Abstract
Two new cassane diterpenoids, 14β-hydroxycassa-11(12),13(15)-dien-12,16-olide (1) and 6′-acetoxypterolobirin B (3), together with a known analogue, identified as 12α,14β-dihydroxycassa-13(15)-en-12,16-olide (2), were isolated from the fruits of Pterolobium macropterum. Compound 1 is a cassane diterpenoid with a Δ11(12) double bond conjugated with an α,β-butenolide-type, whereas compound 3 is a dimeric caged cassane diterpenoid with unique 6/6/6/6/6/5/6/6/6 nonacyclic ring system. The structures of 1 and 3 were characterized by extensive spectroscopic analysis combined with computational ECD analyses. The α-glucosidase inhibitory activity of isolated compounds was evaluated, and compounds 1 and 3 showed significant α-glucosidase inhibitory activity with IC50 values of 66 and 44 μM.

Graphical Abstract
Introduction
Diabetes mellitus is a common metabolic disease that affects how the body uses blood glucose. In 2021, 537 million patients suffered from diabetes worldwide, and the number is feared to increase to 783 million in 2045 [1]. Type 2 diabetes account for the majority of the cases [2]. Currently, inhibition of α-glucosidase, the enzyme responsible for the hydrolysis of carbohydrates in the body, is widely used for the management of type 2 diabetes. The agents, such as acarbose, miglitol, and voglibose, can retard the digestion and absorption of dietary carbohydrates [3,4]. Some cassane-type diterpenoids such as pulcherrimin C and 6β-cinnamoyl-7β-hydroxyvouacapen-5α-ol, have been reported to exhibit significant α-glucosidase inhibitory activity [5].
The genus Pterolobium, comprising approximately 10 species distributed widely in Africa, China, and Thailand [6], is flowering shrubs belonging to Fabaceae. There are only four species known in Thailand [7], and some of them have been applied as antihemorrhoid [8]. Some species of this genus have revealed cassane diterpenoids as mainly secondary metabolites, which have shown interesting biological activities such as cytotoxicity and anti-inflammatory activity [9-11].
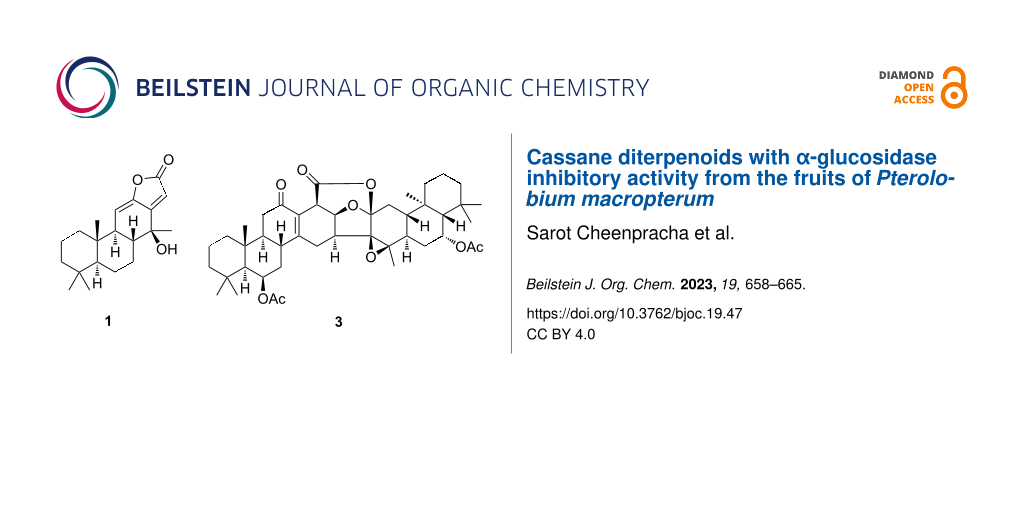
Pterolobium macropterum Kurz is a woody climbing shrub that is mainly distributed in the northern Thailand. Its roots are used in the medicinal plant therapy to treat toothache, fever, and to promote wound healing [11]. Previous phytochemical investigations of P. macropterum have revealed that this plant is a source of cassane diterpenoids featuring the structure of three cyclohexane rings with a constructed furan ring or an α,β-butenolide ring [9,10]. Recently, only two caged cassane diterpenoid dimers isolated from the fruits of this plant have been reported [11]. Some cassane-type diterpenes displayed to exhibit diverse biological properties, including anti-inflammatory [12], antimalarial [13], antitumor [14], antiviral [15], antibacterial [16], antiproliferative [13], and immunomodulatory [17] activities. As part of our studies on Thai medicinal plants, an investigation of the fruits of P. macropterum resulted in the isolation of one new cassane diterpenoid, 14β-hydroxycassa-11(12),13(15)-dien-12,16-olide (1), one new caged cassane diterpenoid dimer featuring a 6/6/6/6/6/5/6/6/6 nonacyclic ring system, 6′-acetoxypterolobirin B (3), and one known compound (Figure 1).
