
Abstract
Four new polyhydroxylated steroids lobophysterols E–H (1–4), together with three known compounds (5–7), were isolated from the soft coral Lobophytum pauciflorum collected at Xisha Island, China. The structures of the new compounds were elucidated by extensive spectroscopic analysis and comparison with NMR data of structurally related compounds reported in the literature. The absolute configuration of 1–3 was determined by X-ray diffraction. All the compounds have assessed the cytotoxicity against HL-60, K562, and Hela cells. Compound 1 showed weak cytotoxicity against K562 cells with an IC50 value of 19.03 μM. In addition, compound 1 also showed a moderate anti-inflammatory effect in zebrafish.
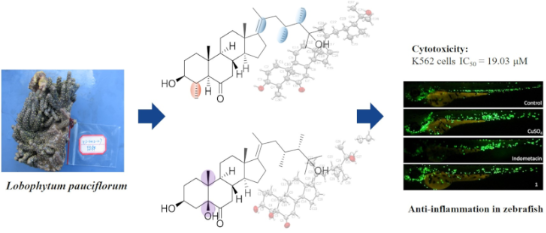
Graphical Abstract
Introduction
The unique and complicated marine environment makes soft corals a treasure-house of secondary metabolites with great variety and bioactivities. Previous chemical studies on soft corals Lobophytum, widely distributed in the world, resulted in the identification of lobane diterpene [1], cembranoids [2], and biscembranoids [3] with different bioactivities. Moreover, structurally specific steroids containing side chains with 23,24-dimethyl groups and (17)20E double bond, have been reported to be frequently isolated from soft corals of this genus, some of them exhibited anti-inflammatory [4], cytotoxic [5,6], and antibacterial activities [7].
To search for bioactive natural products, we have investigated the chemical constituents of the soft coral Lobophytum pauciflorum, collected from Xisha Island in the South China Sea. In the present paper, we describe the isolation of four new polyhydroxylated steroids lobophysterols E–H (1–4), together with three known compounds (5–7) (Figure 1). The structure of the new compounds was established by extensive spectroscopic analysis and comparing with the spectroscopic data of the previously reported structurally-related compounds. Compounds 1 and 2 represented rare examples of steroids with both 23,24-dimethyl groups and 17(20)E double bond. In particular, compound 1 also has a tetracyclic skeleton with a methyl group at C-4. The absolute configuration of 1–3 was determined by X-ray analysis. Herein, we report the isolation, structure elucidation, and bioactivities of these compounds.
![[1860-5397-18-42-1]](/bjoc/content/figures/1860-5397-18-42-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Compound 1 was isolated as a white powder. Its molecular formula was established as C30H50O3 by HRESIMS from the molecular ion peak at m/z 481.3648 [M + Na]+. The 1H NMR data (Table 1) showed 5 methyl singlets (δH 0.76, CH3-19; δH 0.84, CH3-18; δH 1.19, CH3-26; δH 1.19, CH3-27; δH 1.67, CH3-21), 3 methyl doublets (δH 0.81, CH3-29; δH 0.84, CH3-28; δH 1.00, CH3-30), an oxymethine (δH 3.13, 1H, m) and a series of methylene multiplets located between δH 1.25 and δH 2.40. The 13C NMR and DEPT spectra exhibited the presence of 30 carbon signals, including a carbonyl group (δC 210.0), a tetrasubstituted double bond (δC 144.4 and 124.2), one oxygenated sp3 secondary carbon (δC 76.3) and one oxygenated sp3 quaternary carbon (δC 74.2). As two of the six degrees of unsaturation were occupied by the double bond and carbonyl group, the remaining four unsaturations of 1 corresponded to a tetracyclic skeleton.
