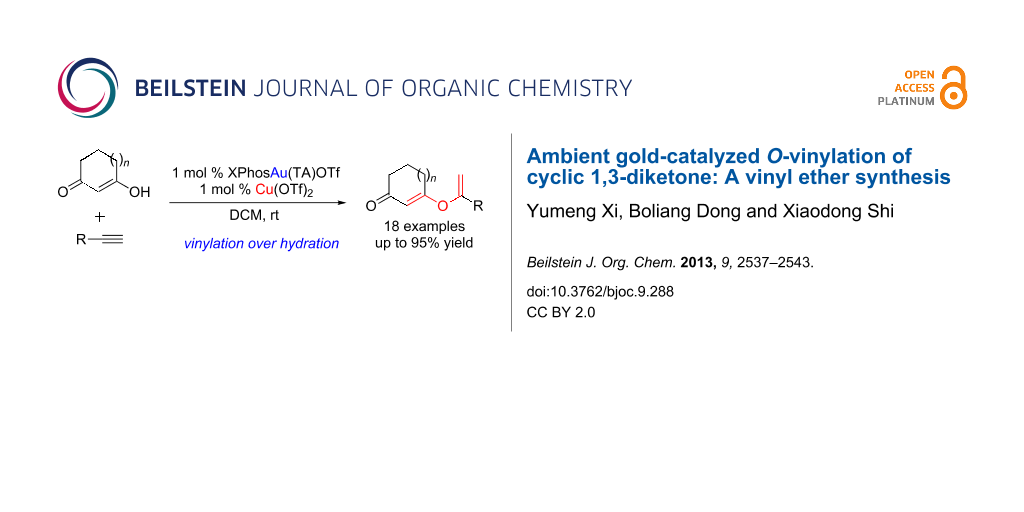
Abstract
Gold-catalyzed O-vinylation of cyclic 1,3-diketones has been achieved for the first time, which provides direct access to various vinyl ethers. A catalytic amount of copper triflate was identified as the significant additive in promoting this transformation. Both aromatic and aliphatic alkynes are suitable substrates with good to excellent yields.

Graphical Abstract
Introduction
The past decade has witnessed the fast growth of homogeneous gold catalysis as one of the important branches in transition-metal chemistry [1-9]. The utility of cationic gold(I) complexes as π-carbophilic acids toward alkyne and allene activation renders them as essential tools in organic synthesis. Generally with the unique reactivity and mild reaction conditions, this type of transformation has found widespread applications in complex molecule synthesis [10,11]. Among those reactions, the gold-catalyzed hydration of alkynes is regarded as one of the signature reactions in the field (known as Teles hydration) [12]. This reaction usually utilized the combination of methanol and water, wherein methanol served as the nucleophile to attack the triple bond, forming the vinyl ether intermediate. This vinyl ether then collapsed to give a ketone as the final product [13-17].
A vinyl ether is a common and versatile building block in organic synthesis as well as polymer chemistry. Typical methods for the preparation of a vinyl ether involve elimination, olefination of esters, addition of alcohols to alkynes, as well as transition metal-mediated cross-coupling reactions [18]. Based on the π-carbophilicity of gold(I), the addition of alcohol to alkyne should provide direct access to the corresponding vinyl ether. However, most of the reported gold-catalyzed O-nucleophile additions to alkynes are intramolecular reactions. No general protocol for vinyl ether synthesis using gold has been reported to date [17]. Nevertheless, there have been examples regarding the intermolecular addition of carboxylic acids [19,20], phenols [21,22] as well as phosphoric acids [23-25] to alkynes. In this context, we report a gold(I)-catalyzed O-vinylation of 1,3-diketones with unactivated alkynes at ambient temperature.
Results and Discussion
As indicated in Scheme 1, the main challenge for the intermolecular O-nucleophile addition to alkynes is the competitive hydration side reaction. Although, theoretically, strictly anhydrous conditions shall prevent the water addition, new effective catalytic systems that can avoid the “precautionary” treatment of solvents and substrates are much more practical and highly desirable.
![[1860-5397-9-288-i1]](/bjoc/content/inline/1860-5397-9-288-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Intermolecular O-addition to alkynes: challenge and opportunity.
