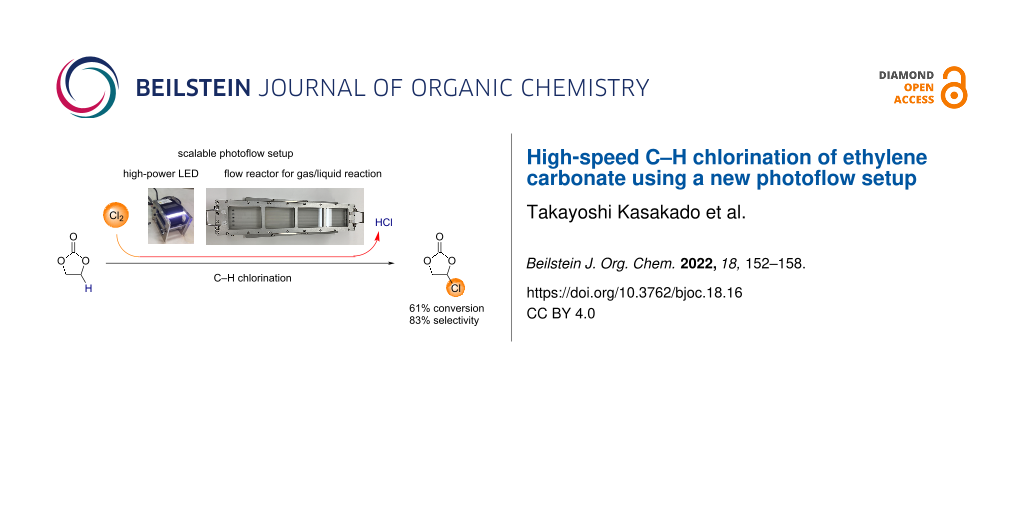
Abstract
We report the high-speed C–H chlorination of ethylene carbonate, which gives chloroethylene carbonate, a precursor to vinylene carbonate. A novel photoflow setup designed for a gas–liquid biphasic reaction turned out to be useful for the direct use of chlorine gas. The setup employed sloped channels so as to make the liquid phase thinner, ensuring a high surface-to-volume ratio. When ethylene carbonate was introduced to the reactor, the residence time was measured to be 15 or 30 s, depending on the slope of the reactor set at 15 or 5°, respectively. Such short time of exposition sufficed the photo C–H chlorination. The partial irradiation of the flow channels also sufficed for the C–H chlorination, which is consistent with the requirement of photoirradiation for the purpose of radical initiation. Near-complete selectivity for single chlorination required the low conversion of ethylene carbonate such as 9%, which was controlled by limited introduction of chlorine gas. At a higher conversion of ethylene carbonate such as 61%, the selectivity for monochlorinated ethylene carbonate over dichlorinated ethylene carbonate was 86%. We found that the substrate contamination with water negatively influenced the performance of the C–H chlorination.

Graphical Abstract
Introduction
The C–H chlorination by molecular chlorine is a highly exothermic reaction that proceeds via a radical chain mechanism as illustrated in Scheme 1 [1-6]. Frequently, photoirradiation is used for radical initiation through homolysis of the Cl–Cl bond to generate chlorine radicals. In a subsequent step, a SH2 reaction by chlorine radicals at C–H bonds generates alkyl radicals and HCl. The second SH2 reaction between alkyl radicals and molecular chlorine then occurs to give the C–H chlorinated product and a chlorine radical, sustaining the radical chain. Chlorine gas is a cheap feedstock since it is formed as a byproduct of the electrolysis of NaCl to produce NaOH in an industrial process [7]. We felt that C–H chlorination would be updated by using scalable flash chemistry [8].
![[1860-5397-18-16-i1]](/bjoc/content/inline/1860-5397-18-16-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Radical chain mechanism for a photo-induced C–H chlorination reaction.
Scheme 1: Radical chain mechanism for a photo-induced C–H chlorination reaction.