Table 1: 1H and 13C NMR data of compounds 1–4 (δ in ppm and J in Hz).
| No. | 1 | 2 | 3 | 4 | ||||
| δCa | δHb | δCc | δHd | δCa | δHb | δCa | δHb | |
| 1 | 36.9, CH2 |
1.80, m;
1.79, m |
25.9, CH2 |
1.79, m;
1.52, m |
25.0, CH2 |
1.79, m;
1.49, m |
36.5, CH2 |
1.22, m;
1.95, m |
| 2 | 30.3, CH2 | 1.83, m | 28.4, CH2 |
1.80, m;
1.66, m |
28.0, CH2 |
1.75, m;
1.66, m |
31.3, CH2 |
1.93, m;
1.61, m |
| 3 | 76.3, CH | 3.13, td (10.7, 4.6) | 67.1, CH | 4.06, m | 65.7, CH | 4.03, m | 70.7, CH | 3.68, m |
| 4 | 34.3, CH | 1.76, m | 38.0, CH2 | 2.45, dd (14.4, 3.5); 1.62, m | 37.3, CH2 |
2.26, m;
1.65, m |
42.0, CH2 |
2.40, m;
2.50, m(br.) |
| 5 | 64.0, CH | 2.08, m | 83.5, C | 82.1, C | 165.3, C | |||
| 6 | 210.0, C | 213.5, C | 212.9, C | 126.2, CH | 5.70, s | |||
| 7 | 48.4, CH2 |
2.06, m;
2.32, m |
43.0, CH2 |
2.41, m;
2.33, dd (14.1, 4.8) |
41.6, CH2 | 2.40, dd (14.1, 3.6); 2.22, m | 202.6, C | |
| 8 | 39.1, CH | 1.85, m | 37.7, CH | 1.83, m | 37.5, CH | 1.76, m | 45.6, CH | 2.23, m |
| 9 | 54.7, CH | 1.26, m | 43.7, CH | 1.94, m | 43.0, CH | 1.75, m | 50.1, CH | 1.53, m |
| 10 | 43.1, C | 45.2, C | 44.2, C | 38.5, C | ||||
| 11 | 22.1, CH2 |
1.36, m;
1.67, m |
23.1, CH2 |
1.49, m;
1.67, m |
21.8, CH2 |
1.36, m;
1.54, m |
21.4, CH2 | 1.57, m |
| 12 | 37.7, CH2 | 2.32, m | 38.8, CH2 | 2.39, m | 39.6, CH2 | 2.05, m | 38.8, CH2 |
1.14, m;
2.03, m |
| 13 | 45.2, C | 46.1, C | 43.3, C | 43.5, C | ||||
| 14 | 56.8, CH | 1.33, m | 57.7, CH | 1.50, m | 57.0, CH | 1.26, m | 49.8, CH | 1.32, m |
| 15 | 24.4, CH2 | 1.56, m | 25.2, CH2 |
1.64, m;
1.23, m |
24.1, CH2 |
1.54, m;
1.08, m |
26.8, CH2 |
2.42, m;
1.23, m |
| 16 | 29.9, CH2 |
2.15, m;
2.33, m |
30.8, CH2 |
2.22, m;
2.38, m |
28.0, CH2 |
1.87, m;
1.52, m |
29.0, CH2 |
1.40, m;
2.13, m |
| 17 | 144.4, C | 145.3, C | 55.9, C | 56.3, CH | 1.24, m | |||
| 18 | 16.6, CH3 | 0.84, s | 16.7, CH3 | 0.88, s | 12.1, CH3 | 0.65, s | 12.0, CH3 | 0.64, s |
| 19 | 14.0, CH3 | 0.76, s | 17.5, CH3 | 0.79, s | 17.2, CH3 | 0.74, s | 17.5, CH3 | 1.20, s |
| 20 | 124.2, C | 125.4, C | 36.3, CH | 40.2, CH | 0.84, m | |||
| 21 | 17.9, CH3 | 1.67, s | 18.1, CH3 | 1.72, s | 19.1, CH3 | 0.93, d (6.6) | 19.5, CH3 | 0.92, d, overlap |
| 22 | 44.3, CH2 |
1.77, m;
1.87, m |
45.4, CH2 |
1.80, m;
1.95, m |
34.9, CH2 | 1.50, m | 25.6, CH | 0.31, m |
| 23 | 30.2, CH | 2.06, m | 31.2, CH | 2.10, m | 28.1, CH2 |
0.77, m;
1.30, m |
24.2, CH | 0.53, m |
| 24 | 45.6, CH | 1.40, m | 46.4, CH | 1.46, m | 45.3, CH | 1.27, m | 45.1, CH | 0.52, m |
| 25 | 74.2, C | 74.5, C | 73.7, C | 33.0, CH | 1.65, m | |||
| 26 | 28.1, CH3 | 1.19, s | 27.7, CH3 | 1.16, s | 26.2, CH3 | 1.14, s | 20.9, CH3 | 0.88, d (6.9) |
| 27 | 28.3, CH3 | 1.19, s | 28.5, CH3 | 1.18, s | 27.5, CH3 | 1.16, s | 18.7, CH3 | 0.85, d (6.8) |
| 28 | 9.3, CH3 | 0.84, d (7.2) | 9.4, CH3 | 0.85, d (7.2) | 15.0, CH3 | 0.88, d (6.7) | 15.9, CH3 | 0.92, d, overlap |
| 29 | 15.7, CH3 | 0.81, d (6.8) | 16.0, CH3 | 0.82, d (6.8) | 10.7, CH2 | 0.12, m | ||
| 30 | 16.6, CH3 | 1.00, d (6.1) | ||||||
aMeasured at 125 MHz in CDCl3. bMeasured at 500 MHz in CDCl3. cMeasured at 125 MHz in CD3OD. dMeasured at 500 MHz in CD3OD.
