydrogenation of 2-phenylquinoline. Although the efficacy of the thiophosphorus acids was disappointing for this reaction, the work should be useful for developing structural design elements.

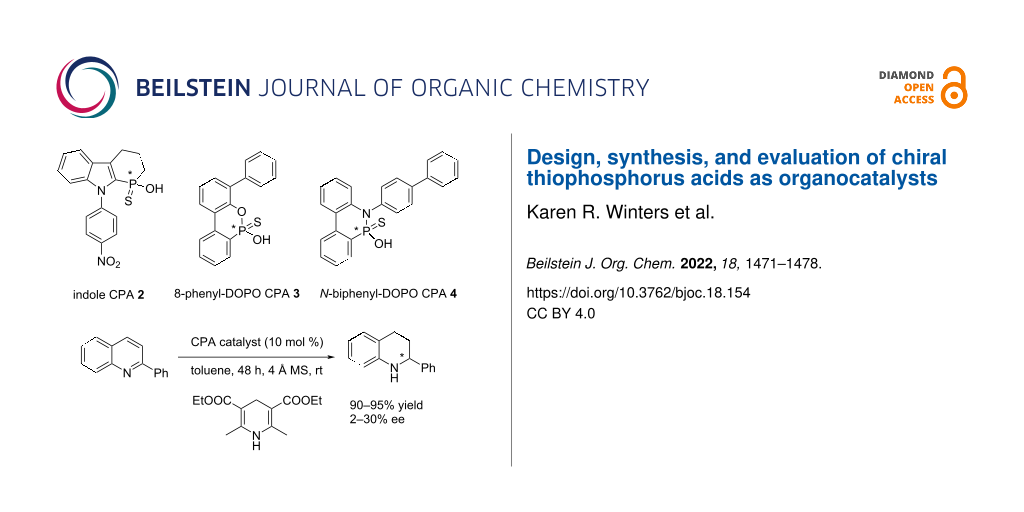
Graphical Abstract
Introduction
The importance of asymmetric organocatalysis was demonstrated by the 2021 Nobel Prize in Chemistry awarded to McMillan and List. A subclass of organocatalysts introduced independently by Akiyama and Terada in 2004 [1,2], are the C2-symmetrical chiral phosphorus acids (CPAs) initially derived from the BINOL scaffold, and later extended to other scaffolds such as VAPOL [3] and SPINOL [4,5] (Figure 1). The great success of these CPAs in asymmetric organocatalysis, is demonstrated by the publication of thousands of articles and reviews [6-17]. In all cases the C2-symmetry is required because of the prototropic tautomeric equilibrium in the hydroxyphosphoryl (P(=O)OH) moiety which renders the phosphorus atom achiral. Substituents can be introduced on the ring system by ortho-functionalization with R groups on each ring. This functionalization helps introduce steric bulk and a range of electron densities extending the C2-symmetry of the BINOL, creating a chiral pocket or environment for enantioselective transformations within the proximity of the acidic proton and phosphoryl oxygen. Additionally, the choice of phosphoric acid diesters also provides a bifunctional catalyst containing both an acidic and basic site (Figure 1).
![[1860-5397-18-154-1]](/bjoc/content/figures/1860-5397-18-154-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chiral phosphorus acids (CPAs) derived from BINOL, VAPOL, and SPINOL. R = H, Ph, 4-PhC6H4-, 4-β-naphthylphenyl, 9-anthryl, 3,5-dimesitylphenyl, 3,5-diphenylphenyl, 4-MeC6H4-, 4-CF3C6H4-, 4-t-BuC6H4-, β-naphthyl, 3,5-t-Bu2C6H3-, 2,4,6-Me3C6H2-, 2,4,6-iPr3C6H2-, Ph3Si-, etc.
Figure 1: Chiral phosphorus acids (CPAs) derived from BINOL, VAPOL, and SPINOL. R = H, Ph, 4-PhC6H4-, 4-β-nap...
Despite the proven value of the CPAs described in the literature, several disadvantages can be identified [18]. As mentioned above, C2-symmetry is required for the catalysts to provide a chiral pocket around the phosphorus. As a result, the CPAs have very high molecular weights (>> 450 g/mol) and require a wasteful duplicative functionalization of the backbone. Moreover, commercially available CPAs are extremely expensive (>> 500,000 $/mol) and immobilizing the CPAs on a solid support is not straightforward [19,20]. In order to avoid this, a significant investment in time must be made to complete the multistep-syntheses that are required [1-4]. Additionally, whereas either enantiomer of BINOL is relatively inexpensive (109 $/mol), it is not the case with SPINOL (17,000 $/mol), and VAPOL is not commercially available. Although one could synthesize these precursors as well, this multistep synthesis is time-consuming and costly. For example, the resolution of racemic SPINOL uses 2.4 equivalents of menthyl chloroformate [21] which itself costs 1,000 $/mol. Furthermore, the R group often needs to be optimized to obtain good enantioselectivities and there does not seem to be a universally successful CPA at this time. Consequently, the availability of each CPA enantiomer requires significant synthetic efforts from the diphenol precursor.
