Abstract
A practical enantioselective N-selective nitroso aldol reaction of α-methylmalonamates with a nitrosoarene is reported. The reaction employs the Takemoto thiourea catalyst for the induction of enantioselectivity, and the corresponding optically active oxyaminated malonamates were obtained in reasonably good yields.

Graphical Abstract
Introduction
Nitrosoarenes are versatile building blocks frequently encountered in organic synthesis as precursors for the synthesis of nitrogen and oxygen-containing molecules [1-5]. The high reactivity caused by the polarization of the N–O bond enables the nitrosoarenes to undergo a wide range of transformations in a chemo- and regioselective manner [6-8]. The noteworthy and widely explored transformations of nitrosoarenes include nitroso ene reactions [9-11], Diels–Alder cycloadditions [12-18], and nitroso aldol reactions [19-23]. Among the various applications of nitrosoarenes, the asymmetric nitroso aldol reaction to achieve optically active α-aminoxy and α-hydroxyamino carbonyl compounds has received considerable attention in the past decades [24]. In 2003, the Yamamoto group demonstrated for the first time that nitrosobenzene could be used as a practical reagent for the catalytic enantioselective α-aminoxylation using a silver-BINAP catalyst combination [25]. Later, the same group could successfully tune the catalytic system to control the regioselectivity in the addition of metal enolate to nitrosoarenes to achieve an α-hydroxyamination [26]. Since then, several groups have shown the use of metal-catalyzed nitroso aldol reactions as a practical tool for the selective introduction of amino or hydroxy moieties at the α-position of a carbonyl function [27-30].
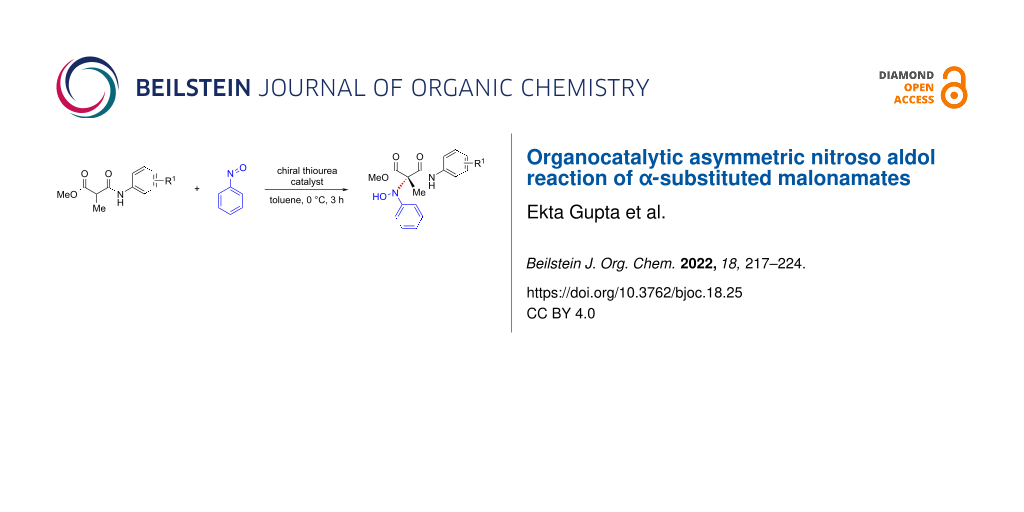
The last two decades have witnessed an upsurge of interest in the development of organocatalyzed nitroso aldol reactions in addition to the metal-catalyzed reactions [31-36]. The most successful among them are the ʟ-proline-catalyzed reactions of enolizable aldehydes with nitrosoarenes [37-43]. Besides ʟ-proline and its derivatives, various secondary amines derived from BINOL and cinchona alkaloids were also found useful in catalyzing the nitroso aldol reaction [44-48]. Surprisingly, the utility of thiourea catalysts in nitroso aldol reactions remains far less developed. The scattered reports where bifunctional thiourea catalysis was found useful for this type of reaction, describe the hydroxyamination of oxindoles and β-ketoamides [49-54]. Recently, it has been shown that malonate derivatives such as malonate half thioesters and malonamides could be effectively used in various enantioselecive addition reactions [55-60]. In this context, Chen and co-workers reported a squaramide-catalyzed asymmetric nitroso aldol reaction of cyclic β-ketoesters and malonamate [61]. Inspired by this, we decided to investigate the use of malonamate in the asymmetric nitroso aldol reaction using thiourea catalysis. Herein, we report a novel nitroso aldol reaction of malonamates with nitrosoarene which provides facile access to chiral hydroxyamino malonamates having a quaternary carbon stereocenter (Scheme 1).
![[1860-5397-18-25-i1]](/bjoc/content/inline/1860-5397-18-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Catalytic asymmetric nitroso aldol reaction.