Scheme 1: Intermolecular O-addition to alkynes: challenge and opportunity.
From literature reported examples, a gold activated water intermediate, [L-Au-(H2O)]+, has been proposed in helping the water addition to the alkyne besides the typical π-acid activation [12-17]. Our group has developed the 1,2,3-triazole coordinated gold complexes (TA-Au) as stable catalysts for alkyne activation in the past several years [26-30]. Considering that TA-Au complexes might have different binding ability toward water activation, we wondered whether the intermolecular O-addition could be achieved through ligand tuning. In this report, we focus on the cyclic 1,3-diketone nucleophiles due to A) the transformation is challenging and has never been reported in the past, B) vinyl ether products are highly functional materials, which can be easily extended to complex molecules through simple transformations.
1,3-Cyclohexanedione and phenylacetylene were used as the model substrates for the evaluation of various gold catalysts. As shown in Table 1, the use of Ph3PAuCl/AgOTf (5%) gave an acceptable yield (59%) of the desired vinyl addition product 3a. Thermally-stable Ph3PAu(TA)OTf (TA-Au, 5%) slightly improved the performance, yielding 63% of the desired product and less hydration byproduct 4. When changing the primary ligand from triphenylphosphine to a NHC, the yield slightly decreased. However, the corresponding TA-Au complexes indicated significantly improved selectivity towards the diketone addition over the hydration (Table 1, entry 4). Finally, the application of the XPhos ligand and the corresponding TA-Au complex largely promoted this reaction, giving 3a in 85% yield with 12% hydration byproduct. Notably, the reaction occurred at room temperature with no need for “careful” condition control (open to the air and untreated solvent). The fact that TA-Au complexes generally gave improved selectivity of diketone addition over hydration (compared with the corresponding [L-Au]+ complexes) supported our hypothesis and made the TA-Au catalysts important for this transformation. Slightly increasing the ratio of alkyne to 1.2 equivalents (relative to the nucleophile) increased the formation of the desired vinyl ester to a near quantitative yield (98% NMR yield, 95% isolated yield), even with only 1% catalyst loading.
Table 1: Screening of gold catalysts.a,b
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-288-i4.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | [Au] cat. | 2a (equiv) | Loading | Time (h) | Conversion of 2a (%) | Yield of 3a (%) | Yield of 4 (%) |
|---|---|---|---|---|---|---|---|
| 1 | PPh3AuCl/AgOTf | 1.0 | 5% | 17 | 81 | 59 | 17 |
| 2 | PPh3Au(TA)OTf | 1.0 | 5% | 17 | 82 | 63 | 11 |
| 3 | IPrAuCl/AgOTf | 1.0 | 5% | 17 | 100 | 55 | 34 |
| 4 | IPrAu(TA)OTf | 1.0 | 5% | 17 | 64 | 50 | 5 |
| 5 | XPhosAuCl/AgOTf | 1.0 | 5% | 10 | 100 | 76 | 22 |
| 6 | XPhosAu(TA)OTf | 1.0 | 5% | 10 | 100 | 85 | 12 |
| 7 | XPhosAu(TA)OTf | 1.2 | 5% | 10 | 100 | 99 | n.d. |
| 8 | XPhosAu(TA)OTf | 1.2 | 3% | 17 | 100 | 99 | n.d. |
| 9 | XPhosAu(TA)OTf | 1.2 | 1% | 20 | 100 | 98 (95) | n.d. |
aGeneral conditions: 1 (0.2 mmol), 2a (1 or 1.2 equiv), and catalyst in CDCl3 (0.4 mL), rt. bYield and conversion are determined by using 1,3,5-trimethoxybenzene as the internal standard. Isolated yield is given in parenthesis.