In order to address these issues, we became interested in exploring C1-symmetrical CPAs, in which the chirality resides exclusively at the phosphorus atom. For this exploratory work, thiophosphorus acids were chosen due to their appropriate acidity and intrinsic chirality. Thiophosphorus acids undergo a tautomeric equilibrium between the thiolic and the thionic forms [22] (Scheme 1). If the substituents R1 and R2 are different, the phosphorus atom is always chiral. Chiral thiophosphorus acids have been obtained by resolution with a chiral amine as early as 1958 [23-27], or from other precursors [28-30].
![[1860-5397-18-154-i1]](/bjoc/content/inline/1860-5397-18-154-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The thiolic/thionic tautomeric equilibrium in thiophosphorus acids.
Scheme 1: The thiolic/thionic tautomeric equilibrium in thiophosphorus acids.
Having selected chiral thiophosphorus acids for our model study, further design requirements were included (Figure 2) to address some issues listed above for the C2-symmetrical catalysts. First and foremost, the compounds must be inexpensive to make, which implies that their syntheses should be easily scaled. A modular synthesis is also desirable if some structure optimization is required. The resolution of the phosphorus center should be straightforward and accomplished late-stage, to avoid carrying the chirality through multiple steps and the possible erosion of enantiomeric excesses. Preferably, both enantiomers of the CPA should also be available and immobilization of the CPA on a solid support should be possible. In this paper, we report our progress towards these objectives.
![[1860-5397-18-154-2]](/bjoc/content/figures/1860-5397-18-154-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Project strategy and requirements for C1-symmetrical CPAs.
Figure 2: Project strategy and requirements for C1-symmetrical CPAs.
It should be noted that a few examples of a "hybrid strategy" in which both the backbone and the phosphorus atom are chiral have been reported by Guinchard [31] and Murai [32].
Results and Discussion
CPA Design
At the outset, we were interested in probing the geometry and influence of the substituent position in the CPAs (Figure 3). In the BINOL-derived CPA, the R-substituent and the phosphorus atom are separated by three bonds. In the indole-based CPAs 1 and 2, the distance is reduced to two bonds, whereas in CPA 3 it is three bonds, and in 4 it is just one bond. Both 3 and 4 are based on 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO) [33].
![[1860-5397-18-154-3]](/bjoc/content/figures/1860-5397-18-154-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: BINOL CPA and C1-symmetrical CPA targets 1–4.
Figure 3: BINOL CPA and C1-symmetrical CPA targets 1–4.
Synthesis
In this section, the syntheses of CPA targets are described. It should be noted that little yield optimization was accomplished since only a small amount of product was needed for the evaluation as an enantioselective catalyst. On the other hand, their successful completions attest to the inexpensive and scalable requirements we had set.
Indole scaffolds
The synthesis of racemic tryptophol CPA 1 is shown in Scheme 2. Commercially available tryptophol (5, 225 $/mol) was N-arylated into 6 via copper-catalyzed cross-coupling [34] in excellent yield. Esterification of 6 with monomethyl H-phosphonate tert-butylamine salt [35] resulted in the mixed H-phosphonate ester 7 in excellent yield.
![[1860-5397-18-154-i2]](/bjoc/content/inline/1860-5397-18-154-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tryptophol-derived thiophosphorus acid 1.
Scheme 2: Synthesis of tryptophol-derived thiophosphorus acid 1.
Cyclization using our homolytic aromatic substitution methodology [36] gave P-heterocycle 8 in modest yield. Other methods based on silver either gave a complex mixture or unreacted starting material. Phosphonate 8 was converted into the corresponding thiophosphonate 9 in moderate yield using Lawesson's reagent. Cleavage of the methyl ester was easily accomplished in quantitative yield, producing racemic tryptophol CPA 1. The resolution of compound 1 was not conducted at this point because its synthesis was deemed problematic. While relatively short (5 steps), the overall yield was only 20% due to a low-yielding key step and a problematic thionation step immediately following. Unfortunately, thionation of 8 with an alternative [37] to Lawesson's reagent did not solve the problem. This prompted our search for alternative methodologies for the synthesis of thiophosphorus acids [38], particularly using the Stec reaction [39,40]. This work also led to the synthesis of CPA 3 [38]. Alternatives to the Stec reaction to prepare chiral thiophosphorus acids have been described [41-43]. Once equipped with our new methods [38], the synthesis of indole-derived 2 was undertaken (Scheme 3).
![[1860-5397-18-154-i3]](/bjoc/content/inline/1860-5397-18-154-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of indole-derived thiophosphorus acid 2.
Scheme 3: Synthesis of indole-derived thiophosphorus acid 2.