Flow C–H chlorination using a compact flow reactor is highly desirable in terms of efficiency and safety in handling highly toxic gases such as chlorine. In 2002, Jähnisch and co-workers reported the first microflow chlorination of 2,4-diisocyano-1-methylbenzene, which used a falling-film reactor developed by IMM [9]. While the flow rate employed was quite low (0.12 mL/min of toluene), the residence time was less than 14 seconds. More recent studies on flow C–H chlorination reactions focused on the use of Cl2 gas in situ generated by photolysis of sulfuryl chloride [10] or by acid treatment of NaOCl [11,12]. We thought that if rationally designed scalable photoflow setups were available, flow C–H chlorination reactions using chlorine gas would be able to focus on production. In this study, we tested a novel photoflow setup consisting of quartz-made straight-line reactors, which are provided from MiChS (LX-1, Figure 1a) and a high-power LED (MiChS LED-s, 365 ± 5 nm, Figure 1b) [13]. Each channel track has a 2 mm depth and 557 mm length, while the width varies from 6 or 13 mm depending on the number of channels 7 or 5, respectively. The flow photoreactor is embedded into an aluminum frame equipped with a heat carrier channel. The design concepts including angle settings to ensure a thin liquid layer are summarized in Figure 1.
![[1860-5397-18-16-1]](/bjoc/content/figures/1860-5397-18-16-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Components for photoflow setup: (a) MiChS LX-1 reactor and (b) MiChS LED-s (365 ± 5 nm, 60–600 W).
Figure 1: Components for photoflow setup: (a) MiChS LX-1 reactor and (b) MiChS LED-s (365 ± 5 nm, 60–600 W).
We chose the C–H chlorination of ethylene carbonate (1) as a model reaction (Scheme 2). Chlorinated ethylene carbonate 2 is a precursor to vinylene carbonate (3), which is used as an electrolyte additive for Li-ion batteries [14-20]. Vinylene carbonate also serves as a useful synthetic building block for Diels–Alder reactions [21-25] and polymerization [26-30].
![[1860-5397-18-16-i2]](/bjoc/content/inline/1860-5397-18-16-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Model reaction: photoflow C–H chlorination of ethylene carbonate (1) to chloroethylene carbonate (2).
Scheme 2: Model reaction: photoflow C–H chlorination of ethylene carbonate (1) to chloroethylene carbonate (2...
Results and Discussion
Using a PTFE tube and PTFE connectors, we connected the photoflow setup with a chlorine gas cylinder through a floating gas level meter in a fume hood (Figure 2). Since ethylene carbonate (1) melts between 34–37 °C, we preheated the container of 1 using an oil bath at 70 °C and pumped it to the photoreactor. In the reactor, hot water (80 °C) was circulated through a hole channel manufactured in an aluminum-made frame to keep the contacted glass reactor warm. The LED lamp was placed on the upper side of the reactor with a 20° angle to the reactor surface. The exiting gases (HCl and unreacted Cl2) were trapped by an aqueous NaOH solution (1.7 M).
![[1860-5397-18-16-2]](/bjoc/content/figures/1860-5397-18-16-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Photoflow setup for the C–H chlorination of ethylene carbonate (1).
Figure 2: Photoflow setup for the C–H chlorination of ethylene carbonate (1).
The reactors are set with a slope of 15 or 5° to achieve a thin substrate layer causing a rapid gas/liquid biphasic reaction. The residence time was estimated to be 15 and 30 seconds, respectively (for the measurement, ethylene carbonate was introduced in the absence of chlorine gas). After the experiments, chlorine gas that remained inside the flow setup was flushed with N2 gas. In general, we used ethylene carbonate (1) with the grade containing less than 0.03% of water. The results are summarized in Table 1.
Table 1: Photoflow C–H chlorination of ethylene carbonate (1) to chloroethylene carbonate (2).a
| entry | angle (°) | flow rate | UV-LED (W) | conversion (%)b | selectivity (%)b | ||
| 1a (mmol/min) | Cl2 (mmol/min) (equiv) | 2 | 2’ | ||||
| 1 | 15 | 74.9 | 12.5 (0.17) | 240 | 9 | 100 | 0 |
| 2 | 15 | 74.9 | 17.4 (0.23) | 240 | 12 | 96 | 4 |
| 3 | 15 | 74.9 | 33.9 (0.45) | 240 | 21 | 91 | 9 |
| 4 | 15 | 74.9 | 75.9 (1.01) | 240 | 39 | 89 | 11 |
| 5c | 15 | 74.9 | 75.9 + 75.9 (2.02) | 240 | 87 | 74 | 26 |
| 6 | 15 | 46.4 | 91.5 (1.97) | 240 | 61 | 86 | 14 |
| 7 | 15 | 29.6 | 91.5 (3.09) | 240 | 76 | 84 | 16 |
| 8 | 15 | 117.6 | 146.5 (1.25) | 240 | 49 | 78 | 22 |
| 9 | 15 | 117.6 | 143.7 (1.22) | 600 | 47 | 78 | 22 |
| 10 | 5 | 117.6 | 146.5 (1.25) | 240 | 61 | 79 | 21 |
aReactions were conducted by using LX-1 with a reactor angle of 15° or 5° (entry 10). Photoirradiation was carried out by using LEDs (365 ± 5 nm at the power of 240 or 600 W). Ethylene carbonate (1) contains 0.03% of H2O. bDetermined by GC analysis. cReaction mixture was circulated twice.