The 1H,1H-COSY experiment (Figure 2) revealed the proton–proton correlations of H-1/H-2/H-3/H-4, H-7/H-8/H-9/H-11/H-12, H-8/H-14/H-15/H-16, and H-22/H-23/H-24/H-28/29. These data, together with the HMBC correlations (Figure 2) from H-19 to C-1/C-5/C-9/C-10, from H3-30 to C-4/C-5, from H-5/H-7 to C-6, from H3-18 to C-12/C-13/C-14/C-17, from H3-21 to C-17/C-20/C-22, and from H3-26 to C-24/C-25 confirmed the establishment of the carbon skeleton of the 23,24-dimethycholestane with a methyl group at C-4. Thus, the planar structure of 1 was established as shown in Figure 1.
![[1860-5397-18-42-2]](/bjoc/content/figures/1860-5397-18-42-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H,1H-COSY and selected key HMBC correlations of 1–4.
Figure 2: 1H,1H-COSY and selected key HMBC correlations of 1–4.
The relative configuration of 1 was deduced by the NOESY spectrum (Figure 3). The NOESY correlations of H-4 with H3-19, H-8 with H3-18 and H3-19 suggested the β-orientation of H-4, H-8, H3-18, and H3-19. Moreover, H3-30 showed NOESY correlations with H-3/H-5, H-5 with H-9, and H-9 with H-14 indicating the α-orientation of H-3, H-5, H-9, H-14, and H3-30. Furthermore, the NOESY correlations of H3-18 with H3-21 suggested the E geometry of Δ17(20). Finally, the absolute configuration of compound 1 was established by single-crystal X-ray diffraction analysis (Figure 4) carried out using Cu Kα radiation with a Flack parameter of 0.0(2).
![[1860-5397-18-42-3]](/bjoc/content/figures/1860-5397-18-42-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected NOESY correlations of compounds 1–4.
Figure 3: Selected NOESY correlations of compounds 1–4.
![[1860-5397-18-42-4]](/bjoc/content/figures/1860-5397-18-42-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: X-ray crystallographic analysis of compounds 1–3.
Figure 4: X-ray crystallographic analysis of compounds 1–3.
Compound 2, was isolated as a white powder with molecular formula C29H48O4, established by HREIMS at m/z 483.3446 [M + Na]+. The 1H and 13C NMR data (Table 1) of 2 exhibited very similar typical features of compound 1, the difference between the two compounds occurred in ring A: an OH was located at C-5, but a missing methyl group at C-4 in 2, which was in agreement with the 13C NMR spectrum and the molecular mass. The hydroxylation at C-5 was deduced from the HMBC correlations (Figure 2) from H3-19/H-4 to C-5. Moreover, the HMBC correlations found from H-4 to C-5/C-6, H-7 to C-6, and H3-18/H3-21 to C-17 confirmed the location of a ketone carbonyl at C-6 and the double bond at C-17, respectively. Thus, the planar structure of 2 was established as shown in Figure 1. The relative configuration of 2 was deduced by the cross-peaks shown by a 2D NOESY spectrum (Figure 3). The NOE correlations of H-8 with H3-18 and H3-19, and H-9 (δH 1.94) with H-14 (δH 1.50) indicated the β-orientation of H-8/H3-18/H3-19, while α-orientation of H-9/H-14. Furthermore, the NOESY correlation of H3-21 with H3-18 suggests the E geometry of ∆17(20). The absolute configuration of 2 was established by single-crystal X-ray diffraction analysis (Figure 4) carried out using Cu Kα radiation with a Flack parameter of 0.3(4).