Scheme 1: Catalytic asymmetric nitroso aldol reaction.
Results and Discussion
Initially, we chose the Takemoto catalyst to promote the model reaction between methyl N-bromophenyl-α-methylmalonamate (1a) and nitrosobenzene (2a) in toluene at 25 °C. Pleasingly, the hydroxyamination reaction proceeded smoothly to give the nitroso aldol product 4a in 80% yield and 60% ee (Table 1, entry 1). The influence of temperature was evaluated subsequently, and the reaction conducted at 0 °C without altering other parameters gave a better result furnishing the product in 90% yield and 90% enantiomeric excess (Table 1, entry 2). Further lowering of the reaction temperature did not improve the enantioselectivity and slowed down the reaction (Table 1, entries 3 and 4). Our next attempts on the improvement of enantioselectivity focused on the screening of various bifunctional H-bonding catalysts, and in this regard, the reaction catalyzed by quinine-derived thiourea catalyst 3b furnished the product 4a in 55% yield and 71% ee (Table 1, entry 5). The other enantiomer was obtained when the reaction was carried out using the squaramide catalyst 3c, however, with low enantioselectivity (Table 1, entry 6). Disappointingly, the reaction catalyzed by ʟ-proline-derived catalysts gave very low enantioselectivity (Table 1, entries 7 and 8). Having identified Takemoto’s catalyst as the most efficient one for this transformation, our attempts to enhance the enantioselectivity centered on the variation of solvents. The reaction was screened using various polar and nonpolar solvents, and toluene was found suitable in terms of the reaction rate, yield, and enantioselectivity (Table 1, entries 9–13). Other solvents such as EtOAc, DCM, chloroform, and hexane provided 4a with moderate enantioselectivities, and the reaction failed when DMF was used as the solvent. When the catalyst loading was reduced to 10 and 5 mol %, the enantioselectivity remained reasonably good, but the reaction yield was substantially affected.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-25-i5.svg?max-width=637&scale=1.0)
|
||||||
| entry | cat. 3 | solvent | temperature (°C) | time (h) | yield (%)b | ee (%)c |
| 1 | 3a | toluene | 25 | 3 | 80 | 60 |
| 2 | 3a | toluene | 0 | 3 | 90 | 90 |
| 3 | 3a | toluene | −10 | 12 | 80 | 86 |
| 4 | 3a | toluene | −20 | 24 | 70 | 78 |
| 5 | 3b | toluene | 0 | 4 | 55 | 71 |
| 6 | 3c | toluene | 0 | 12 | 58 | −41 |
| 7 | 3d | toluene | 0 | 24 | 42 | 15 |
| 8 | 3e | toluene | 0 | 24 | 35 | 13 |
| 9 | 3a | DCM | 0 | 3 | 90 | 77 |
| 10 | 3a | EtOAc | 0 | 3 | 72 | 78 |
| 11 | 3a | CHCl3 | 0 | 3 | 72 | 66 |
| 12 | 3a | hexane | 0 | 3 | 54 | 73 |
| 13 | 3a | DMF | 0 | – | – | – |
| 14d | 3a | toluene | 0 | 5 | 71 | 84 |
| 15e | 3a | toluene | 0 | 8 | 70 | 86 |
aGeneral conditions: 1a (0.20 mmol), 2a (0.24 mmol), 3 (0.04 mmol), solvent (3.0 mL). bIsolated yield after silica gel column chromatography. cDetermined by chiral HPLC analysis. dThe reaction conducted using 10 mol % of the catalyst. eThe reaction conducted using 5 mol % of the catalyst.
Having identified the optimal reaction conditions, we proceeded to evaluate the generality of this nitroso aldol reaction with respect to the amide component of malonamate (Scheme 2). Pleasingly, our strategy was found to be operational with malonamates bearing electronically different substituents such as halo, nitro, acetyl, and alkyl at the para-position of the phenyl ring and the corresponding oxyaminated products were obtained in excellent yields and good enantioselectivities (4a–f). The single crystal X-ray analysis of the product 4a established the absolute stereochemistry which was found to be S (Figure 1) [62]. There was a significant drop in the enantioselectivity when malonamates bearing substitutions at meta- and ortho-positions were used, except for the reaction carried out using methoxy substitution at the 3-position where the corresponding oxyaminated product was obtained in 75% yield and 80% ee (4g–j). The disubstituted malonamates underwent facile oxyamination, and the products were obtained in excellent yields and enantioselectivities (4k, 4l). Of note, the reaction was also feasible with heterocyclic malonamate, albeit with moderate yield and enantioselectivity (4m). Aliphatic malonamate was also found viable for this transformation, giving the oxyaminated product in good yield and moderate enantioselectivity (4n).