![[1860-5397-19-47-1]](/bjoc/content/figures/1860-5397-19-47-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of 1-3 isolated from P. macropterum.
Figure 1: Chemical structures of 1-3 isolated from P. macropterum.
Results and Discussion
The fruits of P. macropterum were extracted using MeOH to give a crude extract. After removal of the organic solvent, the extract was separated by repeated silica gel column chromatography as well as by Sephadex LH-20 to afford two new and one known cassane diterpenoids, identified as 12α,14β-dihydroxycassa-13(15)-en-12,16-olide (2) [18].
Compound 1 was isolated as an amorphous white powder. The molecular formula was determined as C20H28O3 from the HRESI–TOF–MS analysis with a [M + H]+ ion peak at m/z 317.2107 (calcd C20H29O3, 317.2111) and was considered to have 7 degrees of unsaturation. Its IR absorptions revealed the presence of hydroxy (3429 cm−1) and carbonyl (1733 cm−1) groups. The UV absorption band maximum at λmax 283 nm and five downfield-shifted carbon signals at δC 169.8 (C-16), 163.8 (C-13), 149.3 (C-12), 111.6 (C-11), and 109.6 (C-15) in the 13C NMR data suggested the presence of the α,β-butenolide ring conjugated with one extra double bond [12]. In the 1H NMR spectrum (Table 1), the resonances for four methyls [δH 1.30 (3H, s, H3-17), 0.90 (3H, s, H3-18), 0.85 (3H, s, H3-20), and 0.83 (3H, s, H3-19)], two olefinic protons [δH 6.03 (1H, br s, H-15) and 5.86 (1H, br s, H-11)] were observed. The 13C NMR and DEPT spectra, combined with HMQC correlations (Table 1) showed 20 resonances for carbon signals accounting for four methyls, five sp3 methylenes, five methines (two olefinics at δC 111.6, 109.6), and six quaternary carbons (one carbonyl at δC 169.8, two olefinics at δC 163.8, 149.3, and one oxygenated sp3 at δC 72.2). The 1H and 13C NMR spectroscopic data of 1 showed great similarity to those of 12α,14β-dihydroxycassa-13(15)-en-12,16-olide (2) isolated from Caesalpinia bonduc [18]. The difference evident was that compound 1 displayed an extended conjugate π-system with an α,β-unsaturated γ-lactone ring.
Table 1: 1H (500 MHz) and 13C NMR (125 MHz) data for compounds 1 and 3.
| position | 1a | 3a | |||
| δC | δH, mult (J in Hz) | δC | δH, mult (J in Hz) | ||
| 1 | 38.5, CH2 | 1.85 m; 1.06 m | 40.6, CH2 | 1.69 m; 1.61 m | |
| 2 | 18.6, CH2 | 1.64 m; 1.53 m | 18.7, CH2 | 1.64 m; 1.47 m | |
| 3 | 41.8, CH2 | 1.45 m; 1.22 m | 43.8, CH2 | 1.40 m; 1.15 m | |
| 4 | 33.4, C | 33.8, C | |||
| 5 | 54.8, CH | 0.99 dd (11.7, 2.7) | 55.2, CH | 1.03 m | |
| 6 | 21.1, CH2 | 1.80 m; 1.44 m | 68.8, CH | 5.53 br q (2.7) | |
| 7 | 25.1, CH2 | 2.06 m | 37.6, CH2 | 2.72 m | |
| 8 | 46.9, CH | 1.84 br t (11.0) | 35.3, CH | 2.28 m | |
| 9 | 52.2, CH | 1.88 br d (11.0) | 53.6, CH | 1.56 m | |
| 10 | 37.8, C | 37.6, C | |||
| 11 | 111.6, CH | 5.86 br s | 37.7, CH2 |
2.60 dd (14.7, 2.9)
2.37 t (14.7) |
|
| 12 | 149.3, C | 197.9, C | |||
| 13 | 163.8, C | 128.0, C | |||
| 14 | 72.2, C | 155.4, C | |||
| 15 | 109.6, CH | 6.03 br s | 37.0, CH | 4.59 dd (6.6, 1.9) | |
| 16 | 169.8, C | 167.0, C | |||
| 17 | 22.3, CH3 | 1.30 s | 25.3, CH2 |
2.54 dd (20.0, 10)
2.20 m |
|
| 18 | 33.2, CH3 | 0.90 s | 33.2, CH3 | 0.95 s | |
| 19 | 21.5, CH3 | 0.83 s | 23.3, CH3 | 1.00 s | |
| 20 | 15.5, CH3 | 0.85 s | 16.7, CH3 | 1.20 s | |
| 1′ | 40.7, CH2 | 1.69 m; 1.61 m | |||
| 2′ | 18.7, CH2 | 1.64 m; 1.47 m | |||
| 3′ | 43.8, CH2 | 1.40 m; 1.15 m | |||
| 4′ | 33.8, C | ||||
| 5′ | 55.1, CH | 1.03 m | |||
| 6′ | 69.4, CH | 5.56 br q (2.7) | |||
| 7′ | 35.5, CH2 | 2.29 m | |||
| 8′ | 36.0, CH | 2.73 m | |||
| 9′ | 43.1, CH | 1.40 m | |||
| 10′ | 36.8, C | ||||
| 11′ | 30.9, CH2 | 2.18 m | |||
| 12′ | 104.1, C | ||||
| 13′ | 71.3, C | ||||
| 14′ | 65.4, C | ||||
| 15′ | 33.8, CH | 2.73 m | |||
| 16′ | 73.7, CH | 4.55 t (6.6) | |||
| 17′ | 16.2, CH3 | 1.23 s | |||
| 18′ | 33.1, CH3 | 0.95 s | |||
| 19′ | 23.2, CH3 | 0.98 s | |||
| 20′ | 16.3, CH3 | 1.09 s | |||
| 6-OCOCH3 | 21.8, CH3 | 2.06 s | |||
| 6-OCOCH3 | 170.7, C | ||||
| 6′-OCOCH3 | 21.9, CH3 | 2.10 s | |||
| 6′-OCOCH3 | 170.4, C | ||||
aNMR data were recorded in chloroform-d.