Compound 3 was isolated as a white powder with molecular formula C28H48O4, established by HRESIMS at m/z 449.3626 [M + H]+. The 1H and 13C NMR data (Table 1) of 3 were very similar to those of its analog compound 2, showing the identical signals of the tetracyclic parent nucleus. The difference between the two compounds occurred in the side chain: A single bound was located at C-17/C-20 in 3 instead of the double bound in 2 and the absence of a methyl unit at C-23, in agreement with the 13C NMR spectrum and the molecular mass. The HMBC correlations (Figure 2) found from H3-21 to C-17/C-20/C-22, from H3-26 to C-25/C-27/C-24, and from H3-28 to C-24/C-25, together with the 1H,1H-COSY correlation (Figure 2) from H-22 to H-24 confirmed the side chain of compound 3. Thus, the planar structure of compound 3 was established, which was the same as the known compound (3β,5α)-25-trihydroxy-24S-methylcholestan-6-one [8]. The difference was the configuration of C-5, which was established as S by single-crystal X-ray diffraction analysis (Figure 4) carried out using Cu Kα radiation with a Flack parameter of −0.11(9). Thus, the absolute configuration of 3 was established.
Compound 4 was obtained as a yellow powder. Based on the HRESIMS data (m/z 427.3569 [M + H]+), the molecular formula was determined to be C29H46O2, 14 mass units less than compound 5 [6]. By comparing the NMR data (Table 1) of 4 and 5, it is obvious that they possess the same parent nucleus. The major difference between them was that the side chain of 4 lacks a methyl group at C-23, which can also be proved by the molecular mass. The concrete structure of the side chain was established by the COSY and HMBC correlations (Figure 2). The 1H,1H-COSY experiment revealed that the proton–proton correlation of H-17/H-20/H-21/H-22/H-23/H2-29. These data, together with the key HMBC correlations from H3-28 to C-23/C-24/C-25 and from H3-26/H3-27 to C-24/C-25, confirmed the structure of the side chain of compound 4. The NOESY correlations (Figure 3) from H-1a (δH 1.22) to H-3 and H-9, H-9 to H-14, H-1b (δH 1.95) to H3-19, H-8 to H3-18 and H3-19, H3-18 to H-20 indicated that H-3, H-9, H-14, and H-17 were orientated on α-face, while 3-OH, H-8, H3-18, and H3-19 were positioned on the β-face. Further, the NMR data of 4 for the side chain were almost identical to the known compound 8, demethylgorgosterol [9,10]. Thus, the structure of compound 4 was assigned as shown in Figure 1.
Biological activity
The cytotoxic activities of compounds 1–7 were evaluated against three cancer cell lines (HL-60, K562, and Hela), but only compound 1 exhibited weak cytotoxic activity against K562 cells with an IC50 value of 19.03 μM. The investigation of anti-inflammatory activities of lobophysterols E–H with classic transgenic fluorescent zebrafish models (Figure 5) showed that compound 1 exhibited moderate activity, with an inhibition rate of 32% (20 μM).
![[1860-5397-18-42-5]](/bjoc/content/figures/1860-5397-18-42-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Effects of compound 1 on the anti-inflammation of zebrafish internodes. ## Indicates that the CuSO4 model group has a very significant difference compared with the blank group (p < 0.01). * and ** indicate that sample groups have significant differences compared with the CuSO4 model group.
Figure 5: Effects of compound 1 on the anti-inflammation of zebrafish internodes. ## Indicates that the CuSO4...
Conclusion
In conclusion, four new steroids lobophysterols E–H (1–4) and three known analogs were isolated from the soft coral Lobophytum pauciflorum collected at Xisha Island, China. Compounds 1 and 2 represented rare examples of steroids featuring both 23,24-dimethyl groups and 17(20)E double bond. Moreover, compound 1 has a tetracyclic skeleton with a methyl group at C-4. The absolute configuration of 1–3 was determined by X-ray diffraction analyses. Compounds 1–7 were subjected to a cytotoxic activity evaluation against HL-60, K562, and Hela cells, only compound 1 exhibited weak cytotoxic activity against K562 cells with an IC50 value of 19.03 μM. In addition, compound 1 exhibited a moderate anti-inflammatory effect in zebrafish.