![[1860-5397-18-25-i2]](/bjoc/content/inline/1860-5397-18-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Variation of the amide moiety of malonamates. General conditions: 1 (0.20 mmol), 2a (0.24 mmol), 3a (0.04 mmol), toluene (3.0 mL). Yields refer to isolated yields after silica gel column chromatography. Enantioselectivities were determined by chiral HPLC analysis.
Scheme 2: Variation of the amide moiety of malonamates. General conditions: 1 (0.20 mmol), 2a (0.24 mmol), 3a...
![[1860-5397-18-25-1]](/bjoc/content/figures/1860-5397-18-25-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP diagram drawn with 30% ellipsoid probability for non-H atoms of the crystal structure of chiral compound 4a determined at 293 K. The absolute configuration of C7 is S.
Figure 1: ORTEP diagram drawn with 30% ellipsoid probability for non-H atoms of the crystal structure of chir...
Subsequently, the scope of the transformation was investigated with various alkyl esters of malonamate (Scheme 3). In addition to the methyl ester, the reaction was found to proceed smoothly with ethyl, tert-butyl, and p-methoxybenzyl esters of malonamate to furnish the oxyaminated products in good yields and moderate to good enantioselectivities (4o–u). Pleasingly, the reactions carried out using m-methoxybenzyl and 3,4,5-trimethoxybenzyl esters of malonamate afforded the corresponding products in good yields and enantioselectivity (4v, 4w). The scope was further expanded by carrying out a reaction using o-methylnitrosobenzene, and in this case, the reaction proceeded smoothly to afford the corresponding oxyaminated product, albeit with low enantioselectivity (4x, 4y). Disappointingly, the reactions carried out by varying the α-substitution did not afford the desired product.
![[1860-5397-18-25-i3]](/bjoc/content/inline/1860-5397-18-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Variation of ester moiety of malonamates and nitrosoarenes. General conditions: 1 (0.20 mmol), 2a (0.24 mmol), 3a (0.04 mmol), toluene (3.0 mL). Yields refer to isolated yields after silica gel column chromatography. Enantioselectivities were determined by chiral HPLC analysis. aReaction run for 6 h.
Scheme 3: Variation of ester moiety of malonamates and nitrosoarenes. General conditions: 1 (0.20 mmol), 2a (...
In order to demonstrate the synthetic utility of the oxyaminated compounds, the reductive cleavage of the N–O bond was attempted under Zn/AcOH conditions. Pleasingly, the reaction afforded the aniline derivative in good yield, albeit with a considerable diminishment in the ee (Scheme 4).
![[1860-5397-18-25-i4]](/bjoc/content/inline/1860-5397-18-25-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on the literature reports and the stereochemical outcome, a plausible transition state is proposed, as shown in Figure 2. The activation of nitrosobenzene was achieved by the intramolecular hydrogen-bonding of the thiourea moiety with the oxygen of the nitrosobenzene. The tertiary amine, present in the catalyst acts as a base in assisting the deprotonation of the highly acidic malonamate to generate the corresponding enolate. Subsequently, a face-selective nucleophilic addition of the enolate to nitroso selective takes place to afford the nitroso aldol product.
![[1860-5397-18-25-2]](/bjoc/content/figures/1860-5397-18-25-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Proposed transition state for the nitroso aldol reaction.
Figure 2: Proposed transition state for the nitroso aldol reaction.
Conclusion
In summary, an efficient organocatalytic asymmetric nitroso aldol reaction of α-methylmalonamate has been reported. The reaction utilizes the well-known Takemoto catalyst, and this protocol demonstrates for the first time the use of malonamate as a pro-nucleophile in an enantioselective addition reaction. The mild reaction conditions allow the use of various functionalized malonamates. Given the importance of highly functionalized α-amino acid derivatives, the present strategy could be useful in generating a wide range of α-oxyamino malonamates which may serve as a potential platform for the synthesis of medicinally relevant structural units.