Known 3-allylindole (10) [44] was obtained from indole uneventfully. Intermediate 11 was furnished in moderate yield via our palladium-catalyzed hydrophosphinylation [45]. The key heterocyclization of 11 into 12 was accomplished using silver-promoted homolytic aromatic substitution [46], which was superior to our own manganese methodology (43% yield) [36]. Copper-catalyzed arylation [34] of 12 with iodobenzene and 4-nitroiodobenzene gave intermediates 13a and 13b, respectively. Next, conversion of ethyl phosphinate 13 into phosphinamide 14 was accomplished uneventfully [38] with inexpensive (S)-1-phenylethylamine (15 $/mol) as the chiral element. A single diastereoisomer of phosphinamide 14b was easily obtained by crystallization in 20% yield. Subsequent Stec reaction [38-40] gave chiral CPA 2 stereospecifically with retention of configuration [39]. This synthesis accomplishes a few of the requirements that were set inititally (see Figure 2). The chemistry is straightforward and can be scaled easily. The indole N-substituent can be introduced later to make the synthesis more modular, and the resolution is straightforward late in the synthesis. Additionally, the presence of the nitro group in CPA 2 was chosen for two reasons: 1) the possibility to further functionalize at this position through reduction, diazotization, and metal-catalyzed cross-coupling, and 2) immobilization on a solid support via reduction and reaction of the aniline with an electrophile such as polystyrene isocyanate.
DOPO scaffold
We previously reported the syntheses of both enantiomers of 8-phenyl DOPO 3 [38]. The syntheses proceed in only three steps (including the separation of the (S)-1-phenylethylamine-derived phosphonamide diastereoisomers) with SP-3 and RP-3 obtained in 13% and 9% respectively starting from 2,6-diphenylphenol.
Finally, N-biphenyl-DOPO CPA 4 was synthesized in four steps as shown in Scheme 4. Although compound 16 is commercially available, it was synthesized from 2-aminobiphenyl according to the literature [47]. Subsequent reaction with phosphorus trichloride and electrophilic aromatic substitution gave a chlorophosphine intermediate, which was directly reacted with (S)-1-phenylethylamine, then hydrogen peroxide. Phosphonamide diastereoisomers 17 were obtained in moderate yield. Crystallization gave a single diastereoisomer in 20% yield. Stec reaction [38-40] finally gave the desired CPA 4. Although the entire sequence proceeded in only 6% overall yield, it was conducted on a multigram-scale so that more than 0.4 g of 4 was obtained.
![[1860-5397-18-154-i4]](/bjoc/content/inline/1860-5397-18-154-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N-biphenyl-DOPO CPA 4.
Scheme 4: Synthesis of N-biphenyl-DOPO CPA 4.
Evaluation of the catalysts
With our various CPAs 2–4 in hand, their evaluation in asymmetric organocatalysis was conducted. The reaction could have been chosen from a tremendous number of possibilities [1-17]. We selected the one Guinchard used to evaluate his thioacid hybrid-CPAs (Scheme 5) [31]. The transfer hydrogenation of 2-phenylquinoline with a Hantzsch ester 19 is a test reaction commonly used in asymmetric synthesis. The best performing of Guinchard's thiophostones 18 was the pivalate ester (R1 = t-BuC(O)) with an 86% yield of 20 and a 52% ee (19 R2 = Et (2.4 equiv), toluene, 60 °C). Further optimization with the pivalate led to 20 in 82% yield and 68% ee (19 R2 = t-Bu (2 equiv), cyclopentyl methyl ether, 22 °C).
![[1860-5397-18-154-i5]](/bjoc/content/inline/1860-5397-18-154-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Transfer hydrogenation of 2-phenylquinoline and transition-state proposed by Guinchard and coworkers [28].
Scheme 5: Transfer hydrogenation of 2-phenylquinoline and transition-state proposed by Guinchard and coworker...
To account for the best results observed with pivalate 18, Guinchard and coworkers proposed the transition-state shown in Scheme 5 [31]. Based on the fact that the cis-configuration between the sulfur and the pivalate was absolutely required for enantioselectivity, an interaction between both the sulfur and pivalate carbonyl oxygen with the hydrogen of Hantzsch ester's NH was proposed (Scheme 5). Thus, rather weak interactions might still be important in the assembly of a ternary complex and the enantioselectivity of the reaction.
The evaluation of the catalysts is shown in Table 1. CPA 4 was completely ineffective at inducing chirality (Table 1, entry 1) and catalyst 2 was not much better (entry 2). Catalyst 3 on the other hand showed a modest induction (entry 3).
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-154-i6.svg?max-width=637&scale=1.0)
Conclusion
Exploratory efforts toward new C1-symmetrical CPAs were described. Four CPAs were synthesized and three evaluated. The syntheses are straightforward, inexpensive, and scalable. Resolution via the separation of diastereoisomeric phosphorus amides could be accomplished easily, either by chromatography over silica gel or crystallization. Subsequent Stec reaction proved to be a reliable method to convert the resolved amide into the chiral thiophosphorus acids.