The HMBC cross-peaks (Figure 2) from H-11 (δH 5.86) to C-10, C-12, and C-13, and from H-15 (δH 6.03) to C-12, C-13 and C-14 allowed the location of an extended conjugated π-system at C-11 and C-12. Moreover, the downfield shift of C-14 (δC 72.2) and the HMBC correlation between H3-17 and C-14 as well as the appearance of the H3-17 as a singlet signal confirmed the connection of a hydroxy group at C-14.
![[1860-5397-19-47-2]](/bjoc/content/figures/1860-5397-19-47-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Key 1H,1H-COSY, and HMBC correlations of 1 and 3.
Figure 2: Key 1H,1H-COSY, and HMBC correlations of 1 and 3.
The relative configuration of 1 was characterized by NOESY spectra. In the NOESY experiment (Figure 3), the cross-peak between H-8 and H3-20 suggested these protons to be syn oriented. In addition, the cross-peaks between H-5/H-9, H-5/H-7α and H-7α/H3-17 suggested H3-17 to be α-oriented.
![[1860-5397-19-47-3]](/bjoc/content/figures/1860-5397-19-47-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Selected NOESY cross peaks of 1 and 3.
Figure 3: Selected NOESY cross peaks of 1 and 3.
Comparison of the specific rotation was used to establish the absolute configuration of 1. The specific rotation of 1 (![[Graphic 1]](/bjoc/content/inline/1860-5397-19-47-i1.svg?max-width=637&scale=1.18182) −22 (c 0.01, MeOH)) was similar to the reported data of 2 (
−22 (c 0.01, MeOH)) was similar to the reported data of 2 (![[Graphic 2]](/bjoc/content/inline/1860-5397-19-47-i2.svg?max-width=637&scale=1.18182) −36 (c 0.01, MeOH); lit.
−36 (c 0.01, MeOH); lit. ![[Graphic 3]](/bjoc/content/inline/1860-5397-19-47-i3.svg?max-width=637&scale=1.18182) −44 (c 0.05, MeOH)) [18], confirming the same absolute configuration these compounds should be derived from the same biosynthetic pathway. In addition, the ECD spectra of (5S,8R,9S,10R,14S)-1 and its enantiomer were calculated at the B3LYP functional using a TD–DFT method [19]. As illustrated in Figure 4a, the measured ECD curve was compared to the predicted ECD curve of (5S,8R,9S,10R,14S)-1, indicating that the measured and predicted ECD spectra were similar except for a blue-shift in the ECD spectrum. Thus, the structure of 1 was characterized as shown.
−44 (c 0.05, MeOH)) [18], confirming the same absolute configuration these compounds should be derived from the same biosynthetic pathway. In addition, the ECD spectra of (5S,8R,9S,10R,14S)-1 and its enantiomer were calculated at the B3LYP functional using a TD–DFT method [19]. As illustrated in Figure 4a, the measured ECD curve was compared to the predicted ECD curve of (5S,8R,9S,10R,14S)-1, indicating that the measured and predicted ECD spectra were similar except for a blue-shift in the ECD spectrum. Thus, the structure of 1 was characterized as shown.
![[1860-5397-19-47-4]](/bjoc/content/figures/1860-5397-19-47-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Measured and predicted ECD spectra of 1 and 3.
Figure 4: Measured and predicted ECD spectra of 1 and 3.