Experimental
General experimental procedure for the thiourea-catalyzed nitroso aldol reaction of malonamates: To an oven-dried round-bottomed flask equipped with a magnetic stirring bar were added α-methylmalonamate 1a (57 mg, 0.20 mmol, 1 equiv), nitrosobenzene 2a (26 mg, 0.24 mmol, 1.2 equiv) and (R,R)-TUC 3a (17 mg, 0.04 mmol, 0.2 equiv). Then, the round-bottomed flask was sealed, evacuated, and backfilled with nitrogen. The mixture was dissolved in 3 mL of anhydrous toluene and was kept stirring at 0 °C for the specified time. After the completion of the reaction, as indicated by TLC, the solvent was evaporated and the residue extracted using ethyl acetate and water. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was purified using column chromatography (100–200 mesh silica gel) using EtOAc/hexane as the eluent to afford product 4a as white solid (71 mg, 90%). Rf 0.20 (EtOAc/hexane 3:7); mp 115–117 °C; 1H NMR (400 MHz, CDCl3) δ 9.04 (s, 1H), 7.47–7.44 (m, 5H), 7.31–7.26 (m, 2H), 7.20–7.14 (m, 3H), 3.81 (s, 3H), 1.59 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3) δ 171.5 (C), 167.0 (C), 147.1 (C), 136.5 (C), 132.1 (CH), 132.1(CH), 128.9 (CH), 128.9 (CH), 126.1 (CH), 122.2 (CH), 122.2 (CH), 121.6 (CH), 121.6 (CH), 117.4 (C), 76.8 (C), 53.5 (CH3), 17.9 (CH3) ppm. The enantiomeric excess was determined by HPLC on a Chiralpak IC column (hexane/ethanol 90:10 v/v, flow rate 1.0 mL/min, 254 nm, τminor = 13.0 min, τmajor = 13.9 min, 90% ee). HRMS (m/z): [M + Na]+ calcd for C17H17BrN2NaO4+, 415.0264; found, 415.0278.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, complete characterization data for all compounds, single-crystal X-ray data of 4a, copies of NMR spectra, and HPLC chromatograms. | ||
| Format: PDF | Size: 5.8 MB | Download |
References
-
Huang, J.; Chen, Z.; Yuan, J.; Peng, Y. Asian J. Org. Chem. 2016, 5, 951–960. doi:10.1002/ajoc.201600242
Return to citation in text: [1] -
Yamamoto, H.; Momiyama, N. Chem. Commun. 2005, 3514. doi:10.1039/b503212c
Return to citation in text: [1] -
Janey, J. M. Angew. Chem., Int. Ed. 2005, 44, 4292–4300. doi:10.1002/anie.200462314
Return to citation in text: [1] -
Merino, P.; Tejero, T. Angew. Chem., Int. Ed. 2004, 43, 2995–2997. doi:10.1002/anie.200301760
Return to citation in text: [1] -
The Chemistry of Amino, Nitroso, Nitro and Related Groups. In The Chemistry of Functional Groups; Patai, S., Ed.; John Wiley & Sons: Chichester, United Kingdom, 1996. doi:10.1002/047085720x
Return to citation in text: [1] -
Yamamoto, H.; Kawasaki, M. Bull. Chem. Soc. Jpn. 2007, 80, 595–607. doi:10.1246/bcsj.80.595
Return to citation in text: [1] -
Lee, J.; Chen, L.; West, A. H.; Richter-Addo, G. B. Chem. Rev. 2002, 102, 1019–1066. doi:10.1021/cr0000731
Return to citation in text: [1] -
Zuman, P.; Shah, B. Chem. Rev. 1994, 94, 1621–1641. doi:10.1021/cr00030a007
Return to citation in text: [1] -
Baidya, M.; Yamamoto, H. Synthesis 2013, 45, 1931–1938. doi:10.1055/s-0033-1339175
Return to citation in text: [1] -
Malkov, A. V. Chem. Heterocycl. Compd. 2012, 48, 39–43. doi:10.1007/s10593-012-0966-6
Return to citation in text: [1] -
Adam, W.; Krebs, O. Chem. Rev. 2003, 103, 4131–4146. doi:10.1021/cr030004x
Return to citation in text: [1] -
Bodnar, B. S.; Miller, M. J. Angew. Chem., Int. Ed. 2011, 50, 5630–5647. doi:10.1002/anie.201005764
Return to citation in text: [1] -
Jana, C. K.; Studer, A. Angew. Chem., Int. Ed. 2007, 46, 6542–6544. doi:10.1002/anie.200701631
Return to citation in text: [1] -