The CPAs synthesized clearly failed to induce any significant asymmetry. It is interesting to note, however, that the enantiomeric excess increases with an increase in bond length separation between the phosphorus and the R group. From the reaction evaluation we found that dual activation might be required from the catalyst in certain enantioselective reactions. Thus, CPA platforms that reintroduce a dual donor–acceptor role, such as P-stereogenic triflamide CPAs P(O)NHSO2CF3, is currently under investigation since BINOL-derived triflamides have been successful [48,49]. Another possibility would be to look at reactions in which the catalyst would not require a Brønsted basic site. Both directions are currently under investigation and results will be shared in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures and copies of spectra. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. doi:10.1002/anie.200353240
Return to citation in text: [1] [2] [3] -
Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356–5357. doi:10.1021/ja0491533
Return to citation in text: [1] [2] [3] -
Rowland, G. B.; Zhang, H.; Rowland, E. B.; Chennamadhavuni, S.; Wang, Y.; Antilla, J. C. J. Am. Chem. Soc. 2005, 127, 15696–15697. doi:10.1021/ja0533085
Return to citation in text: [1] [2] [3] -
Xu, F.; Huang, D.; Han, C.; Shen, W.; Lin, X.; Wang, Y. J. Org. Chem. 2010, 75, 8677–8680. doi:10.1021/jo101640z
Return to citation in text: [1] [2] [3] -
Terada, M. Curr. Org. Chem. 2011, 15, 2227–2256. doi:10.2174/138527211796150732
Return to citation in text: [1] [2] -
Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Chem. Rev. 2014, 114, 9047–9153. doi:10.1021/cr5001496
Return to citation in text: [1] [2] -
Maji, R.; Mallojjala, S. C.; Wheeler, S. E. Chem. Soc. Rev. 2018, 47, 1142–1158. doi:10.1039/c6cs00475j
Return to citation in text: [1] [2] -
Lin, X.; Wang, L.; Han, Z.; Chen, Z. Chin. J. Chem. 2021, 39, 802–824. doi:10.1002/cjoc.202000446
Return to citation in text: [1] [2] -
Fang, G.-C.; Cheng, Y.-F.; Yu, Z.-L.; Li, Z.-L.; Liu, X.-Y. Top. Curr. Chem. 2019, 377, 23. doi:10.1007/s41061-019-0249-0
Return to citation in text: [1] [2] -
Mitra, R.; Niemeyer, J. ChemCatChem 2018, 10, 1221–1234. doi:10.1002/cctc.201701698
Return to citation in text: [1] [2] -
Li, X.; Song, Q. Chin. Chem. Lett. 2018, 29, 1181–1192. doi:10.1016/j.cclet.2018.01.045
Return to citation in text: [1] [2] -
Rahman, A.; Lin, X. Org. Biomol. Chem. 2018, 16, 4753–4777. doi:10.1039/c8ob00900g
Return to citation in text: [1] [2] -
Lv, F.; Liu, S.; Hu, W. Asian J. Org. Chem. 2013, 2, 824–836. doi:10.1002/ajoc.201300097
Return to citation in text: [1] [2] -
Biaggi, C.; Benaglia, M.; Annunziata, R.; Rossi, S. Chirality 2010, 22, 369–378. doi:10.1002/chir.20754
Return to citation in text: [1] [2] -
Zamfir, A.; Schenker, S.; Freund, M.; Tsogoeva, S. B. Org. Biomol. Chem. 2010, 8, 5262–5276. doi:10.1039/c0ob00209g
Return to citation in text: [1] [2] -
Terada, M. Bull. Chem. Soc. Jpn. 2010, 83, 101–119. doi:10.1246/bcsj.20090268
Return to citation in text: [1] [2] -
Terada, M. Synthesis 2010, 1929–1982. doi:10.1055/s-0029-1218801
Return to citation in text: [1] [2] -
Antenucci, A.; Dughera, S.; Renzi, P. ChemSusChem 2021, 14, 2785–2853. doi:10.1002/cssc.202100573
Return to citation in text: [1] -
Rueping, M.; Sugiono, E.; Steck, A.; Theissmann, T. Adv. Synth. Catal. 2010, 352, 281–287. doi:10.1002/adsc.200900746
Return to citation in text: [1] -