When the reaction of ethylene carbonate (1, flow rate: 74.9 mmol/min, containing 0.03% of H2O) with 0.17 equiv of Cl2 gas (flow rate: 12.5 mmol/min) was carried out under irradiation by UV-LED (240 W) with a 15° reactor angle, the desired chloroethylene carbonate (2) was formed selectively with a 9% conversion of 1 (Table 1, entry 1). When 0.23 equiv of Cl2 was used, the selectivity became 96% with 12% conversion of 1, in which a small amount of undesired 1,2-dichloroethylene carbonate (2’) was detected by GC (Table 1, entry 2). When 0.45 equiv of Cl2 was used, the conversion of 1 increased to 21% and the selectivity of 2 became 91% (Table 1, entry 3). The reaction of 1 with one equivalent of Cl2 gave 2 and 2’ in a ratio of 89:11 with 39% conversion of 1 (Table 1, entry 4). When the reaction mixture was circulated twice, we observed a higher conversion of 1 (87%) and obtained a 74:26 mixture of 2 and 2’ (Table 1, entry 5). Then, we limited the feeding of 1 (flow rate: 46.4 mmol/min) in order to increase conversion, which worked well. The reaction of 1 with 1.97 equiv of Cl2 resulted in 61% conversion of 1 and an 86:14 ratio of 2 and 2’ (Table 1, entry 6). When a lower feeding of 1 (29.6 mmol/min) and an excess amount of Cl2 (3.09 equiv) were used, higher conversion of 1 (76%) was attained with the selectivity of 84:16 (Table 1, entry 7). The irradiation at 600 W gave an almost similar result (Table 1, entries 8 and 9), which suggested that 240 W sufficed the reaction. Indeed, when the reaction was carried out with a shallow reactor angle such as 5°, the conversion of 1 increased from 49 to 61% (Table 1, entries 8 and 10). This is due to the extended residence time from 15 to 30 s. Flow gas/liquid reactions are often carried out using a tubular reactor and mixer under slug flow conditions. However, it is not easy to apply such conditions to the present photochlorination reaction since the volume of the Cl2 gas is ca. 400 times larger than that of ethylene carbonate (for entry 8 in Table 1). In addition, a much longer tubular reactor would be required to ensure 15–30 s residence time.
We then investigated the effect of contamination with water on the reaction, since Cl2 gas is known to react with H2O under irradiation conditions [31] and the results are summarized in Table 2. The flow rate of 1 and the equivalents of chlorine to 1 were set to be 187 mmol/min and 0.60–0.69, respectively. The reactor angle and light power were 15° and 240 W, respectively. The chlorination reaction using an ordinary grade of the substrate 1 containing 0.03% of water gave a 96:4 ratio of products 2 and 2’ with 26% conversion of 1 (Table 2, entry 1). In contrast, when we used substrate 1 containing 0.15% of water, the conversion decreased to 11% (Table 2, entry 2). With 0.76% of water, the conversion decreased further to 9% (Table 2, entry 3). These results suggest that the reaction has to be carried out carefully under dry conditions.
Table 2: Effect of contamination of water.a
| entry | water contamination | flow rate | conversion (%)b | selectivity (%)b | ||
| 1a (mmol/min) | Cl2 (mmol/min) (equiv) | 2 | 2’ | |||
| 1 | 0.03% | 187.0 | 126.8 (0.68) | 26 | 96 | 4 |
| 2 | 0.15% | 187.0 | 112.7 (0.60) | 11 | 92 | 8 |
| 3 | 0.76% | 187.0 | 118.3 (0.63) | 9 | 100 | 0 |
aReactions were conducted by using LX-1 with a rector angle of 15° and LEDs (240 W). bMeasured by GC.