Yamamoto, Y.; Yamamoto, H. Eur. J. Org. Chem. 2006, 2031–2043. doi:10.1002/ejoc.200500847
Return to citation in text: [1] -
Leach, A. G.; Houk, K. N. Chem. Commun. 2002, 1243–1255. doi:10.1039/b111251c
Return to citation in text: [1] -
Vogt, P. F.; Miller, M. J. Tetrahedron 1998, 54, 1317–1348. doi:10.1016/s0040-4020(97)10072-2
Return to citation in text: [1] -
Waldmann, H. Synthesis 1994, 535–551. doi:10.1055/s-1994-25519
Return to citation in text: [1] -
Streith, J.; Defoin, A. Synthesis 1994, 1107–1117. doi:10.1055/s-1994-25647
Return to citation in text: [1] -
Ramakrishna, I.; Sahoo, H.; Baidya, M. Chem. Commun. 2016, 52, 3215–3218. doi:10.1039/c5cc10102f
Return to citation in text: [1] -
Maji, B.; Yamamoto, H. Angew. Chem., Int. Ed. 2014, 53, 8714–8717. doi:10.1002/anie.201311069
Return to citation in text: [1] -
Shen, K.; Liu, X.; Wang, G.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2011, 50, 4684–4688. doi:10.1002/anie.201100758
Return to citation in text: [1] -
Yanagisawa, A.; Takeshita, S.; Izumi, Y.; Yoshida, K. J. Am. Chem. Soc. 2010, 132, 5328–5329. doi:10.1021/ja910588w
Return to citation in text: [1] -
Momiyama, N.; Yamamoto, H. Org. Lett. 2002, 4, 3579–3582. doi:10.1021/ol026443k
Return to citation in text: [1] -
Merino, P.; Tejero, T.; Delso, I.; Matute, R. Synthesis 2016, 48, 653–676. doi:10.1055/s-0035-1561505
Return to citation in text: [1] -
Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2003, 125, 6038–6039. doi:10.1021/ja0298702
Return to citation in text: [1] -
Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2004, 126, 5360–5361. doi:10.1021/ja039103i
Return to citation in text: [1] -
Maji, B.; Yamamoto, H. Angew. Chem., Int. Ed. 2014, 53, 14472–14475. doi:10.1002/anie.201408893
Return to citation in text: [1] -
Yu, C.; Song, A.; Zhang, F.; Wang, W. ChemCatChem 2014, 6, 1863–1865. doi:10.1002/cctc.201402121
Return to citation in text: [1] -
Frazier, C. P.; Sandoval, D.; Palmer, L. I.; Read de Alaniz, J. Chem. Sci. 2013, 4, 3857. doi:10.1039/c3sc51658j
Return to citation in text: [1] -
Baidya, M.; Griffin, K. A.; Yamamoto, H. J. Am. Chem. Soc. 2012, 134, 18566–18569. doi:10.1021/ja309311z
Return to citation in text: [1] -
Chen, W.; Wang, Y.; Mi, X.; Luo, S. Org. Lett. 2019, 21, 8178–8182. doi:10.1021/acs.orglett.9b02636
Return to citation in text: [1] -
Ding, X.; Tang, W.; Zhu, C.; Cheng, Y. Adv. Synth. Catal. 2010, 352, 108–112. doi:10.1002/adsc.200900652
Return to citation in text: [1] -
Lu, M.; Zhu, D.; Lu, Y.; Zeng, X.; Tan, B.; Xu, Z.; Zhong, G. J. Am. Chem. Soc. 2009, 131, 4562–4563. doi:10.1021/ja8088907
Return to citation in text: [1] -
Huang, K.; Huang, Z.-Z.; Li, X.-L. J. Org. Chem. 2006, 71, 8320–8323. doi:10.1021/jo061507g
Return to citation in text: [1] -
Kumarn, S.; Shaw, D. M.; Longbottom, D. A.; Ley, S. V. Org. Lett. 2005, 7, 4189–4191. doi:10.1021/ol051577u
Return to citation in text: [1] -
Sundén, H.; Dahlin, N.; Ibrahem, I.; Adolfsson, H.; Córdova, A. Tetrahedron Lett. 2005, 46, 3385–3389. doi:10.1016/j.tetlet.2005.03.085
Return to citation in text: [1] -
Ramakrishna, I.; Ramaraju, P.; Baidya, M. Org. Lett. 2018, 20, 1023–1026. doi:10.1021/acs.orglett.7b03968
Return to citation in text: [1] -
Lin, H.; Tan, Y.; Sun, X.-W.; Lin, G.-Q. Org. Lett. 2012, 14, 3818–3821. doi:10.1021/ol301218x
Return to citation in text: [1] -
Font, D.; Bastero, A.; Sayalero, S.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2007, 9, 1943–1946. doi:10.1021/ol070526p
Return to citation in text: [1] -
Ramachary, D. B.; Barbas, C. F., III. Org. Lett. 2005, 7, 1577–1580. doi:10.1021/ol050246e