Experimental
General experimental procedures
NMR spectra were measured via an Agilent DD2-500 spectrometer (500 MHz for 1H NMR and 125 MHz for 13C NMR). Chemical shifts are reported in parts per million (ppm) with coupling constants (J) in hertz relative to the solvent peaks; δH 3.31 and δC 49.0 for CD3OD; δH 7.26 and δC 77.16 for CDCl3. HRESIMS data were surveyed on a Thermo LTQ-Orbitrap mass spectrometer. IR spectra were recorded on a Nicolet NEXUS 470 spectrophotometer using KBr pellets. UV spectra were recorded on a Jasco J-815 CD spectropolarimeter. Optical rotations were measured with a Jasco P-1020 polarimeter. Semi-preparative HPLC (Agilent Technologies 1260 Infinity II) equipped with a reversed-phase column ((YMC-packed C18, 5 µm, 250 × 10 mm, 2.0 mL/min) was used to purify samples. Silica gel (300–400 mesh, Qingdao) was used for column chromatography (CC), and precoated silica gel plates (GF254, Qingdao) were used for TLC.
Soft coral material
The soft coral Lobophytum pauciflorum was collected from Yongle Islands of Xisha Islands of the South China Sea in May 2012. The sample was identified by Pingjyun Sung, National Museum of Marine Biology and Aquarium (NMMBA), Checheng, Pingtung 944, Taiwan, China. The voucher specimen (No. XS-2012-27), frozen at −20 °C, was deposited at the School of Medicine and Pharmacy, Ocean University of China, P. R. China.
Extraction and isolation
The frozen bodies of Lobophytum pauciflorum (3.6 kg, wet weight; 1.1 kg, dry weight) were cut into pieces and exhaustively extracted with MeOH five times at room temperature. The solvent was removed under reduced pressure and the combined organic extract was desalted three times by anhydrous methanol. The desalted residue (110 g) was subjected to silica gel column chromatography (CC) eluted with two gradient systems, PE/acetone (1:0 to 1:1) and subsequently CH2Cl2/MeOH (15:1 to 1:1) to afford 8 fractions. Fraction 4 (14.6 g) was split (chromatographed on) by silica gel eluting with a gradient of PE/acetone (30:1 to 1:1) to give three subfractions (F41–F43). Subfraction F41 (2.1 g) was chromatographed over silica gel column (PE/acetone, 50:1 to 2:1) to give seven subfractions (F411–F417), F411 (330 mg) was purified by semi-preparative HPLC (ODS, 5 µm, 250 × 10 mm; methanol/water, 95:5, v/v; 2.0 mL/min) to afford compound 4 (2.3 mg), 5 (4.7 mg), 6 (15.5 mg) and 7 (10.2 mg). Fr.412 (210 mg) was chromatographed on semi-preparative HPLC (ODS, 5 µm, 250 × 10 mm; methanol/water, 85:15, v/v; 2.0 mL/min) to give compound 1 (2.5 mg), 2 (5.8 mg) and 3 (13.3 mg).
Identification of new compounds
Compound 1: colorless crystals; [α]D25 −19.28 (c 0.13, MeOH); IR (KBr) νmax: 3389, 2944, 2871, 1707, 1603, 1467, 1380 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Table 1; HRESIMS (m/z) [M + Na]+ calcd for C30H50O3Na, 481.3652; found, 481.3648.
Compound 2: colorless crystals; [α]D25 −12.77 (c 0.3, MeOH); IR (KBr) νmax: 3361, 2926, 2855, 1702, 1651, 1459, 1376 cm−1; 1H and 13C NMR data (CD3OD, 500 and 125 MHz) see Table 1; HRESIMS (m/z) [M + Na]+ calcd for C29H48O4Na, 483.3445; found, 483.3446.
Compound 3: colorless crystals; [α]D25 −18.36 (c 0.5, MeOH); IR (KBr) νmax: 3390, 2938, 1702, 1459, 1376 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Table 1; HRESIMS (m/z) [M + H]+ calcd for C28H49O4, 449.3625; found, 449.3626.
Compound 4: yelllow crystals; [α]D25 −27.83 (c 0.13, MeOH); IR (KBr) νmax: 3391, 2957, 2872, 1683, 1650, 1558, 1540, 1357 cm−1; 1H and 13C NMR data (CDCl3, 500 and 125 MHz) see Table 1; HRESIMS (m/z) [M + H]+ calcd for C29H47O2, 427.3571; found, 427.3569.