Compound 3 was obtained as a colorless oil. The molecular formula was assigned to be C44H60O9 based on the HRESI–TOF–MS analysis with a [M + H]+ ion peak at m/z 733.4305 (calcd for C44H61O9, 733.4310) and NMR data, implying 15 degrees of unsaturation. The IR absorption band at 1724 cm−1 suggested the presence of α,β-unsaturated γ-lactone functionality. The 13C NMR and DEPT spectra in combination with HMQC data (Table 1) showed resonances of 44 carbons which were classified as nine methyls, 11 methylenes, 11 methines (three oxygenated sp3 at δC 73.7, 69.4, 68.8), and 13 quaternary carbons (four carbonyls at δC 197.9, 170.7, 170.4, 167.0, two olefinics at δC 155.4, 128.0, and three oxygenated at δC 104.1, 71.3, 65.4). Assuming a cassane-type diterpene skeleton, the 1H and 13C NMR spectra (Table 1) displayed two sets (A and B) of the characteristic signals of seven methyl singlets [set A: δH/δC 1.20 (s, H3-20)/16.7, 1.00 (s, H3-19)/23.3, 0.95 (s, H3-18)/33.2; set B: δH/δC1.23 (s, H3-17′)/16.2, 1.09 (s, H3-20′)/16.3, 0.98 (s, H3-19′)/23.2, 0.95 (s, H3-18′)/33.1], three oxygenated methines [set A : δH/δC 5.53 (br q, J = 2.7 Hz, H-6)/68.8; set B : δH/δC 5.56 (br q, J = 2.7 Hz, H-6′)/69.4, 4.55 (t, J = 6.6 Hz, H-16′)/73.7], and two acetoxy groups [set A: δH 2.06 (s, 6-OCOCH3)/δC 21.8, and 170.7; set B: δH 2.10 (s, 6′-OCOCH3)/δC 21.9 and 170.4]. Careful analysis of the NMR data indicated the presence of a dimeric cassane-type diterpene skeleton whose NMR spectra resembled those of pterolobirin B [11], an unprecedented caged cassane diterpenoid dimer with unique 6/6/6/6/6/5/6/6/6 nonacyclic ring system. The minor difference was the additional acetoxy group (δH 2.10) at C-6′. This conclusion was suggested by the HMBC correlations (Figure 2) from H-6′ to 6′-OCOCH3 (δC 170.4) and C-10′ (δC 36.8), and from H3-20′ to C-5′ (δC 55.1) and C-10′, combined with the spin system CH(5′)-CH(6′)-CH2(7′)-CH(8′)-CH(9′)-CH2(11′) found in the COSY spectrum (Figure 2). The COSY correlations between H-15/H-16′, H-16′/H-15′ and H-15′/H2-17, and the HMBC cross-peaks from H-15′ to C-14 (δC 155.4), C-15 (δC 37.0), C-12′ (δC 104.1), and C-14′ (δC 65.4), and from H-16′ to C-13 (δC 128.0), C-16 (δC 167.0), C-17 (δC 25.3), C-12′, and C-13′ (δC 71.3) clearly indicated the two C–C bond linkages of both units through the C-15/C-16′ and C-17/C-15′ bonds. Furthermore, the aforementioned ring structure and functionalities accounting for 13 out of 15 degrees of unsaturation required the presence of two heterocyclic rings in the molecule. The presence of an ester carbonyl signal (δC 167.0) and a deshielded oxygenated carbon resonance at C-12′ (δC 104.1) implied the formation of six-membered ring via an ester bond between C-16 and C-12′. In addition, an epoxide moiety at C-13′ and C-14′ was further supported by the downfield shift of C-13′ (δC 71.1) and C-14′ (δC 65.4) and the cross-peaks from H3-17′/H-15′ to C-13′ and C-14′ in the HMBC spectrum.
In the NOESY experiments of 3 (Figure 3), the interactions of H3-20 to H3-18 and H-8 indicated the β-orientations, while the cross-peaks of H3-20′ to H3-18′ and H-8′ revealed the α-orientations of these protons. The protons H-6 and H-6′ are α- and β-oriented, respectively, as indicated by NOESY cross peaks from H-5 to H-6 and H-9; and from H-5′ to H-6′ and H3-19′, combined with the small coupling constants of H-6 (J = 2.7 Hz) and H-6′ (J = 2.7 Hz). The syn orientations of H-15, H-15′, H-16′ and H3-17′ were established from the NOESY correlations of H-15′ to H3-17′, H-16′ and of H-15 to H-16′. From above information, the relative configuration of C-12′ was assigned and supported by the biosynthetic pathway based on a Diels–Alder adduct, thus displaying the same relative configuration found in pterolobirin B [11]. The absolute configuration of 3 was thus elucidated as 5S,6R,8R,9S,10R,15R,5′S,6′R,8′R,9′S,10′R,12′S,13′R,14′R,15′S,16′S and the measured ECD spectrum (Figure 4b) with the positive at 243 nm and negative at 330 nm CEs, is very well matched with the ECD curve of pterolobirin B [11]. Although, the predicted ECD data is not in good agreement with the measured ECD data. It is noted that the calculation could not completely simulate the experimental results depend on the level of theory and basis set as well as the polarity of solvent. Finally, comparison of the specific rotation was used to establish the absolute configuration. Pterolobirin B showed ![[Graphic 4]](/bjoc/content/inline/1860-5397-19-47-i4.svg?max-width=637&scale=1.18182) −72 (c 0.1, CHCl3) [11] and 3 showed
−72 (c 0.1, CHCl3) [11] and 3 showed ![[Graphic 5]](/bjoc/content/inline/1860-5397-19-47-i5.svg?max-width=637&scale=1.18182) −87 (c 0.01, MeOH), which also supports the absolute configuration. Thus, the structure of 3 was assigned as shown.