Given the encouraging results associated with the XPhosAu-TA catalysts, we explored the reaction scope. However, surprisingly, significantly slower reaction was observed when conducting the reaction in dichloromethane (DCM) or 1,2-dichloroethane (DCE), though selectivity for the diketone addition over hydration was maintained as shown in Table 1. Solvent screening shown in Scheme 2 proved that the trace amount of acid from chloroform decomposition was crucial for the optimal performance, which likely helped the protodeauration. Other Lewis acids have also been screened and the Cu(OTf)2 was identified [31] as the optimal choice due to the practical reasons (easy to weigh and less hygroscopic) and excellent reactivity. Cu(OTf)2 and HOTf itself could not catalyze the reaction, suggesting that it is indeed a gold-catalyzed process. Finally, it is worth noting that the use of 1 mol % Echavarren catalyst t-BuXPhosAu(MeCN)SbF6 gave a slightly lower yield (71%), and the combination of 1 mol % t-BuXPhosAu(MeCN)SbF6 and 1 mol % Cu(OTf)2 gave 86% yield (under otherwise identical conditions). This result highlights the synthetic utility of TA-Au catalyst in the transformation.
![[1860-5397-9-288-i2]](/bjoc/content/inline/1860-5397-9-288-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Acid as the critical additive for optimal performance.
Scheme 2: Acid as the critical additive for optimal performance.
With this optimal conditions in hand, we embarked on the evaluation of the substrate scope for this transformation. A variety of aromatic alkynes were initially tested as summarized in Table 2.
Table 2: Reaction scope of alkynes.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-288-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | Yieldb | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-288-i6.svg?max-width=637&scale=1.0)
|
2a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-288-i7.svg?max-width=637&scale=1.0)
|
3a | 95% |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-288-i8.svg?max-width=637&scale=1.0)
|
2b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-288-i9.svg?max-width=637&scale=1.0)
|
3b | 88% |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-288-i10.svg?max-width=637&scale=1.0)
|
2c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-288-i11.svg?max-width=637&scale=1.0)
|
3c | 89% |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-9-288-i12.svg?max-width=637&scale=1.0)
|
2d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-9-288-i13.svg?max-width=637&scale=1.0)
|
3d | 87% |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-9-288-i14.svg?max-width=637&scale=1.0)
|
2e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-9-288-i15.svg?max-width=637&scale=1.0)
|
3e | 59% |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-9-288-i16.svg?max-width=637&scale=1.0)
|
2f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-9-288-i17.svg?max-width=637&scale=1.0)
|
3f | 68% |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-9-288-i18.svg?max-width=637&scale=1.0)
|
2g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-9-288-i19.svg?max-width=637&scale=1.0)
|
3g | 86% |
| 8c |
![[Graphic 17]](/bjoc/content/inline/1860-5397-9-288-i20.svg?max-width=637&scale=1.0)
|
2h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-9-288-i21.svg?max-width=637&scale=1.0)
|
3h | 70% |
aGeneral conditions: 1 (0.4 mmol), 2 (1.2 equiv), 1 mol % XPhosAu(TA)OTf and 1 mol % Cu(OTf)2 in dry DCM (0.8 mL), rt. bIsolated yield. c1 (0.4 mmol), 2 (2 equiv), 3 mol % XPhosAu(TA)OTf and 3 mol % Cu(OTf)2.
This reaction tolerated both electron-rich (2b) and electron-deficient (2c) alkynes. Aromatic alkynes with substitutents at meta (2e) and ortho (2f) positions also worked well, although giving slightly lower yields. Electron-rich heterocycle-containing alkynes (2g) were also suitable substrates for this transformation. However, electron-poor alkynes, such as 2- and 3-pyridylacetylenes, did not undergo the reaction, likely caused by the low reactivity of the C–C triple bonds. Interestingly, the addition to the ethynylferrocene gave the corresponding vinyl ether in good yield, which highlighted the mild conditions of this catalytic system and potential applications of this gold catalyst in other metal containing compound syntheses. A series of aliphatic alkynes were also tested for this reaction. Alkynes containing 6-, 5- and 3-membered rings worked well, giving good to excellent yields (Table 3). Notably, the cyclopropylacetylene formed the direct O-addition adduct (6c) with no ring opening product observed. In addition, the conjugate enyne could undergo this reaction, giving the interesting electron-rich conjugated diene (6d). In some cases, thermodynamically more stable internal alkenes were also observed along with the kinetic product terminal alkene, likely through olefin isomerization [22].
