Abstract
Anion–π catalysis, introduced in 2013, stands for the stabilization of anionic transition states on π-acidic aromatic surfaces. Anion–π catalysis on carbon allotropes is particularly attractive because high polarizability promises access to really strong anion–π interactions. With these expectations, anion–π catalysis on fullerenes has been introduced in 2017, followed by carbon nanotubes in 2019. Consistent with expectations from theory, anion–π catalysis on carbon allotropes generally increases with polarizability. Realized examples reach from enolate addition chemistry to asymmetric Diels–Alder reactions and autocatalytic ether cyclizations. Currently, anion–π catalysis on carbon allotropes gains momentum because the combination with electric-field-assisted catalysis promises transformative impact on organic synthesis.
Graphical Abstract
Introduction
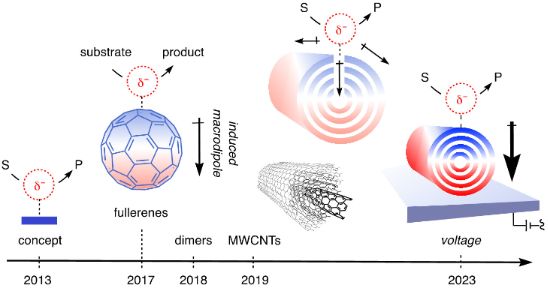
Anion–π catalysis was introduced ten years ago [1]. The idea is to stabilize anionic transition states on electron-deficient, π-acidic aromatic surfaces (Figure 1A). The true beginning is arguably in 2015 because it took some time to find the benchmark reaction needed to develop the catalysts (Figure 2) [2]. With this operational enolate chemistry in hand, it quickly became clear that increasing π acidity at the same time decreases the stability of the catalyst [3-5]. This suggested that induced rather than intrinsic anion–π interactions should provide access to really strong catalysts [3]. They have been predicted theoretically to occur on π-stacks [6], and confirmed recently to exist and apply to anion–π catalysis on π-stacked foldamers (Figure 1B) [7] and micelles [8]. However, due to their unique polarizability [9-11], the dream scaffolds for induced anion–π interactions are carbon allotropes. Anionic transition states placed on C60 fullerenes 1 will drive the 60 π electrons toward the other side, thus inducing a transient macrodipole that will stabilize the same transition state that induced its formation (Figure 1C) [12]. This intriguing mechanism of catalysis should be further intensified on single-walled carbon nanotubes 2 (SWCNTs, Figure 1D) and multi-walled carbon nanotubes 3 (MWCNTs, Figure 1E) [13]. Multiple substrate/transition-state binding should reduce particularly in-plane polarization of the π system and thus induced anion–π interactions. Since the polarization caused by substrate/transition-state binding hinders additional binding, this effect should occur only at high concentrations.
![[1860-5397-19-140-1]](/bjoc/content/figures/1860-5397-19-140-1.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (A) Anion–π catalysis: Stabilization of anionic transition states from substrate S to product P on π-acidic aromatic surfaces. (B) Anion–(π)n–π catalysis: Stabilization of anionic transition states by polarization of π stacks to induce oriented macrodipoles. (C–E) Anion–π catalysis on (C) C60 fullerenes 1, (D) SWCNTs 2 and (E) MWCNTs 3. Part E of Figure 1 was adapted from [44] (© 2023 M. A. Gutiérrez López et al., published by the American Association for the Advancement of Science, distributed under the terms of the Creative Commons Attribution 4.0 International License, https://creativecommons.org/licenses/by/4.0).
Figure 1: (A) Anion–π catalysis: Stabilization of anionic transition states from substrate S to product P on ...
![[1860-5397-19-140-2]](/bjoc/content/figures/1860-5397-19-140-2.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Bioinspired enolate addition chemistry to benchmark anion–π catalysts: Stabilization of “enol” intermediate II over “keto” intermediate I and nitronate transition state III by anion–π interactions (blue) selectively catalyzes the formation of the intrinsically disfavored enolate addition (A) product 6 over decarboxylation (D) product 7. A/D product ratios are used to characterize anion–π catalysis, increasing A/D ratios indicate increasing anion–π catalysis. PMP = para-methoxyphenyl.
Figure 2: Bioinspired enolate addition chemistry to benchmark anion–π catalysts: Stabilization of “enol” inte...
These expectations were first explored with anion–π catalysis on fullerenes in 2017 [12], followed by SWCNTs and MWCNTs two years later [13]. Particularly MWCNTs have the potential to couple anion–π and cation–π catalysis with electric-field-assisted catalysis [14]. While anion–π (and cation–π [15,16]) catalysis, compared to other unorthodox interactions, has been less impactful than expected [3,4,17-31], this combination has the potential to revolutionize organic catalysis [32-43]. These high expectations have never been realized for technical reasons. However, recent breakthroughs suggest that these methodological problems are now solved [44]. In electrochemical microfluidic reactors, electric-field-assisted anion–π catalysis on MWCNTs 3 is in place to lift anion–π catalysis on a level of general practical significance. For this reason, it appeared timely to recapitulate the results available so far on anion–π catalysis on carbon allotropes.