Conclusion
In this work, we reported that a novel photoflow setup designed for a gas–liquid biphasic reaction turned out to be useful for the C–H chlorination using chlorine gas in flow. Two decades after the first report on the microflow chlorination of a toluene derivative by Jähnisch and co-workers, we propose a new photoflow setup for C–H chlorination using chlorine gas, applicable to scalable flow C–H chlorination. In our test reaction using C–H chlorination of ethylene carbonate (1), chloroethylene carbonate (2) was obtained in good to excellent selectivity by tuning the flow rates of 1 and chlorine gas. Partial irradiation of the flow channel is sufficient for the C–H chlorination, consistent with the requirement for light irradiation for the radical initiation step. If we apply the conditions to give 80% selectivity with 60% conversion with 30 s residence time, around 15 kilograms of chloroethylene carbonate (2) can be synthesized per day, which suggests the high potential of the present photoflow setup. We also demonstrated that the contamination with water had a negative impact on the reaction and the system should be kept dry for continuous production. We are now investigating some other photo gas–liquid flow reactions, which will be reported in due course.
Experimental
The photoflow setup consisting of a flow photoreactor LX-1 and UV-LEDs were supplied from MiChS Inc., Ltd. (http://www.michs.jp). The angle of the photoflow reactor was set to be 15 or 5° and heated water at 80 °C was circulated in a channel of an aluminum-made frame to avoid solidification of ethylene carbonate (1), whose melting point is 34–37 ºC. The UV-LED (365 ± 5 nm) was set with an angle of 20° to the reactor surface. Ethylene carbonate (1) preheated to 70 °C was fed into each channel of the flow photoreactor by using a diaphragm pump. At the same time, chlorine gas was fed into the reactor from the top-side inlet. Evolved HCl gas and unreacted Cl2 gas were trapped by an aqueous 1.7 M NaOH solution. The first eluted solution was discarded for 3 min after which the eluted solution was collected for analysis. GC analysis was performed on a Shimadzu GC-2014 equipped with an FID detector using an Agilent J&W DB-1 column (Ø 0.25 mm × 30 m) under the following conditions: initial oven temperature: 40 °C, temperature change rate of 5 °C/min to 250 °C, hold at this temperature for 10 min. Yields were determined by using the percentage peak area method with compensation for the relative sensitivities of each component. Product 2 and byproduct 2’ were confirmed by 1H and 13C NMR analysis (see Supporting Information File 1).
Supporting Information
| Supporting Information File 1: GC analysis and NMR spectra of the crude reaction mixture for the chlorination of compound 1. | ||
| Format: PDF | Size: 562.4 KB | Download |
References
-
Ingold, K. U.; Lusztyk, J.; Raner, K. D. Acc. Chem. Res. 1990, 23, 219–225. doi:10.1021/ar00175a003
Return to citation in text: [1] -
Fletcher, B.; Suleman, N. K.; Tanko, J. M. J. Am. Chem. Soc. 1998, 120, 11839–11844. doi:10.1021/ja982289e
Return to citation in text: [1] -
Sun, N.; Klabunde, K. J. J. Am. Chem. Soc. 1999, 121, 5587–5588. doi:10.1021/ja990084f
Return to citation in text: [1] -
Pease, R. N.; Walz, G. F. J. Am. Chem. Soc. 1931, 53, 3728–3737. doi:10.1021/ja01361a016
Return to citation in text: [1] -
Brown, H. C.; Kharasch, M. S.; Chao, T. H. J. Am. Chem. Soc. 1940, 62, 3435–3439. doi:10.1021/ja01869a040
Return to citation in text: [1] -
Kharasch, M. S.; Berkman, M. G. J. Org. Chem. 1941, 6, 810–817. doi:10.1021/jo01206a004
Return to citation in text: [1] -
Wang, Y.; Liu, Y.; Wiley, D.; Zhao, S.; Tang, Z. J. Mater. Chem. A 2021, 9, 18974–18993. doi:10.1039/d1ta02745j
See for a recent review.
Return to citation in text: [1] -
Yoshida, J.-i. Flash Chemistry: Fast Organic Synthesis in Microsystems; John Wiley & Sons: Chichester, UK, 2008.