Clot-Almenara, L.; Rodríguez-Escrich, C.; Osorio-Planes, L.; Pericàs, M. A. ACS Catal. 2016, 6, 7647–7651. doi:10.1021/acscatal.6b02621
Return to citation in text: [1] -
Birman, V. B.; Rheingold, A. L.; Lam, K.-C. Tetrahedron: Asymmetry 1999, 10, 125–131. doi:10.1016/s0957-4166(98)00481-9
Return to citation in text: [1] -
Kabachnik, M. I.; Mastrukova, T. A.; Shipov, A. E.; Melentyeva, T. A. Tetrahedron 1960, 9, 10–28. doi:10.1016/0040-4020(60)80048-8
Return to citation in text: [1] -
Ribeiro, N.; Kobayashi, Y.; Maeda, J.; Saigo, K. Chirality 2011, 23, 438–448. doi:10.1002/chir.20702
Return to citation in text: [1] -
Boter, H. L.; Platenburg, D. H. J. M. Recl. Trav. Chim. Pays-Bas 1967, 86, 399–404. doi:10.1002/recl.19670860408
Return to citation in text: [1] -
Stamatia, V. Curr. Org. Chem. 2011, 15, 2469–2480. doi:10.2174/138527211796150642
Return to citation in text: [1] -
Aaron, H. S.; Shryne, T. M.; Miller, J. I. J. Am. Chem. Soc. 1958, 80, 107–110. doi:10.1021/ja01534a029
Return to citation in text: [1] -
Aaron, H. S.; Braun, J.; Shryne, T. M.; Frack, H. F.; Smith, G. E.; Uyeda, R. T.; Miller, J. I. J. Am. Chem. Soc. 1960, 82, 596–598. doi:10.1021/ja01488a024
Return to citation in text: [1] -
Michalski, J.; Radziejewski, C.; Skrzypczynski, Z.; Dabkowski, W. J. Am. Chem. Soc. 1980, 102, 7974–7976. doi:10.1021/ja00547a044
Return to citation in text: [1] [2] -
Skrzypczynski, Z.; Michalski, J. J. Org. Chem. 1988, 53, 4549–4551. doi:10.1021/jo00254a024
Return to citation in text: [1] -
Haynes, R. K.; Au-Yeung, T.-L.; Chan, W.-K.; Lam, W.-L.; Li, Z.-Y.; Yeung, L.-L.; Chan, A. S. C.; Li, P.; Koen, M.; Mitchell, C. R.; Vonwiller, S. C. Eur. J. Org. Chem. 2000, 3205–3216. doi:10.1002/1099-0690(200009)2000:18<3205::aid-ejoc3205>3.0.co;2-d
Return to citation in text: [1] -
Ferry, A.; Stemper, J.; Marinetti, A.; Voituriez, A.; Guinchard, X. Eur. J. Org. Chem. 2014, 188–193. doi:10.1002/ejoc.201301253
Return to citation in text: [1] [2] [3] -
Kuwabara, K.; Maekawa, Y.; Minoura, M.; Murai, T. Org. Lett. 2018, 20, 1375–1379. doi:10.1021/acs.orglett.8b00147
Return to citation in text: [1] -
Salmeia, K. A.; Gaan, S. Polym. Degrad. Stab. 2015, 113, 119–134. doi:10.1016/j.polymdegradstab.2014.12.014
Return to citation in text: [1] -
Antilla, J. C.; Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 11684–11688. doi:10.1021/ja027433h
Return to citation in text: [1] [2] -
Bryant, D. E.; Kilner, C.; Kee, T. P. Inorg. Chim. Acta 2009, 362, 614–616. doi:10.1016/j.ica.2008.04.028
Return to citation in text: [1] -
Berger, O.; Montchamp, J.-L. J. Org. Chem. 2019, 84, 9239–9256. doi:10.1021/acs.joc.9b01239
Return to citation in text: [1] [2] -
Bergman, J.; Pettersson, B.; Hasimbegovic, V.; Svensson, P. H. J. Org. Chem. 2011, 76, 1546–1553. doi:10.1021/jo101865y
Return to citation in text: [1] -
Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Wozniak, L. A.; Okruszek, A. Chem. Soc. Rev. 2003, 32, 158–169. doi:10.1039/b207207f
Return to citation in text: [1] [2] [3] [4] -
Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1962, 84, 1316–1317. doi:10.1021/ja00866a060
Return to citation in text: [1] [2] [3] -
Xu, Q.; Zhao, C.-Q.; Han, L.-B. J. Am. Chem. Soc. 2008, 130, 12648–12655. doi:10.1021/ja804412k
Return to citation in text: [1] -
Berger, O.; Montchamp, J.-L. Angew. Chem., Int. Ed. 2013, 52, 11377–11380. doi:10.1002/anie.201306628
Return to citation in text: [1] -
Varga, B.; Szemesi, P.; Nagy, P.; Herbay, R.; Holczbauer, T.; Fogassy, E.; Keglevich, G.; Bagi, P. J. Org. Chem. 2021, 86, 14493–14507. doi:10.1021/acs.joc.1c01364