Return to citation in text: [1] -
Zhong, G. Angew. Chem., Int. Ed. 2003, 42, 4247–4250. doi:10.1002/anie.200352097
Return to citation in text: [1] -
Brown, S. P.; Brochu, M. P.; Sinz, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2003, 125, 10808–10809. doi:10.1021/ja037096s
Return to citation in text: [1] -
Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M. Tetrahedron Lett. 2003, 44, 8293–8296. doi:10.1016/j.tetlet.2003.09.057
Return to citation in text: [1] -
Kano, T.; Shirozu, F.; Maruoka, K. J. Am. Chem. Soc. 2013, 135, 18036–18039. doi:10.1021/ja4099627
Return to citation in text: [1] -
Mielgo, A.; Velilla, I.; Gómez-Bengoa, E.; Palomo, C. Chem. – Eur. J. 2010, 16, 7496–7502. doi:10.1002/chem.201000376
Return to citation in text: [1] -
Kano, T.; Yamamoto, A.; Shirozu, F.; Maruoka, K. Synthesis 2009, 1557–1563. doi:10.1055/s-0029-1216635
Return to citation in text: [1] -
López-Cantarero, J.; Cid, M. B.; Poulsen, T. B.; Bella, M.; Ruano, J. L. G.; Jørgensen, K. A. J. Org. Chem. 2007, 72, 7062–7065. doi:10.1021/jo071186o
Return to citation in text: [1] -
Kano, T.; Ueda, M.; Takai, J.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 6046–6047. doi:10.1021/ja0604515
Return to citation in text: [1] -
Zhang, X.-X.; Gao, Y.; Hu, X.-S.; Ji, C.-B.; Liu, Y.-L.; Yu, J.-S. Adv. Synth. Catal. 2020, 362, 4763–4793. doi:10.1002/adsc.202000966
Return to citation in text: [1] -
Ji, S.-P.; Liu, L.-W.; Chen, F.; Ren, H.-X.; Yang, Y.; Zhang, Z.-B.; Peng, L.; Wang, L.-X. Eur. J. Org. Chem. 2016, 5437–5444. doi:10.1002/ejoc.201600884
Return to citation in text: [1] -
Mailhol, D.; Castillo, J.-C.; Mohanan, K.; Abonia, R.; Coquerel, Y.; Rodriguez, J. ChemCatChem 2013, 5, 1192–1199. doi:10.1002/cctc.201200723
Return to citation in text: [1] -
Jia, L.-N.; Huang, J.; Peng, L.; Wang, L.-L.; Bai, J.-F.; Tian, F.; He, G.-Y.; Xu, X.-Y.; Wang, L.-X. Org. Biomol. Chem. 2012, 10, 236–239. doi:10.1039/c1ob06413d
Return to citation in text: [1] -
Companyó, X.; Valero, G.; Pineda, O.; Calvet, T.; Font-Bardía, M.; Moyano, A.; Rios, R. Org. Biomol. Chem. 2012, 10, 431–439. doi:10.1039/c1ob06503c
Return to citation in text: [1] -
Han, X.; Kwiatkowski, J.; Xue, F.; Huang, K.-W.; Lu, Y. Angew. Chem., Int. Ed. 2009, 48, 7604–7607. doi:10.1002/anie.200903635
Return to citation in text: [1] -
Saadi, J.; Wennemers, H. Nat. Chem. 2016, 8, 276–280. doi:10.1038/nchem.2437
Return to citation in text: [1] -
Cosimi, E.; Engl, O. D.; Saadi, J.; Ebert, M.-O.; Wennemers, H. Angew. Chem., Int. Ed. 2016, 55, 13127–13131. doi:10.1002/anie.201607146
Return to citation in text: [1] -
Bahlinger, A.; Fritz, S. P.; Wennemers, H. Angew. Chem., Int. Ed. 2014, 53, 8779–8783. doi:10.1002/anie.201310532
Return to citation in text: [1] -
Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307
Return to citation in text: [1] -
Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536
Return to citation in text: [1] -
Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187
Return to citation in text: [1] -
Yang, H.-J.; Dai, L.; Yang, S.-Q.; Chen, F.-E. Synlett 2012, 23, 948–950. doi:10.1055/s-0031-1290613
Return to citation in text: [1] -
Crystallographic data for 4a: CCDC 1898632 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1]
| 1. | Huang, J.; Chen, Z.; Yuan, J.; Peng, Y. Asian J. Org. Chem. 2016, 5, 951–960. doi:10.1002/ajoc.201600242 |
| 2. | Yamamoto, H.; Momiyama, N. Chem. Commun. 2005, 3514. doi:10.1039/b503212c |