Return to citation in text: [1] -
Ehrich, H.; Linke, D.; Morgenschweis, K.; Baerns, M.; Jähnisch, K. Chimia 2002, 56, 647–653. doi:10.2533/000942902777680063
Return to citation in text: [1] -
Matsubara, H.; Hino, Y.; Tokizane, M.; Ryu, I. Chem. Eng. J. 2011, 167, 567–571. doi:10.1016/j.cej.2010.08.086
Return to citation in text: [1] -
Fukuyama, T.; Tokizane, M.; Matsui, A.; Ryu, I. React. Chem. Eng. 2016, 1, 613–615. doi:10.1039/c6re00159a
Return to citation in text: [1] -
Strauss, F. J.; Cantillo, D.; Guerra, J.; Kappe, C. O. React. Chem. Eng. 2016, 1, 472–476. doi:10.1039/c6re00135a
Return to citation in text: [1] -
Hyodo, M.; Iwano, H.; Kasakado, T.; Fukuyama, T.; Ryu, I. Micromachines 2021, 12, 1307. doi:10.3390/mi12111307
Return to citation in text: [1] -
Zhang, S. S. J. Power Sources 2006, 162, 1379–1394. doi:10.1016/j.jpowsour.2006.07.074
See for a review on electrolyte additives for lithium ion batteries.
Return to citation in text: [1] -
Ivanov, S.; Sauerteig, D.; Dimitrova, A.; Krischok, S.; Bund, A. J. Power Sources 2020, 457, 228020. doi:10.1016/j.jpowsour.2020.228020
Return to citation in text: [1] -
Michan, A. L.; Parimalam, B. S.; Leskes, M.; Kerber, R. N.; Yoon, T.; Grey, C. P.; Lucht, B. L. Chem. Mater. 2016, 28, 8149–8159. doi:10.1021/acs.chemmater.6b02282
Return to citation in text: [1] -
Liu, Y.-H.; Takeda, S.; Kaneko, I.; Yoshitake, H.; Yanagida, M.; Saito, Y.; Sakai, T. RSC Adv. 2016, 6, 75777–75781. doi:10.1039/c6ra15168j
Return to citation in text: [1] -
Wang, Y.; Nakamura, S.; Tasaki, K.; Balbuena, P. B. J. Am. Chem. Soc. 2002, 124, 4408–4421. doi:10.1021/ja017073i
Return to citation in text: [1] -
Burns, J. C.; Petibon, R.; Nelson, K. J.; Sinha, N. N.; Kassam, A.; Way, B. M.; Dahn, J. R. J. Electrochem. Soc. 2013, 160, A1668–A1674. doi:10.1149/2.031310jes
Return to citation in text: [1] -
Xiong, D.; Burns, J. C.; Smith, A. J.; Sinha, N.; Dahn, J. R. J. Electrochem. Soc. 2011, 158, A1431–A1435. doi:10.1149/2.100112jes
Return to citation in text: [1] -
Aotake, T.; Tanimoto, H.; Hotta, H.; Kuzuhara, D.; Okujima, T.; Uno, H.; Yamada, H. Chem. Commun. 2013, 49, 3661–3663. doi:10.1039/c3cc40827b
Return to citation in text: [1] -
Geiseler, O.; Müller, M.; Podlech, J. Tetrahedron 2013, 69, 3683–3689. doi:10.1016/j.tet.2013.03.013
Return to citation in text: [1] -
Revés, M.; Lledó, A.; Ji, Y.; Blasi, E.; Riera, A.; Verdaguer, X. Org. Lett. 2012, 14, 3534–3537. doi:10.1021/ol301545e
Return to citation in text: [1] -
Dong, S.; Cahill, K. J.; Kang, M.-I.; Colburn, N. H.; Henrich, C. J.; Wilson, J. A.; Beutler, J. A.; Johnson, R. P.; Porco, J. A., Jr. J. Org. Chem. 2011, 76, 8944–8954. doi:10.1021/jo201658y
Return to citation in text: [1] -