−87 (c 0.01, MeOH), which also supports the absolute configuration. Thus, the structure of 3 was assigned as shown.
The isolated compounds were evaluated for their α-glucosidase inhibitory activity [20]. Compounds 1 and 3 exhibited significant α-glucosidase inhibitory activity with IC50 values of 66 and 44 µM, respectively, which showed stronger inhibitory activity than the positive control, acarbose (IC50 178 μM). Compound 2 was inactive in this assay, with IC50 value >200 μM, which suggested that a Δ11(12) double bond might be important for the α-glucosidase inhibitory activity.
Conclusion
In conclusion, two new cassane diterpenoids, 14β-hydroxycassa-11(12),13(15)-dien-12,16-olide (1) and 6′-acetoxypterolobirin B (3) together with one known analogue were isolated from the MeOH extract of P. macropterum fruits. Their structures and absolute configurations of 1 and 3 were established by spectroscopic analyses and ECD data. Compound 1 has an extended conjugated π-system with an α,β-unsaturated γ-lactone ring at the Δ11(12) double bond, while compound 3 is caged cassane diterpenoid dimers with unique 6/6/6/6/6/5/6/6/6 nonacyclic ring system. Only two caged cassane diterpenoid dimers with unique 6/6/6/6/6/5/6/6/6 nonacyclic ring system were isolated previously from the same plant [11]. Biological evaluation revealed that compounds 1 and 3 exhibited significant α-glucosidase inhibitory activity with IC50 values of 66 and 44 μM, respectively.
Experimental
General experimental procedures
Optical rotations were measured on a JASCO P-2000 polarimeter in MeOH. The UV spectra were recorded on a PerkinElmer UV–vis spectrophotometer. ECD spectra were acquired on a JASCO J-1500 circular dichroism spectrometer. FTIR spectra were obtained using a PerkinElmer FTS FT-IR spectrophotometer. NMR spectra were obtained on a Bruker NEO 500 MHz NMR Ultra Shield. Chemical shifts are referenced in parts per million (δ) in the deuterated solvents (CDCl3) using TMS as an internal standard. An Agilent 1290 infinity II/Q-TOFMS mass spectrometer was employed to acquire HRESI–TOF–MS spectra. Column chromatography (CC) was carried out on silica gel 60 (70–230 mesh, Merck, Darmstadt, Germany), and Sephadex LH-20 (GE Healthcare). Thin-layer chromatography (TLC) was performed on silica gel 60 F254 plates (Merck, Darmstadt, Germany) using precoated aluminum plates for analytical purposes.
Plant material
Fresh fruits of Pterolobium macropterum Kurz were collected from Song Khwae District, Nan Province, Thailand (GPS: 19°16'51.5"N 100°43'30.3"E) in August 2021 and identified by Mr. Martin van de Bult, Doi Tung Development Project. A voucher specimen (UP-CNP003) was deposited at the Chemistry of Natural Products for Sustainability Laboratory, School of Science, University of Phayao.
Extraction and isolation
The fresh fruits of P. macropterum (0.2 kg) were ground and soaked with MeOH (3 × 2 L) at room temperature for 3 days. The solvent was evaporated under reduced pressure at 40 °C, affording MeOH extract (10.5 g). The extract was subjected to silica gel column chromatography (CC) (70–230 mesh, 2 × 60 cm) eluting with hexanes–acetone (100:0 → 0:100, v/v) to afford 10 fractions (Fr.1–Fr.10). Fr.5 (142.0 mg) was further fractionated over a Sephadex LH-20 column with a mixture of CH2Cl2–MeOH (1:1, v/v) affording five subfractions (Fr.5.1–Fr5.5). Subfraction Fr.5.2 (17.2 mg) was chromatographed over silica gel CC eluting with acetone–hexanes (1:19, v/v) to give compound 3 (1.1 mg). Fr.6 (842.0 mg) was chromatographed over silica gel CC eluting with acetone–hexanes (1:19, v/v), and then purified by silica gel CC using 100% CH2Cl2 to afford compound 1 (12.5 mg). Chromatographic purification of Fr.8 (1.2 g) over a Sephadex LH-20 column with a mixture of CH2Cl2–MeOH (1:1, v/v) afforded three subfractions (Fr.8.1–Fr8.3). Fr.8.2 (257.2 mg) was purified by silica gel CC eluting with acetone–hexanes (1:9, v/v), to provide five subfractions (Fr.8.2.1–Fr.8.2.5). Subfraction Fr.8.2.5 (28.8 mg), was chromatographed over silica gel CC eluting with 100% CH2Cl2 afforded compound 2 (6.9 mg).