Cytotoxicity assays
In vitro cytotoxicity was determined by the MTT method against K562 (chronic myeloid leukemia) and HL-60 (human promyelocytic leukemia) cell lines, and by the SRB method against the Hela cell line.
Zebrafish maintenance
Adult zebrafish were cultivated by Qilu University of Technology (Jinan, China). Transgenic zebrafish [Tg: zlyz-EGFP and Tg (vegfr2: GFP)] expressing enhanced green fluorescent protein (EGFP) was used in this article. The conditions of the maintenance complied with guidelines of the Organization for Economic Co-operation and Development (OECD). The zebrafish were maintained under a 14/10 h light/dark cycle at the temperature (28 ± 0.5°C) in a closed flow-through system with charcoal-filtered tap water to ensure normal spawning.
CuSO4-induced model of zebrafish
In a manner similar to literature reference [11], healthy zebrafish larvae were selected into 24-well plates (n = 10/well) in a 2 mL final volume of embryo medium at 3 dpf and divided into five groups: a control group (fresh fish water), a model group: 40 μM CuSO4, a positive drug group: 50 μM indomethacin (Solarbio, China) and drug groups: 20 μM CuSO4 (Sigma-Aldrich, St. Louis, MO, USA) was added to the drug groups and incubated for 1 h after treatment with different compounds for 2 h. All treatments were performed in triplicate. Each zebrafish larva was photographed by a fluorescence microscope (AXIO, Zoom. V16), and the number of macrophages was counted by using the Image-Pro Plus software.
Supporting Information
| Supporting Information File 1: Crystal data and structure refinement for compounds 1–3 and NMR, MS, and IR spectra of compounds 1–4. | ||
| Format: PDF | Size: 5.8 MB | Download |
References
-
Ahmed, A. F.; Teng, W.-T.; Huang, C.-Y.; Dai, C.-F.; Hwang, T.-L.; Sheu, J.-H. Mar. Drugs 2017, 15, 300. doi:10.3390/md15100300
Return to citation in text: [1] -
Lu, Y.; Lin, Y.-C.; Wen, Z.-H.; Su, J.-H.; Sung, P.-J.; Hsu, C.-H.; Kuo, Y.-H.; Chiang, M. Y.; Dai, C.-F.; Sheu, J.-H. Tetrahedron 2010, 66, 7129–7135. doi:10.1016/j.tet.2010.06.094
Return to citation in text: [1] -
Yan, P.; Lv, Y.; van Ofwegen, L.; Proksch, P.; Lin, W. Org. Lett. 2010, 12, 2484–2487. doi:10.1021/ol100567d
Return to citation in text: [1] -
Huang, C.-Y.; Tseng, W.-R.; Ahmed, A. F.; Chiang, P.-L.; Tai, C.-J.; Hwang, T.-L.; Dai, C.-F.; Sheu, J.-H. Mar. Drugs 2018, 16, 93. doi:10.3390/md16030093
Return to citation in text: [1] -
Zhang, Q.; Liang, L.-F.; Miao, Z.-H.; Wu, B.; Guo, Y.-W. Steroids 2019, 141, 76–80. doi:10.1016/j.steroids.2018.11.015
Return to citation in text: [1] -
Thanh, N. V.; Ngoc, N. T.; Anh, H. L. T.; Thung, D. C.; Thao, D. T.; Cuong, N. X.; Nam, N. H.; Kiem, P. V.; Minh, C. V. J. Asian Nat. Prod. Res. 2016, 18, 938–944. doi:10.1080/10286020.2016.1173676
Return to citation in text: [1] [2] -