Return to citation in text: [1] -
Kimura, M.; Futamata, M.; Mukai, R.; Tamaru, Y. J. Am. Chem. Soc. 2005, 127, 4592–4593. doi:10.1021/ja0501161
Return to citation in text: [1] -
Deprèle, S.; Montchamp, J.-L. J. Am. Chem. Soc. 2002, 124, 9386–9387. doi:10.1021/ja0261978
Return to citation in text: [1] -
Wang, H.; Li, X.; Wan, B. Synthesis 2012, 6, 941–945. doi:10.1055/s-0031-1289700
Return to citation in text: [1] -
Riedmüller, S.; Kaufhold, O.; Spreitzer, H.; Nachtsheim, B. J. Eur. J. Org. Chem. 2014, 1391–1394. doi:10.1002/ejoc.201400046
Return to citation in text: [1] -
Nakashima, D.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 9626–9627. doi:10.1021/ja062508t
Return to citation in text: [1] -
Rueping, M.; Nachtsheim, B. J.; Moreth, S. A.; Bolte, M. Angew. Chem., Int. Ed. 2008, 47, 593–596. doi:10.1002/anie.200703668
Return to citation in text: [1]
| 34. | Antilla, J. C.; Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 11684–11688. doi:10.1021/ja027433h |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
| 39. | Wozniak, L. A.; Okruszek, A. Chem. Soc. Rev. 2003, 32, 158–169. doi:10.1039/b207207f |
| 40. | Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1962, 84, 1316–1317. doi:10.1021/ja00866a060 |
| 1. | Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. doi:10.1002/anie.200353240 |
| 2. | Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356–5357. doi:10.1021/ja0491533 |
| 18. | Antenucci, A.; Dughera, S.; Renzi, P. ChemSusChem 2021, 14, 2785–2853. doi:10.1002/cssc.202100573 |
| 34. | Antilla, J. C.; Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 11684–11688. doi:10.1021/ja027433h |
| 28. | Michalski, J.; Radziejewski, C.; Skrzypczynski, Z.; Dabkowski, W. J. Am. Chem. Soc. 1980, 102, 7974–7976. doi:10.1021/ja00547a044 |
| 6. | Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Chem. Rev. 2014, 114, 9047–9153. doi:10.1021/cr5001496 |
| 7. | Maji, R.; Mallojjala, S. C.; Wheeler, S. E. Chem. Soc. Rev. 2018, 47, 1142–1158. doi:10.1039/c6cs00475j |
| 8. | Lin, X.; Wang, L.; Han, Z.; Chen, Z. Chin. J. Chem. 2021, 39, 802–824. doi:10.1002/cjoc.202000446 |
| 9. | Fang, G.-C.; Cheng, Y.-F.; Yu, Z.-L.; Li, Z.-L.; Liu, X.-Y. Top. Curr. Chem. 2019, 377, 23. doi:10.1007/s41061-019-0249-0 |
| 10. | Mitra, R.; Niemeyer, J. ChemCatChem 2018, 10, 1221–1234. doi:10.1002/cctc.201701698 |
| 11. | Li, X.; Song, Q. Chin. Chem. Lett. 2018, 29, 1181–1192. doi:10.1016/j.cclet.2018.01.045 |
| 12. | Rahman, A.; Lin, X. Org. Biomol. Chem. 2018, 16, 4753–4777. doi:10.1039/c8ob00900g |
| 13. | Lv, F.; Liu, S.; Hu, W. Asian J. Org. Chem. 2013, 2, 824–836. doi:10.1002/ajoc.201300097 |
| 14. | Biaggi, C.; Benaglia, M.; Annunziata, R.; Rossi, S. Chirality 2010, 22, 369–378. doi:10.1002/chir.20754 |
| 15. | Zamfir, A.; Schenker, S.; Freund, M.; Tsogoeva, S. B. Org. Biomol. Chem. 2010, 8, 5262–5276. doi:10.1039/c0ob00209g |
| 16. | Terada, M. Bull. Chem. Soc. Jpn. 2010, 83, 101–119. doi:10.1246/bcsj.20090268 |
| 17. | Terada, M. Synthesis 2010, 1929–1982. doi:10.1055/s-0029-1218801 |
| 35. | Bryant, D. E.; Kilner, C.; Kee, T. P. Inorg. Chim. Acta 2009, 362, 614–616. doi:10.1016/j.ica.2008.04.028 |
| 31. | Ferry, A.; Stemper, J.; Marinetti, A.; Voituriez, A.; Guinchard, X. Eur. J. Org. Chem. 2014, 188–193. doi:10.1002/ejoc.201301253 |
| 4. | Xu, F.; Huang, D.; Han, C.; Shen, W.; Lin, X.; Wang, Y. J. Org. Chem. 2010, 75, 8677–8680. doi:10.1021/jo101640z |