14β-Hydroxycassa-11(12),13(15)-dien-12,16-olide (1): amorphous, white powder; ![[Graphic 6]](/bjoc/content/inline/1860-5397-19-47-i6.svg?max-width=637&scale=1.18182) −22 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 283 (4.20) nm; ECD (0.001, MeOH) λmax (Δε) 241 (+11.5), 271 (−10.4), 297 (+2.5) nm; IR (neat) νmax 3429, 2926, 1733, 1664, 1603, 1263 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, see Table 1; HRESI–TOF–MS m/z: 317.2107 [M + H]+ (calcd for C20H29O3, 317.2111).
−22 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 283 (4.20) nm; ECD (0.001, MeOH) λmax (Δε) 241 (+11.5), 271 (−10.4), 297 (+2.5) nm; IR (neat) νmax 3429, 2926, 1733, 1664, 1603, 1263 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, see Table 1; HRESI–TOF–MS m/z: 317.2107 [M + H]+ (calcd for C20H29O3, 317.2111).
6′-Acetoxypterolobirin B (3): colorless oil; ![[Graphic 7]](/bjoc/content/inline/1860-5397-19-47-i7.svg?max-width=637&scale=1.18182) −87 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 243 (3.63) nm; ECD (0.001, MeOH) λmax (Δε) 243 (+21.1), 330 (−4.70) nm; IR (neat) νmax: 2973, 1724, 1660, 1508, 1733 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, see Table 1; HRESI–TOF–MS m/z: 733.4305 [M + H]+ (calcd for C44H61O9, 733.4310).
−87 (c 0.01, MeOH); UV (MeOH) λmax (log ε) 243 (3.63) nm; ECD (0.001, MeOH) λmax (Δε) 243 (+21.1), 330 (−4.70) nm; IR (neat) νmax: 2973, 1724, 1660, 1508, 1733 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) spectra, see Table 1; HRESI–TOF–MS m/z: 733.4305 [M + H]+ (calcd for C44H61O9, 733.4310).
α-Glucosidase inhibitory assay
α-Glucosidase inhibitory activity was performed according to experimental literature with slight modification [20]. α-Glucosidase (0.05 U/mL) and substrate, p-nitrophenyl-α-ᴅ-glucopyronoside (p-NPG) (1 mM) were dissolved in 0.1 M sodium phosphate buffer (pH 6.9). Fifty μL of sample (1 mg/mL in 10% DMSO) and 50 μL of α-glucosidase were preincubated at 37 °C for 10 min in a 96 well plate. The substrate solution (50 μL) was added to the mixture to start the reaction, with further incubation at 37 °C for 20 min. The reaction was terminated by adding 1 mL of 0.3 M Na2CO3. Enzymatic activity was quantified by measuring the absorbance at 405 nm. The percent inhibition of activity was calculated as (A0 − A1)/A0 × 100, where A0 is the absorbance of control, and A1 is the absorbance with the sample. Acarbose was used as a standard drug and all experiments were evaluated in triplicate.
Supporting Information
| Supporting Information File 1: Copies of NMR spectra for compounds 1 and 3. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021.
Return to citation in text: [1] -
Forouhi, N. G.; Wareham, N. J. Medicine 2014, 42, 698–702. doi:10.1016/j.mpmed.2014.09.007
Return to citation in text: [1] -
Van de Laar, F. A. Vasc. Health Risk Manage. 2008, 4, 1189–1195. doi:10.2147/vhrm.s3119
Return to citation in text: [1] -
Sohrabi, M.; Binaeizadeh, M. R.; Iraji, A.; Larijani, B.; Saeedi, M.; Mahdavi, M. RSC Adv. 2022, 12, 12011–12052. doi:10.1039/d2ra00067a
Return to citation in text: [1] -
Yun, X.; Chen, X.-M.; Wang, J.-Y.; Lu, W.; Zhang, Z.-H.; Kim, Y. H.; Zong, S.-C.; Li, C.-H.; Gao, J.-M. Nat. Prod. Res. 2022, 36, 4630–4638. doi:10.1080/14786419.2021.2007096
Return to citation in text: [1] -
Chen, D.; Zhang, D.; Hou, D. Flora of China; Science Press: Beijing, China, 2010; pp 47–48.