Cheng, Z.-B.; Xiao, H.; Fan, C.-Q.; Lu, Y.-N.; Zhang, G.; Yin, S. Steroids 2013, 78, 1353–1358. doi:10.1016/j.steroids.2013.10.004
Return to citation in text: [1] -
Putra, M. Y.; Bavestrello, G.; Cerrano, C.; Renga, B.; D’Amore, C.; Fiorucci, S.; Fattorusso, E.; Taglialatela-Scafati, O. Steroids 2012, 77, 433–440. doi:10.1016/j.steroids.2011.12.026
Return to citation in text: [1] -
Kobayashi, M.; Mrishna, M. M.; Haribabu, B.; Anjaneyulu, V. Chem. Pharm. Bull. 1993, 41, 87–89. doi:10.1248/cpb.41.87
Return to citation in text: [1] -
Ling, N. C.; Hale, R. L.; Djerassi, C. J. Am. Chem. Soc. 1970, 92, 5281–5282. doi:10.1021/ja00720a082
Return to citation in text: [1] -
Gui, Y.-H.; Liu, L.; Wu, W.; Zhang, Y.; Jia, Z.-L.; Shi, Y.-P.; Kong, H.-T.; Liu, K.-C.; Jiao, W.-H.; Lin, H.-W. Bioorg. Chem. 2020, 94, 103435. doi:10.1016/j.bioorg.2019.103435
Return to citation in text: [1]
| 1. | Ahmed, A. F.; Teng, W.-T.; Huang, C.-Y.; Dai, C.-F.; Hwang, T.-L.; Sheu, J.-H. Mar. Drugs 2017, 15, 300. doi:10.3390/md15100300 |
| 5. | Zhang, Q.; Liang, L.-F.; Miao, Z.-H.; Wu, B.; Guo, Y.-W. Steroids 2019, 141, 76–80. doi:10.1016/j.steroids.2018.11.015 |
| 6. | Thanh, N. V.; Ngoc, N. T.; Anh, H. L. T.; Thung, D. C.; Thao, D. T.; Cuong, N. X.; Nam, N. H.; Kiem, P. V.; Minh, C. V. J. Asian Nat. Prod. Res. 2016, 18, 938–944. doi:10.1080/10286020.2016.1173676 |
| 4. | Huang, C.-Y.; Tseng, W.-R.; Ahmed, A. F.; Chiang, P.-L.; Tai, C.-J.; Hwang, T.-L.; Dai, C.-F.; Sheu, J.-H. Mar. Drugs 2018, 16, 93. doi:10.3390/md16030093 |
| 3. | Yan, P.; Lv, Y.; van Ofwegen, L.; Proksch, P.; Lin, W. Org. Lett. 2010, 12, 2484–2487. doi:10.1021/ol100567d |
| 2. | Lu, Y.; Lin, Y.-C.; Wen, Z.-H.; Su, J.-H.; Sung, P.-J.; Hsu, C.-H.; Kuo, Y.-H.; Chiang, M. Y.; Dai, C.-F.; Sheu, J.-H. Tetrahedron 2010, 66, 7129–7135. doi:10.1016/j.tet.2010.06.094 |
| 9. | Kobayashi, M.; Mrishna, M. M.; Haribabu, B.; Anjaneyulu, V. Chem. Pharm. Bull. 1993, 41, 87–89. doi:10.1248/cpb.41.87 |
| 10. | Ling, N. C.; Hale, R. L.; Djerassi, C. J. Am. Chem. Soc. 1970, 92, 5281–5282. doi:10.1021/ja00720a082 |
| 6. | Thanh, N. V.; Ngoc, N. T.; Anh, H. L. T.; Thung, D. C.; Thao, D. T.; Cuong, N. X.; Nam, N. H.; Kiem, P. V.; Minh, C. V. J. Asian Nat. Prod. Res. 2016, 18, 938–944. doi:10.1080/10286020.2016.1173676 |
| 8. | Putra, M. Y.; Bavestrello, G.; Cerrano, C.; Renga, B.; D’Amore, C.; Fiorucci, S.; Fattorusso, E.; Taglialatela-Scafati, O. Steroids 2012, 77, 433–440. doi:10.1016/j.steroids.2011.12.026 |
| 7. | Cheng, Z.-B.; Xiao, H.; Fan, C.-Q.; Lu, Y.-N.; Zhang, G.; Yin, S. Steroids 2013, 78, 1353–1358. doi:10.1016/j.steroids.2013.10.004 |
| 11. | Gui, Y.-H.; Liu, L.; Wu, W.; Zhang, Y.; Jia, Z.-L.; Shi, Y.-P.; Kong, H.-T.; Liu, K.-C.; Jiao, W.-H.; Lin, H.-W. Bioorg. Chem. 2020, 94, 103435. doi:10.1016/j.bioorg.2019.103435 |
© 2022 Zhang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.