| 5. | Terada, M. Curr. Org. Chem. 2011, 15, 2227–2256. doi:10.2174/138527211796150732 |
| 32. | Kuwabara, K.; Maekawa, Y.; Minoura, M.; Murai, T. Org. Lett. 2018, 20, 1375–1379. doi:10.1021/acs.orglett.8b00147 |
| 1. | Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. doi:10.1002/anie.200353240 |
| 2. | Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356–5357. doi:10.1021/ja0491533 |
| 3. | Rowland, G. B.; Zhang, H.; Rowland, E. B.; Chennamadhavuni, S.; Wang, Y.; Antilla, J. C. J. Am. Chem. Soc. 2005, 127, 15696–15697. doi:10.1021/ja0533085 |
| 4. | Xu, F.; Huang, D.; Han, C.; Shen, W.; Lin, X.; Wang, Y. J. Org. Chem. 2010, 75, 8677–8680. doi:10.1021/jo101640z |
| 5. | Terada, M. Curr. Org. Chem. 2011, 15, 2227–2256. doi:10.2174/138527211796150732 |
| 6. | Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Chem. Rev. 2014, 114, 9047–9153. doi:10.1021/cr5001496 |
| 7. | Maji, R.; Mallojjala, S. C.; Wheeler, S. E. Chem. Soc. Rev. 2018, 47, 1142–1158. doi:10.1039/c6cs00475j |
| 8. | Lin, X.; Wang, L.; Han, Z.; Chen, Z. Chin. J. Chem. 2021, 39, 802–824. doi:10.1002/cjoc.202000446 |
| 9. | Fang, G.-C.; Cheng, Y.-F.; Yu, Z.-L.; Li, Z.-L.; Liu, X.-Y. Top. Curr. Chem. 2019, 377, 23. doi:10.1007/s41061-019-0249-0 |
| 10. | Mitra, R.; Niemeyer, J. ChemCatChem 2018, 10, 1221–1234. doi:10.1002/cctc.201701698 |
| 11. | Li, X.; Song, Q. Chin. Chem. Lett. 2018, 29, 1181–1192. doi:10.1016/j.cclet.2018.01.045 |
| 12. | Rahman, A.; Lin, X. Org. Biomol. Chem. 2018, 16, 4753–4777. doi:10.1039/c8ob00900g |
| 13. | Lv, F.; Liu, S.; Hu, W. Asian J. Org. Chem. 2013, 2, 824–836. doi:10.1002/ajoc.201300097 |
| 14. | Biaggi, C.; Benaglia, M.; Annunziata, R.; Rossi, S. Chirality 2010, 22, 369–378. doi:10.1002/chir.20754 |
| 15. | Zamfir, A.; Schenker, S.; Freund, M.; Tsogoeva, S. B. Org. Biomol. Chem. 2010, 8, 5262–5276. doi:10.1039/c0ob00209g |
| 16. | Terada, M. Bull. Chem. Soc. Jpn. 2010, 83, 101–119. doi:10.1246/bcsj.20090268 |
| 17. | Terada, M. Synthesis 2010, 1929–1982. doi:10.1055/s-0029-1218801 |
| 3. | Rowland, G. B.; Zhang, H.; Rowland, E. B.; Chennamadhavuni, S.; Wang, Y.; Antilla, J. C. J. Am. Chem. Soc. 2005, 127, 15696–15697. doi:10.1021/ja0533085 |
| 33. | Salmeia, K. A.; Gaan, S. Polym. Degrad. Stab. 2015, 113, 119–134. doi:10.1016/j.polymdegradstab.2014.12.014 |
| 31. | Ferry, A.; Stemper, J.; Marinetti, A.; Voituriez, A.; Guinchard, X. Eur. J. Org. Chem. 2014, 188–193. doi:10.1002/ejoc.201301253 |
| 22. | Kabachnik, M. I.; Mastrukova, T. A.; Shipov, A. E.; Melentyeva, T. A. Tetrahedron 1960, 9, 10–28. doi:10.1016/0040-4020(60)80048-8 |
| 28. | Michalski, J.; Radziejewski, C.; Skrzypczynski, Z.; Dabkowski, W. J. Am. Chem. Soc. 1980, 102, 7974–7976. doi:10.1021/ja00547a044 |
| 29. | Skrzypczynski, Z.; Michalski, J. J. Org. Chem. 1988, 53, 4549–4551. doi:10.1021/jo00254a024 |
| 30. | Haynes, R. K.; Au-Yeung, T.-L.; Chan, W.-K.; Lam, W.-L.; Li, Z.-Y.; Yeung, L.-L.; Chan, A. S. C.; Li, P.; Koen, M.; Mitchell, C. R.; Vonwiller, S. C. Eur. J. Org. Chem. 2000, 3205–3216. doi:10.1002/1099-0690(200009)2000:18<3205::aid-ejoc3205>3.0.co;2-d |
| 47. | Riedmüller, S.; Kaufhold, O.; Spreitzer, H.; Nachtsheim, B. J. Eur. J. Org. Chem. 2014, 1391–1394. doi:10.1002/ejoc.201400046 |
| 21. | Birman, V. B.; Rheingold, A. L.; Lam, K.-C. Tetrahedron: Asymmetry 1999, 10, 125–131. doi:10.1016/s0957-4166(98)00481-9 |
| 31. | Ferry, A.; Stemper, J.; Marinetti, A.; Voituriez, A.; Guinchard, X. Eur. J. Org. Chem. 2014, 188–193. doi:10.1002/ejoc.201301253 |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