| 3. | Janey, J. M. Angew. Chem., Int. Ed. 2005, 44, 4292–4300. doi:10.1002/anie.200462314 |
| 4. | Merino, P.; Tejero, T. Angew. Chem., Int. Ed. 2004, 43, 2995–2997. doi:10.1002/anie.200301760 |
| 5. | The Chemistry of Amino, Nitroso, Nitro and Related Groups. In The Chemistry of Functional Groups; Patai, S., Ed.; John Wiley & Sons: Chichester, United Kingdom, 1996. doi:10.1002/047085720x |
| 19. | Ramakrishna, I.; Sahoo, H.; Baidya, M. Chem. Commun. 2016, 52, 3215–3218. doi:10.1039/c5cc10102f |
| 20. | Maji, B.; Yamamoto, H. Angew. Chem., Int. Ed. 2014, 53, 8714–8717. doi:10.1002/anie.201311069 |
| 21. | Shen, K.; Liu, X.; Wang, G.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2011, 50, 4684–4688. doi:10.1002/anie.201100758 |
| 22. | Yanagisawa, A.; Takeshita, S.; Izumi, Y.; Yoshida, K. J. Am. Chem. Soc. 2010, 132, 5328–5329. doi:10.1021/ja910588w |
| 23. | Momiyama, N.; Yamamoto, H. Org. Lett. 2002, 4, 3579–3582. doi:10.1021/ol026443k |
| 61. | Yang, H.-J.; Dai, L.; Yang, S.-Q.; Chen, F.-E. Synlett 2012, 23, 948–950. doi:10.1055/s-0031-1290613 |
| 12. | Bodnar, B. S.; Miller, M. J. Angew. Chem., Int. Ed. 2011, 50, 5630–5647. doi:10.1002/anie.201005764 |
| 13. | Jana, C. K.; Studer, A. Angew. Chem., Int. Ed. 2007, 46, 6542–6544. doi:10.1002/anie.200701631 |
| 14. | Yamamoto, Y.; Yamamoto, H. Eur. J. Org. Chem. 2006, 2031–2043. doi:10.1002/ejoc.200500847 |
| 15. | Leach, A. G.; Houk, K. N. Chem. Commun. 2002, 1243–1255. doi:10.1039/b111251c |
| 16. | Vogt, P. F.; Miller, M. J. Tetrahedron 1998, 54, 1317–1348. doi:10.1016/s0040-4020(97)10072-2 |
| 17. | Waldmann, H. Synthesis 1994, 535–551. doi:10.1055/s-1994-25519 |
| 18. | Streith, J.; Defoin, A. Synthesis 1994, 1107–1117. doi:10.1055/s-1994-25647 |
| 62. | Crystallographic data for 4a: CCDC 1898632 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 9. | Baidya, M.; Yamamoto, H. Synthesis 2013, 45, 1931–1938. doi:10.1055/s-0033-1339175 |
| 10. | Malkov, A. V. Chem. Heterocycl. Compd. 2012, 48, 39–43. doi:10.1007/s10593-012-0966-6 |
| 11. | Adam, W.; Krebs, O. Chem. Rev. 2003, 103, 4131–4146. doi:10.1021/cr030004x |
| 49. | Zhang, X.-X.; Gao, Y.; Hu, X.-S.; Ji, C.-B.; Liu, Y.-L.; Yu, J.-S. Adv. Synth. Catal. 2020, 362, 4763–4793. doi:10.1002/adsc.202000966 |
| 50. | Ji, S.-P.; Liu, L.-W.; Chen, F.; Ren, H.-X.; Yang, Y.; Zhang, Z.-B.; Peng, L.; Wang, L.-X. Eur. J. Org. Chem. 2016, 5437–5444. doi:10.1002/ejoc.201600884 |
| 51. | Mailhol, D.; Castillo, J.-C.; Mohanan, K.; Abonia, R.; Coquerel, Y.; Rodriguez, J. ChemCatChem 2013, 5, 1192–1199. doi:10.1002/cctc.201200723 |
| 52. | Jia, L.-N.; Huang, J.; Peng, L.; Wang, L.-L.; Bai, J.-F.; Tian, F.; He, G.-Y.; Xu, X.-Y.; Wang, L.-X. Org. Biomol. Chem. 2012, 10, 236–239. doi:10.1039/c1ob06413d |
| 53. | Companyó, X.; Valero, G.; Pineda, O.; Calvet, T.; Font-Bardía, M.; Moyano, A.; Rios, R. Org. Biomol. Chem. 2012, 10, 431–439. doi:10.1039/c1ob06503c |
| 54. | Han, X.; Kwiatkowski, J.; Xue, F.; Huang, K.-W.; Lu, Y. Angew. Chem., Int. Ed. 2009, 48, 7604–7607. doi:10.1002/anie.200903635 |
| 6. | Yamamoto, H.; Kawasaki, M. Bull. Chem. Soc. Jpn. 2007, 80, 595–607. doi:10.1246/bcsj.80.595 |
| 7. | Lee, J.; Chen, L.; West, A. H.; Richter-Addo, G. B. Chem. Rev. 2002, 102, 1019–1066. doi:10.1021/cr0000731 |
| 8. | Zuman, P.; Shah, B. Chem. Rev. 1994, 94, 1621–1641. doi:10.1021/cr00030a007 |
| 55. | Saadi, J.; Wennemers, H. Nat. Chem. 2016, 8, 276–280. doi:10.1038/nchem.2437 |
| 56. | Cosimi, E.; Engl, O. D.; Saadi, J.; Ebert, M.-O.; Wennemers, H. Angew. Chem., Int. Ed. 2016, 55, 13127–13131. doi:10.1002/anie.201607146 |