Return to citation in text: [1] -
Vidal, J. E.; Larsen, S. S.; Larsen, K. Flora of Thailand; The Forest Herbarium, National Park, Wildlife and Plant Conservation Department: Bangkok, Thailand, 1984; Vol. 4, pp 56–61.
Return to citation in text: [1] -
Chuakul, W.; Saralamp, P. J. Natl. Res. Counc. Thailand 2002, 34, 47–73.
Return to citation in text: [1] -
Suthiwong, J.; Pitchuanchom, S.; Wattanawongdon, W.; Hahnvajanawong, C.; Yenjai, C. J. Nat. Prod. 2014, 77, 2432–2437. doi:10.1021/np500476h
Return to citation in text: [1] [2] -
Raksat, A.; Choodej, S.; Aree, T.; Nejad Ebrahimi, S.; Pudhom, K. Phytochemistry 2022, 196, 113074. doi:10.1016/j.phytochem.2021.113074
Return to citation in text: [1] [2] -
Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Wang, M.; Yu, S.; Qi, S.; Zhang, B.; Song, K.; Liu, T.; Gao, H. J. Nat. Prod. 2021, 84, 2175–2188. doi:10.1021/acs.jnatprod.1c00233
Return to citation in text: [1] [2] -
Ma, G.; Wu, H.; Chen, D.; Zhu, N.; Zhu, Y.; Sun, Z.; Li, P.; Yang, J.; Yuan, J.; Xu, X. J. Nat. Prod. 2015, 78, 2364–2371. doi:10.1021/acs.jnatprod.5b00317
Return to citation in text: [1] [2] -
Bao, H.; Zhang, L.-L.; Liu, Q.-Y.; Feng, L.; Ye, Y.; Lu, J.-J.; Lin, L.-G. Molecules 2016, 21, 791. doi:10.3390/molecules21060791
Return to citation in text: [1] -
Jiang, R.-W.; Ma, S.-C.; But, P. P.-H.; Mak, T. C. W. J. Nat. Prod. 2001, 64, 1266–1272. doi:10.1021/np010174+
Return to citation in text: [1] -
Dickson, R. A.; Houghton, P. J.; Hylands, P. J. Phytochemistry 2007, 68, 1436–1441. doi:10.1016/j.phytochem.2007.03.008
Return to citation in text: [1] -
Shukla, S.; Mehta, A.; John, J.; Mehta, P.; Vyas, S. P.; Shukla, S. J. Ethnopharmacol. 2009, 125, 252–256. doi:10.1016/j.jep.2009.07.002
Return to citation in text: [1] -
Yadav, P. P.; Maurya, R.; Sarkar, J.; Arora, A.; Kanojiya, S.; Sinha, S.; Srivastava, M. N.; Raghubir, R. Phytochemistry 2009, 70, 256–261. doi:10.1016/j.phytochem.2008.12.008
Return to citation in text: [1] [2] [3] -
Chokchaisiri, R.; Chaichompoo, W.; Chunglok, W.; Cheenpracha, S.; Ganranoo, L.; Phutthawong, N.; Bureekaew, S.; Suksamrarn, A. J. Nat. Prod. 2020, 83, 14–19. doi:10.1021/acs.jnatprod.9b00307
Return to citation in text: [1] -
Meesakul, P.; Richardson, C.; Pyne, S. G.; Laphookhieo, S. J. Nat. Prod. 2019, 82, 741–747. doi:10.1021/acs.jnatprod.8b00581
Return to citation in text: [1] [2]
| 20. | Meesakul, P.; Richardson, C.; Pyne, S. G.; Laphookhieo, S. J. Nat. Prod. 2019, 82, 741–747. doi:10.1021/acs.jnatprod.8b00581 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 20. | Meesakul, P.; Richardson, C.; Pyne, S. G.; Laphookhieo, S. J. Nat. Prod. 2019, 82, 741–747. doi:10.1021/acs.jnatprod.8b00581 |
| 1. | IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. |
| 6. | Chen, D.; Zhang, D.; Hou, D. Flora of China; Science Press: Beijing, China, 2010; pp 47–48. |
| 15. | Jiang, R.-W.; Ma, S.-C.; But, P. P.-H.; Mak, T. C. W. J. Nat. Prod. 2001, 64, 1266–1272. doi:10.1021/np010174+ |
| 5. | Yun, X.; Chen, X.-M.; Wang, J.-Y.; Lu, W.; Zhang, Z.-H.; Kim, Y. H.; Zong, S.-C.; Li, C.-H.; Gao, J.-M. Nat. Prod. Res. 2022, 36, 4630–4638. doi:10.1080/14786419.2021.2007096 |