| 39. | Wozniak, L. A.; Okruszek, A. Chem. Soc. Rev. 2003, 32, 158–169. doi:10.1039/b207207f |
| 40. | Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1962, 84, 1316–1317. doi:10.1021/ja00866a060 |
| 1. | Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. doi:10.1002/anie.200353240 |
| 2. | Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356–5357. doi:10.1021/ja0491533 |
| 3. | Rowland, G. B.; Zhang, H.; Rowland, E. B.; Chennamadhavuni, S.; Wang, Y.; Antilla, J. C. J. Am. Chem. Soc. 2005, 127, 15696–15697. doi:10.1021/ja0533085 |
| 4. | Xu, F.; Huang, D.; Han, C.; Shen, W.; Lin, X.; Wang, Y. J. Org. Chem. 2010, 75, 8677–8680. doi:10.1021/jo101640z |
| 39. | Wozniak, L. A.; Okruszek, A. Chem. Soc. Rev. 2003, 32, 158–169. doi:10.1039/b207207f |
| 19. | Rueping, M.; Sugiono, E.; Steck, A.; Theissmann, T. Adv. Synth. Catal. 2010, 352, 281–287. doi:10.1002/adsc.200900746 |
| 20. | Clot-Almenara, L.; Rodríguez-Escrich, C.; Osorio-Planes, L.; Pericàs, M. A. ACS Catal. 2016, 6, 7647–7651. doi:10.1021/acscatal.6b02621 |
| 23. | Ribeiro, N.; Kobayashi, Y.; Maeda, J.; Saigo, K. Chirality 2011, 23, 438–448. doi:10.1002/chir.20702 |
| 24. | Boter, H. L.; Platenburg, D. H. J. M. Recl. Trav. Chim. Pays-Bas 1967, 86, 399–404. doi:10.1002/recl.19670860408 |
| 25. | Stamatia, V. Curr. Org. Chem. 2011, 15, 2469–2480. doi:10.2174/138527211796150642 |
| 26. | Aaron, H. S.; Shryne, T. M.; Miller, J. I. J. Am. Chem. Soc. 1958, 80, 107–110. doi:10.1021/ja01534a029 |
| 27. | Aaron, H. S.; Braun, J.; Shryne, T. M.; Frack, H. F.; Smith, G. E.; Uyeda, R. T.; Miller, J. I. J. Am. Chem. Soc. 1960, 82, 596–598. doi:10.1021/ja01488a024 |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
| 36. | Berger, O.; Montchamp, J.-L. J. Org. Chem. 2019, 84, 9239–9256. doi:10.1021/acs.joc.9b01239 |
| 48. | Nakashima, D.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 9626–9627. doi:10.1021/ja062508t |
| 49. | Rueping, M.; Nachtsheim, B. J.; Moreth, S. A.; Bolte, M. Angew. Chem., Int. Ed. 2008, 47, 593–596. doi:10.1002/anie.200703668 |
| 37. | Bergman, J.; Pettersson, B.; Hasimbegovic, V.; Svensson, P. H. J. Org. Chem. 2011, 76, 1546–1553. doi:10.1021/jo101865y |
| 46. | Wang, H.; Li, X.; Wan, B. Synthesis 2012, 6, 941–945. doi:10.1055/s-0031-1289700 |
| 36. | Berger, O.; Montchamp, J.-L. J. Org. Chem. 2019, 84, 9239–9256. doi:10.1021/acs.joc.9b01239 |
| 44. | Kimura, M.; Futamata, M.; Mukai, R.; Tamaru, Y. J. Am. Chem. Soc. 2005, 127, 4592–4593. doi:10.1021/ja0501161 |
| 45. | Deprèle, S.; Montchamp, J.-L. J. Am. Chem. Soc. 2002, 124, 9386–9387. doi:10.1021/ja0261978 |
| 41. | Xu, Q.; Zhao, C.-Q.; Han, L.-B. J. Am. Chem. Soc. 2008, 130, 12648–12655. doi:10.1021/ja804412k |
| 42. | Berger, O.; Montchamp, J.-L. Angew. Chem., Int. Ed. 2013, 52, 11377–11380. doi:10.1002/anie.201306628 |
| 43. | Varga, B.; Szemesi, P.; Nagy, P.; Herbay, R.; Holczbauer, T.; Fogassy, E.; Keglevich, G.; Bagi, P. J. Org. Chem. 2021, 86, 14493–14507. doi:10.1021/acs.joc.1c01364 |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
| 39. | Wozniak, L. A.; Okruszek, A. Chem. Soc. Rev. 2003, 32, 158–169. doi:10.1039/b207207f |
| 40. | Wadsworth, W. S., Jr.; Emmons, W. D. J. Am. Chem. Soc. 1962, 84, 1316–1317. doi:10.1021/ja00866a060 |
| 38. | Winters, K. R.; Montchamp, J.-L. J. Org. Chem. 2020, 85, 14545–14558. doi:10.1021/acs.joc.0c01151 |
© 2022 Winters and Montchamp; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.