Taffin, C.; Kreutler, G.; Bourgeois, D.; Clot, E.; Périgaud, C. New J. Chem. 2010, 34, 517–525. doi:10.1039/b9nj00536f
Return to citation in text: [1] -
Huang, X.; Wu, J.; Wang, X.; Tian, Y.; Zhang, F.; Yang, M.; Xu, B.; Wu, B.; Liu, X.; Li, H. ACS Appl. Energy Mater. 2021, 4, 9368–9375. doi:10.1021/acsaem.1c01570
Return to citation in text: [1] -
Zhang, Y.; Chen, S.; Chen, Y.; Li, L. Mater. Chem. Front. 2021, 5, 3681–3691. doi:10.1039/d1qm00004g
Return to citation in text: [1] -
Li, H.; Yang, J.; Xu, Z.; Lu, H.; Zhang, T.; Chen, S.; Wang, J.; NuLi, Y.; Hirano, S.-i. ACS Appl. Energy Mater. 2020, 3, 8552–8561. doi:10.1021/acsaem.0c01173
Return to citation in text: [1] -
Chai, J.; Liu, Z.; Zhang, J.; Sun, J.; Tian, Z.; Ji, Y.; Tang, K.; Zhou, X.; Cui, G. ACS Appl. Mater. Interfaces 2017, 9, 17897–17905. doi:10.1021/acsami.7b02844
Return to citation in text: [1] -
Zhao, H.; Zhou, X.; Park, S.-J.; Shi, F.; Fu, Y.; Ling, M.; Yuca, N.; Battaglia, V.; Liu, G. J. Power Sources 2014, 263, 288–295. doi:10.1016/j.jpowsour.2014.04.063
Return to citation in text: [1] -
Allmand, A. J.; Cunliffe, P. W.; Maddison, R. E. W. J. Chem. Soc., Trans. 1925, 127, 822–840. doi:10.1039/ct9252700822
Return to citation in text: [1]
| 1. | Ingold, K. U.; Lusztyk, J.; Raner, K. D. Acc. Chem. Res. 1990, 23, 219–225. doi:10.1021/ar00175a003 |
| 2. | Fletcher, B.; Suleman, N. K.; Tanko, J. M. J. Am. Chem. Soc. 1998, 120, 11839–11844. doi:10.1021/ja982289e |
| 3. | Sun, N.; Klabunde, K. J. J. Am. Chem. Soc. 1999, 121, 5587–5588. doi:10.1021/ja990084f |
| 4. | Pease, R. N.; Walz, G. F. J. Am. Chem. Soc. 1931, 53, 3728–3737. doi:10.1021/ja01361a016 |
| 5. | Brown, H. C.; Kharasch, M. S.; Chao, T. H. J. Am. Chem. Soc. 1940, 62, 3435–3439. doi:10.1021/ja01869a040 |
| 6. | Kharasch, M. S.; Berkman, M. G. J. Org. Chem. 1941, 6, 810–817. doi:10.1021/jo01206a004 |
| 10. | Matsubara, H.; Hino, Y.; Tokizane, M.; Ryu, I. Chem. Eng. J. 2011, 167, 567–571. doi:10.1016/j.cej.2010.08.086 |
| 9. | Ehrich, H.; Linke, D.; Morgenschweis, K.; Baerns, M.; Jähnisch, K. Chimia 2002, 56, 647–653. doi:10.2533/000942902777680063 |
| 8. | Yoshida, J.-i. Flash Chemistry: Fast Organic Synthesis in Microsystems; John Wiley & Sons: Chichester, UK, 2008. |
| 7. |
Wang, Y.; Liu, Y.; Wiley, D.; Zhao, S.; Tang, Z. J. Mater. Chem. A 2021, 9, 18974–18993. doi:10.1039/d1ta02745j
See for a recent review. |
| 21. | Aotake, T.; Tanimoto, H.; Hotta, H.; Kuzuhara, D.; Okujima, T.; Uno, H.; Yamada, H. Chem. Commun. 2013, 49, 3661–3663. doi:10.1039/c3cc40827b |
| 22. | Geiseler, O.; Müller, M.; Podlech, J. Tetrahedron 2013, 69, 3683–3689. doi:10.1016/j.tet.2013.03.013 |
| 23. | Revés, M.; Lledó, A.; Ji, Y.; Blasi, E.; Riera, A.; Verdaguer, X. Org. Lett. 2012, 14, 3534–3537. doi:10.1021/ol301545e |