| 57. | Bahlinger, A.; Fritz, S. P.; Wennemers, H. Angew. Chem., Int. Ed. 2014, 53, 8779–8783. doi:10.1002/anie.201310532 |
| 58. | Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Chem. – Eur. J. 2013, 19, 15856–15860. doi:10.1002/chem.201303307 |
| 59. | Ricci, A.; Pettersen, D.; Bernardi, L.; Fini, F.; Fochi, M.; Herrera, R. P.; Sgarzani, V. Adv. Synth. Catal. 2007, 349, 1037–1040. doi:10.1002/adsc.200600536 |
| 60. | Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187 |
| 27. | Maji, B.; Yamamoto, H. Angew. Chem., Int. Ed. 2014, 53, 14472–14475. doi:10.1002/anie.201408893 |
| 28. | Yu, C.; Song, A.; Zhang, F.; Wang, W. ChemCatChem 2014, 6, 1863–1865. doi:10.1002/cctc.201402121 |
| 29. | Frazier, C. P.; Sandoval, D.; Palmer, L. I.; Read de Alaniz, J. Chem. Sci. 2013, 4, 3857. doi:10.1039/c3sc51658j |
| 30. | Baidya, M.; Griffin, K. A.; Yamamoto, H. J. Am. Chem. Soc. 2012, 134, 18566–18569. doi:10.1021/ja309311z |
| 37. | Ramakrishna, I.; Ramaraju, P.; Baidya, M. Org. Lett. 2018, 20, 1023–1026. doi:10.1021/acs.orglett.7b03968 |
| 38. | Lin, H.; Tan, Y.; Sun, X.-W.; Lin, G.-Q. Org. Lett. 2012, 14, 3818–3821. doi:10.1021/ol301218x |
| 39. | Font, D.; Bastero, A.; Sayalero, S.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2007, 9, 1943–1946. doi:10.1021/ol070526p |
| 40. | Ramachary, D. B.; Barbas, C. F., III. Org. Lett. 2005, 7, 1577–1580. doi:10.1021/ol050246e |
| 41. | Zhong, G. Angew. Chem., Int. Ed. 2003, 42, 4247–4250. doi:10.1002/anie.200352097 |
| 42. | Brown, S. P.; Brochu, M. P.; Sinz, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2003, 125, 10808–10809. doi:10.1021/ja037096s |
| 43. | Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M. Tetrahedron Lett. 2003, 44, 8293–8296. doi:10.1016/j.tetlet.2003.09.057 |
| 26. | Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2004, 126, 5360–5361. doi:10.1021/ja039103i |
| 44. | Kano, T.; Shirozu, F.; Maruoka, K. J. Am. Chem. Soc. 2013, 135, 18036–18039. doi:10.1021/ja4099627 |
| 45. | Mielgo, A.; Velilla, I.; Gómez-Bengoa, E.; Palomo, C. Chem. – Eur. J. 2010, 16, 7496–7502. doi:10.1002/chem.201000376 |
| 46. | Kano, T.; Yamamoto, A.; Shirozu, F.; Maruoka, K. Synthesis 2009, 1557–1563. doi:10.1055/s-0029-1216635 |
| 47. | López-Cantarero, J.; Cid, M. B.; Poulsen, T. B.; Bella, M.; Ruano, J. L. G.; Jørgensen, K. A. J. Org. Chem. 2007, 72, 7062–7065. doi:10.1021/jo071186o |
| 48. | Kano, T.; Ueda, M.; Takai, J.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 6046–6047. doi:10.1021/ja0604515 |
| 25. | Momiyama, N.; Yamamoto, H. J. Am. Chem. Soc. 2003, 125, 6038–6039. doi:10.1021/ja0298702 |
| 24. | Merino, P.; Tejero, T.; Delso, I.; Matute, R. Synthesis 2016, 48, 653–676. doi:10.1055/s-0035-1561505 |
| 31. | Chen, W.; Wang, Y.; Mi, X.; Luo, S. Org. Lett. 2019, 21, 8178–8182. doi:10.1021/acs.orglett.9b02636 |
| 32. | Ding, X.; Tang, W.; Zhu, C.; Cheng, Y. Adv. Synth. Catal. 2010, 352, 108–112. doi:10.1002/adsc.200900652 |
| 33. | Lu, M.; Zhu, D.; Lu, Y.; Zeng, X.; Tan, B.; Xu, Z.; Zhong, G. J. Am. Chem. Soc. 2009, 131, 4562–4563. doi:10.1021/ja8088907 |
| 34. | Huang, K.; Huang, Z.-Z.; Li, X.-L. J. Org. Chem. 2006, 71, 8320–8323. doi:10.1021/jo061507g |
| 35. | Kumarn, S.; Shaw, D. M.; Longbottom, D. A.; Ley, S. V. Org. Lett. 2005, 7, 4189–4191. doi:10.1021/ol051577u |
| 36. | Sundén, H.; Dahlin, N.; Ibrahem, I.; Adolfsson, H.; Córdova, A. Tetrahedron Lett. 2005, 46, 3385–3389. doi:10.1016/j.tetlet.2005.03.085 |
© 2022 Gupta et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.