Table 3: Reaction scope with aliphatic alkynes.a
![[Graphic 19]](/bjoc/content/inline/1860-5397-9-288-i22.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Substrate | Product | Yieldb | ||
|---|---|---|---|---|---|
| 1 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-9-288-i23.svg?max-width=637&scale=1.0)
|
5a |
![[Graphic 21]](/bjoc/content/inline/1860-5397-9-288-i24.svg?max-width=637&scale=1.0)
|
6a | 89% |
| 2c |
![[Graphic 22]](/bjoc/content/inline/1860-5397-9-288-i25.svg?max-width=637&scale=1.0)
|
5b |
![[Graphic 23]](/bjoc/content/inline/1860-5397-9-288-i26.svg?max-width=637&scale=1.0)
|
6b | 80% |
| 3 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-9-288-i27.svg?max-width=637&scale=1.0)
|
5c |
![[Graphic 25]](/bjoc/content/inline/1860-5397-9-288-i28.svg?max-width=637&scale=1.0)
|
6c | 86% |
| 4 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-9-288-i29.svg?max-width=637&scale=1.0)
|
5d |
![[Graphic 27]](/bjoc/content/inline/1860-5397-9-288-i30.svg?max-width=637&scale=1.0)
|
6d | 86% |
| 5 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-9-288-i31.svg?max-width=637&scale=1.0)
|
5e |
![[Graphic 29]](/bjoc/content/inline/1860-5397-9-288-i32.svg?max-width=637&scale=1.0)
|
6e | 67% |
aGeneral conditions: 1 (0.4 mmol), 5 (2 equiv), 1 mol % XPhosAu(TA)OTf and 1 mol % Cu(OTf)2 in dry DCM (0.8 mL), rt. bIsolated yield. cA ratio of 1:0.4 is observed for terminal/internal alkene mixtures. Combined yield.
The internal alkyne (1-phenyl-1-propyne), which was usually much less reactive than the terminal alkyne, was also tested. As expected, no reaction occurred at room temperature under the optimal conditions. To our delight, the desired products 8 were obtained while refluxing at 60 °C for 48 h, though in low yield. Two regioisomers were isolated, which were assigned as 8a and 8b (Scheme 3A). Diphenylacetylene gave trace amounts of the desired product under the identical conditions.
![[1860-5397-9-288-i3]](/bjoc/content/inline/1860-5397-9-288-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of internal alkyne and other O-nucleophiles. Isolated yields are given in paranthesis. aA ratio of 1:2.6 is observed for terminal/internal alkene mixtures. Combined yield.
Scheme 3: Reactions of internal alkyne and other O-nucleophiles. Isolated yields are given in paranthesis. aA...
Several diketones were tested to explore the scope of nucleophiles. The 1,3-cyclohexanedione derivative worked well, giving the vinyl ether 10a in good yield. The five-membered diketone, 1,3-cyclopentadione could also yield the desired O-vinylation product (10b–10d) in excellent yield, under similar conditions. Elevated temperature (50 °C) was required for good results due to the poor solubility of 1,3-cyclopentadione in DCM at room temperature. Finally and notably, all tested acyclic 1,3-diketones as well as 1,2-diketones gave no O-addition products under the current reaction conditions, likely caused by the intramolecular H-bonding.
Conclusion
In this letter, we report the first successful gold(I)-catalyzed intermolecular O-vinylation of cyclic 1,3-diketones with unactivated alkynes. The reaction tolerates a large scope of alkynes, giving the desired O-addition products in good to excellent yields. The triazole coordinated gold catalysts gave improved reactivity compared with the typical [L-Au]+ by overcoming the undesired hydration. This discovery will likely benefit many future developments that currently suffer from the common hydration side reaction. The application of copper(II) triflate as the effective additive not only improves the reactivity, but also provides another example for plausible bimetallic catalysis, which is a very active research area in the gold catalysis community. Evaluation of distinct O-nucleophiles toward intermolecular addition to alkynes is currently underway in our group.