| 24. | Dong, S.; Cahill, K. J.; Kang, M.-I.; Colburn, N. H.; Henrich, C. J.; Wilson, J. A.; Beutler, J. A.; Johnson, R. P.; Porco, J. A., Jr. J. Org. Chem. 2011, 76, 8944–8954. doi:10.1021/jo201658y |
| 25. | Taffin, C.; Kreutler, G.; Bourgeois, D.; Clot, E.; Périgaud, C. New J. Chem. 2010, 34, 517–525. doi:10.1039/b9nj00536f |
| 31. | Allmand, A. J.; Cunliffe, P. W.; Maddison, R. E. W. J. Chem. Soc., Trans. 1925, 127, 822–840. doi:10.1039/ct9252700822 |
| 14. |
Zhang, S. S. J. Power Sources 2006, 162, 1379–1394. doi:10.1016/j.jpowsour.2006.07.074
See for a review on electrolyte additives for lithium ion batteries. |
| 15. | Ivanov, S.; Sauerteig, D.; Dimitrova, A.; Krischok, S.; Bund, A. J. Power Sources 2020, 457, 228020. doi:10.1016/j.jpowsour.2020.228020 |
| 16. | Michan, A. L.; Parimalam, B. S.; Leskes, M.; Kerber, R. N.; Yoon, T.; Grey, C. P.; Lucht, B. L. Chem. Mater. 2016, 28, 8149–8159. doi:10.1021/acs.chemmater.6b02282 |
| 17. | Liu, Y.-H.; Takeda, S.; Kaneko, I.; Yoshitake, H.; Yanagida, M.; Saito, Y.; Sakai, T. RSC Adv. 2016, 6, 75777–75781. doi:10.1039/c6ra15168j |
| 18. | Wang, Y.; Nakamura, S.; Tasaki, K.; Balbuena, P. B. J. Am. Chem. Soc. 2002, 124, 4408–4421. doi:10.1021/ja017073i |
| 19. | Burns, J. C.; Petibon, R.; Nelson, K. J.; Sinha, N. N.; Kassam, A.; Way, B. M.; Dahn, J. R. J. Electrochem. Soc. 2013, 160, A1668–A1674. doi:10.1149/2.031310jes |
| 20. | Xiong, D.; Burns, J. C.; Smith, A. J.; Sinha, N.; Dahn, J. R. J. Electrochem. Soc. 2011, 158, A1431–A1435. doi:10.1149/2.100112jes |
| 13. | Hyodo, M.; Iwano, H.; Kasakado, T.; Fukuyama, T.; Ryu, I. Micromachines 2021, 12, 1307. doi:10.3390/mi12111307 |
| 11. | Fukuyama, T.; Tokizane, M.; Matsui, A.; Ryu, I. React. Chem. Eng. 2016, 1, 613–615. doi:10.1039/c6re00159a |
| 12. | Strauss, F. J.; Cantillo, D.; Guerra, J.; Kappe, C. O. React. Chem. Eng. 2016, 1, 472–476. doi:10.1039/c6re00135a |
| 26. | Huang, X.; Wu, J.; Wang, X.; Tian, Y.; Zhang, F.; Yang, M.; Xu, B.; Wu, B.; Liu, X.; Li, H. ACS Appl. Energy Mater. 2021, 4, 9368–9375. doi:10.1021/acsaem.1c01570 |
| 27. | Zhang, Y.; Chen, S.; Chen, Y.; Li, L. Mater. Chem. Front. 2021, 5, 3681–3691. doi:10.1039/d1qm00004g |
| 28. | Li, H.; Yang, J.; Xu, Z.; Lu, H.; Zhang, T.; Chen, S.; Wang, J.; NuLi, Y.; Hirano, S.-i. ACS Appl. Energy Mater. 2020, 3, 8552–8561. doi:10.1021/acsaem.0c01173 |
| 29. | Chai, J.; Liu, Z.; Zhang, J.; Sun, J.; Tian, Z.; Ji, Y.; Tang, K.; Zhou, X.; Cui, G. ACS Appl. Mater. Interfaces 2017, 9, 17897–17905. doi:10.1021/acsami.7b02844 |
| 30. | Zhao, H.; Zhou, X.; Park, S.-J.; Shi, F.; Fu, Y.; Ling, M.; Yuca, N.; Battaglia, V.; Liu, G. J. Power Sources 2014, 263, 288–295. doi:10.1016/j.jpowsour.2014.04.063 |
© 2022 Kasakado et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.