| 16. | Dickson, R. A.; Houghton, P. J.; Hylands, P. J. Phytochemistry 2007, 68, 1436–1441. doi:10.1016/j.phytochem.2007.03.008 |
| 3. | Van de Laar, F. A. Vasc. Health Risk Manage. 2008, 4, 1189–1195. doi:10.2147/vhrm.s3119 |
| 4. | Sohrabi, M.; Binaeizadeh, M. R.; Iraji, A.; Larijani, B.; Saeedi, M.; Mahdavi, M. RSC Adv. 2022, 12, 12011–12052. doi:10.1039/d2ra00067a |
| 13. | Ma, G.; Wu, H.; Chen, D.; Zhu, N.; Zhu, Y.; Sun, Z.; Li, P.; Yang, J.; Yuan, J.; Xu, X. J. Nat. Prod. 2015, 78, 2364–2371. doi:10.1021/acs.jnatprod.5b00317 |
| 2. | Forouhi, N. G.; Wareham, N. J. Medicine 2014, 42, 698–702. doi:10.1016/j.mpmed.2014.09.007 |
| 14. | Bao, H.; Zhang, L.-L.; Liu, Q.-Y.; Feng, L.; Ye, Y.; Lu, J.-J.; Lin, L.-G. Molecules 2016, 21, 791. doi:10.3390/molecules21060791 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 9. | Suthiwong, J.; Pitchuanchom, S.; Wattanawongdon, W.; Hahnvajanawong, C.; Yenjai, C. J. Nat. Prod. 2014, 77, 2432–2437. doi:10.1021/np500476h |
| 10. | Raksat, A.; Choodej, S.; Aree, T.; Nejad Ebrahimi, S.; Pudhom, K. Phytochemistry 2022, 196, 113074. doi:10.1016/j.phytochem.2021.113074 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 12. | Wang, M.; Yu, S.; Qi, S.; Zhang, B.; Song, K.; Liu, T.; Gao, H. J. Nat. Prod. 2021, 84, 2175–2188. doi:10.1021/acs.jnatprod.1c00233 |
| 7. | Vidal, J. E.; Larsen, S. S.; Larsen, K. Flora of Thailand; The Forest Herbarium, National Park, Wildlife and Plant Conservation Department: Bangkok, Thailand, 1984; Vol. 4, pp 56–61. |
| 9. | Suthiwong, J.; Pitchuanchom, S.; Wattanawongdon, W.; Hahnvajanawong, C.; Yenjai, C. J. Nat. Prod. 2014, 77, 2432–2437. doi:10.1021/np500476h |
| 10. | Raksat, A.; Choodej, S.; Aree, T.; Nejad Ebrahimi, S.; Pudhom, K. Phytochemistry 2022, 196, 113074. doi:10.1016/j.phytochem.2021.113074 |
| 18. | Yadav, P. P.; Maurya, R.; Sarkar, J.; Arora, A.; Kanojiya, S.; Sinha, S.; Srivastava, M. N.; Raghubir, R. Phytochemistry 2009, 70, 256–261. doi:10.1016/j.phytochem.2008.12.008 |
| 13. | Ma, G.; Wu, H.; Chen, D.; Zhu, N.; Zhu, Y.; Sun, Z.; Li, P.; Yang, J.; Yuan, J.; Xu, X. J. Nat. Prod. 2015, 78, 2364–2371. doi:10.1021/acs.jnatprod.5b00317 |
| 17. | Shukla, S.; Mehta, A.; John, J.; Mehta, P.; Vyas, S. P.; Shukla, S. J. Ethnopharmacol. 2009, 125, 252–256. doi:10.1016/j.jep.2009.07.002 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 11. | Raksat, A.; Aree, T.; Pudhom, K. J. Nat. Prod. 2020, 83, 2241–2245. doi:10.1021/acs.jnatprod.0c00354 |
| 18. | Yadav, P. P.; Maurya, R.; Sarkar, J.; Arora, A.; Kanojiya, S.; Sinha, S.; Srivastava, M. N.; Raghubir, R. Phytochemistry 2009, 70, 256–261. doi:10.1016/j.phytochem.2008.12.008 |
| 19. | Chokchaisiri, R.; Chaichompoo, W.; Chunglok, W.; Cheenpracha, S.; Ganranoo, L.; Phutthawong, N.; Bureekaew, S.; Suksamrarn, A. J. Nat. Prod. 2020, 83, 14–19. doi:10.1021/acs.jnatprod.9b00307 |
| 12. | Wang, M.; Yu, S.; Qi, S.; Zhang, B.; Song, K.; Liu, T.; Gao, H. J. Nat. Prod. 2021, 84, 2175–2188. doi:10.1021/acs.jnatprod.1c00233 |
| 18. | Yadav, P. P.; Maurya, R.; Sarkar, J.; Arora, A.; Kanojiya, S.; Sinha, S.; Srivastava, M. N.; Raghubir, R. Phytochemistry 2009, 70, 256–261. doi:10.1016/j.phytochem.2008.12.008 |
© 2023 Cheenpracha et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.