Supporting Information
| Supporting Information File 1: General methods, characterization data and NMR spectra of synthesized compounds. | ||
| Format: PDF | Size: 15.3 MB | Download |
References
-
Hashmi, A. S. K.; Toste, F. D. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527646869
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Jiménez-Núñez, E. S.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c
Return to citation in text: [1] -
Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454
Return to citation in text: [1] -
Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368
Return to citation in text: [1] -
Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k
Return to citation in text: [1] -
Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c
Return to citation in text: [1] -
Teles, J. H.; Brode, S.; Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. doi:10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.3.CO;2-E
Return to citation in text: [1] [2] -
Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Angew. Chem., Int. Ed. 2002, 41, 4563–4565. doi:10.1002/1521-3773(20021202)41:23<4563::AID-ANIE4563>3.0.CO;2-U
Return to citation in text: [1] [2] -
Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067–2074. doi:10.1021/jo802558e
Return to citation in text: [1] [2] -
Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e
Return to citation in text: [1] [2] -
Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136
Return to citation in text: [1] [2] -
Corma, A.; Ruiz, V. R.; Leyva-Pérez, A.; Sabater, M. J. Adv. Synth. Catal. 2010, 352, 1701–1710. doi:10.1002/adsc.201000094
Return to citation in text: [1] [2] [3] -
Winternheimer, D. J.; Shade, R. E.; Merlic, C. A. Synthesis 2010, 2497–2511. doi:10.1055/s-0030-1258166
Return to citation in text: [1] -
Chary, B. C.; Kim, S. J. Org. Chem. 2010, 75, 7928–7931. doi:10.1021/jo101543q
Return to citation in text: [1] -
Luo, T.; Dai, M.; Zheng, S.-L.; Schreiber, S. L. Org. Lett. 2011, 13, 2834–2836. doi:10.1021/ol200794w
Return to citation in text: [1] -
Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. J. Org. Chem. 2010, 75, 2247–2258. doi:10.1021/jo1000143
Return to citation in text: [1] -
Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182
Return to citation in text: [1] [2] -
Lee, P. H.; Kim, S.; Park, A.; Chary, B. C.; Kim, S. Angew. Chem., Int. Ed. 2010, 49, 6806–6809. doi:10.1002/anie.201001799
Return to citation in text: [1] -
Nun, P.; Egbert, J. D.; Oliva-Madrid, M.-J.; Nolan, S. P. Chem.–Eur. J. 2012, 18, 1064–1067. doi:10.1002/chem.201103304
Return to citation in text: [1] -
Mo, J.; Kang, D.; Eom, D.; Kim, S. H.; Lee, P. H. Org. Lett. 2013, 15, 26–29. doi:10.1021/ol3029274
Return to citation in text: [1] -
Duan, H.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2009, 131, 12100–12102. doi:10.1021/ja9041093
Return to citation in text: [1] -
Chen, Y.; Yan, W.; Akhmedov, N. G.; Shi, X. Org. Lett. 2010, 12, 344–347. doi:10.1021/ol902680k
Return to citation in text: [1] -
Wang, D.; Ye, X.; Shi, X. Org. Lett. 2010, 12, 2088–2091. doi:10.1021/ol100576m
Return to citation in text: [1] -
Wang, D.; Gautam, L. N. S.; Bollinger, C.; Harris, A.; Li, M.; Shi, X. Org. Lett. 2011, 13, 2618–2621. doi:10.1021/ol200714h
Return to citation in text: [1] -
Wang, Q.; Aparaj, S.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Org. Lett. 2012, 14, 1334–1337. doi:10.1021/ol300227a
Return to citation in text: [1] -
Guérinot, A.; Fang, W.; Sircoglou, M.; Bour, C.; Bezzenine-Lafollée, S.; Gandon, V. Angew. Chem., Int. Ed. 2013, 52, 5848–5852. doi:10.1002/anie.201300600
Return to citation in text: [1]
| 1. | Hashmi, A. S. K.; Toste, F. D. Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, Germany, 2012. doi:10.1002/9783527646869 |
| 2. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 3. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 4. | Jiménez-Núñez, E. S.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326–3350. doi:10.1021/cr0684319 |
| 5. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 6. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 7. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c |
| 8. | Hashmi, A. S. K.; Hutchings, G. J. Angew. Chem., Int. Ed. 2006, 45, 7896–7936. doi:10.1002/anie.200602454 |
| 9. | Zhang, L.; Sun, J.; Kozmin, S. A. Adv. Synth. Catal. 2006, 348, 2271–2296. doi:10.1002/adsc.200600368 |
| 18. | Winternheimer, D. J.; Shade, R. E.; Merlic, C. A. Synthesis 2010, 2497–2511. doi:10.1055/s-0030-1258166 |
| 13. | Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Angew. Chem., Int. Ed. 2002, 41, 4563–4565. doi:10.1002/1521-3773(20021202)41:23<4563::AID-ANIE4563>3.0.CO;2-U |
| 14. | Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067–2074. doi:10.1021/jo802558e |
| 15. | Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e |
| 16. | Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136 |
| 17. | Corma, A.; Ruiz, V. R.; Leyva-Pérez, A.; Sabater, M. J. Adv. Synth. Catal. 2010, 352, 1701–1710. doi:10.1002/adsc.201000094 |
| 12. | Teles, J. H.; Brode, S.; Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. doi:10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.3.CO;2-E |
| 22. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 10. | Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766–1775. doi:10.1039/b615629k |
| 11. | Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. doi:10.1039/c1cs15279c |
| 23. | Lee, P. H.; Kim, S.; Park, A.; Chary, B. C.; Kim, S. Angew. Chem., Int. Ed. 2010, 49, 6806–6809. doi:10.1002/anie.201001799 |
| 24. | Nun, P.; Egbert, J. D.; Oliva-Madrid, M.-J.; Nolan, S. P. Chem.–Eur. J. 2012, 18, 1064–1067. doi:10.1002/chem.201103304 |
| 25. | Mo, J.; Kang, D.; Eom, D.; Kim, S. H.; Lee, P. H. Org. Lett. 2013, 15, 26–29. doi:10.1021/ol3029274 |
| 26. | Duan, H.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Am. Chem. Soc. 2009, 131, 12100–12102. doi:10.1021/ja9041093 |
| 27. | Chen, Y.; Yan, W.; Akhmedov, N. G.; Shi, X. Org. Lett. 2010, 12, 344–347. doi:10.1021/ol902680k |
| 28. | Wang, D.; Ye, X.; Shi, X. Org. Lett. 2010, 12, 2088–2091. doi:10.1021/ol100576m |
| 29. | Wang, D.; Gautam, L. N. S.; Bollinger, C.; Harris, A.; Li, M.; Shi, X. Org. Lett. 2011, 13, 2618–2621. doi:10.1021/ol200714h |
| 30. | Wang, Q.; Aparaj, S.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Org. Lett. 2012, 14, 1334–1337. doi:10.1021/ol300227a |
| 21. | Kuram, M. R.; Bhanuchandra, M.; Sahoo, A. K. J. Org. Chem. 2010, 75, 2247–2258. doi:10.1021/jo1000143 |
| 22. | Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2013, 52, 9767–9771. doi:10.1002/anie.201304182 |
| 31. | Guérinot, A.; Fang, W.; Sircoglou, M.; Bour, C.; Bezzenine-Lafollée, S.; Gandon, V. Angew. Chem., Int. Ed. 2013, 52, 5848–5852. doi:10.1002/anie.201300600 |
| 19. | Chary, B. C.; Kim, S. J. Org. Chem. 2010, 75, 7928–7931. doi:10.1021/jo101543q |
| 20. | Luo, T.; Dai, M.; Zheng, S.-L.; Schreiber, S. L. Org. Lett. 2011, 13, 2834–2836. doi:10.1021/ol200794w |
| 17. | Corma, A.; Ruiz, V. R.; Leyva-Pérez, A.; Sabater, M. J. Adv. Synth. Catal. 2010, 352, 1701–1710. doi:10.1002/adsc.201000094 |
| 12. | Teles, J. H.; Brode, S.; Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. doi:10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.3.CO;2-E |
| 13. | Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Angew. Chem., Int. Ed. 2002, 41, 4563–4565. doi:10.1002/1521-3773(20021202)41:23<4563::AID-ANIE4563>3.0.CO;2-U |
| 14. | Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067–2074. doi:10.1021/jo802558e |
| 15. | Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e |
| 16. | Krauter, C. M.; Hashmi, A. S. K.; Pernpointner, M. ChemCatChem 2010, 2, 1226–1230. doi:10.1002/cctc.201000136 |
| 17. | Corma, A.; Ruiz, V. R.; Leyva-Pérez, A.; Sabater, M. J. Adv. Synth. Catal. 2010, 352, 1701–1710. doi:10.1002/adsc.201000094 |
© 2013 Xi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)