Review
Anion–π catalysis on fullerenes
The use of fullerenes in catalysis is surprisingly underdeveloped [45-51]. Anion–π and cation–π interactions on fullerenes attract similarly little attention until today [52-57]. Anion–π catalysis on fullerenes has been introduced in 2017 [12]. Fullerene anion–π catalysts were developed with the benchmark reaction introduced two years earlier (Figure 2) [2]. In this reaction, at the beginning of all biosynthesis, finetuned malonic acid half thioesters 4 [58-60] are deprotonated, and tautomers I and II add to an enolate acceptor. For anion–π catalysis, nitroolefins like 5 [60] were convenient acceptors because they are compatible with asymmetric catalysis, and because stabilization of intermediate III by privileged nitronate-π interactions drives the reaction forward. Decarboxylation of the resulting intermediate IV then affords the chiral addition product 6. This enolate addition is in kinetic competition with simple decarboxylation, yielding thioacetate 7. Under most conditions, this decarboxylation is favored. Anion–π catalysis selectively accelerates the intrinsically disfavored but significant enolate addition by stabilizing the planar sp2 intermediate II that has to add before decarboxylation can occur [2,61]. The non-planar sp3 keto intermediate I that can decarboxylate through intermediate V without preceding enolate addition is less stabilized on the planar π surfaces of anion–π catalysts. The A/D product ratio is thus a convenient measure for anion–π catalysis, the larger the better, with A/D > 1, the intrinsic selectivity for decarboxylation has been inverted [2,3,18].
Applying lessons from simpler anion–π catalysts, trialkylamines were tethered to C60 fullerenes [12]. These amines function as bases, their position next to the aromatic surface is essential to turn on anion–π interactions as soon as substrate 4 is deprotonated. Fullerene derivatization with the Bingel reaction installs a cyclopropane that continues with one or two acid derivatives. In fullerene 8, this is an amide with a short ethylene tether for the tertiary amine (Figure 3).
![[1860-5397-19-140-3]](/bjoc/content/figures/1860-5397-19-140-3.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Structure and activity of fullerene-amine dyads to catalyze the intrinsically disfavored but biologically relevant enolate addition chemistry in Figure 2. A/D ratios of fullerene catalysts are normalized against the A/D0 ratios of the fullerene-free controls (9, 12, 18, 20, 23), increasing A/D ratios indicate increasing anion–π catalysis on fullerenes.
Figure 3: Structure and activity of fullerene-amine dyads to catalyze the intrinsically disfavored but biolog...
Fullerene 8 catalyzed the formation of the addition product 6 with high selectivity over the decarboxylation product 7 [12]. The experimental product ratio A/D8 was divided by the product ratio A/D9 measured with the fullerene-free 9. The resulting A/D8/9 = 2.6 reports the isolate contribution of the fullerene to catalysis in a comparable manner. This high value supported that the high polarizability of fullerenes provides access to strong induced anion–π interactions for efficient anion–π catalysis (Figure 1C).
Elongation of the tether in fullerenes 10 and 11 reduced product ratios to still significant A/D10/9 ~ A/D11/9 = 2.1. The origin of this decrease in anion–π catalysis is likely to include increasing entropy losses in pseudo-macrocyclic transition states. Normalized against the fullerene-free control 12, a secondary Bingel amide in 13 caused a drop to A/D13/12 = 1.6. Similarly, low A/D14/12 = 1.5 for an ester in 14 supported that removal of the hydrogen-bond donor in 5 rather than steric constraints account for the decrease in anion–π catalysis. Elongation of the tether in the ester series 14–16 was as in the amide series from 8 and thus supported entropic contributions to anion–π catalysis. Steric increase of the secondary amide in 17 impeded anion–π catalysis, presumably because the catalytic π surface next to the ammonium cation became inaccessible for anions paired with the tethered ammonium cation. Normalized against the fullerene-free control 18, an electron-withdrawing cyano group on the cyclopropane in 19 gave similarly poor A/D19/18 = 1.5.
At constant tether length as short as possible, strong increases in anion–π catalysis were found by further minimizing entropy losses in pseudo-macrocyclic transition states, bound non-covalently to both the active π surface and the ammonium cation of the tether. Normalized against the fullerene-free control 20, the most preorganized dyad 21 gave a new record A/D21/20 = 4.1. This value was much higher than the previous best A/D8/9 = 2.6 with a freely rotating tether. Further increasing activity with fullerene 22 should not be overrated because A/D22/23 = 4.6 was recorded against triethylamine (23), which is a less precise control compared to the exactly matching 20.
Asymmetric anion–π catalysis on fullerenes
The best fullerene catalyst 21 was applied to other reactions. Diels–Alder reactions are of special interest for anion–π catalysis because of the promise to accelerate an intrinsically disfavored but relevant pathway, like in the benchmark enolate addition (Figure 2) [62]. Namely, in solution, the endo transition state VI is preferred to maximize orbital overlap (Figure 4) [63]. For π-acidic surfaces, the exo transition state VII is more completely accessible (Figure 4). The 3-hydroxy-2-pyrone (24) was selected as representative diene for the anionic [4 + 2] cycloaddition with maleimide 25 as standard dienophile to afford endo product 26 and exo product 27 [64-66]. With the less powerful fullerene catalyst 14, the increase of the exo/endo selectivity compared to the fullerene-free control 12, i.e., exo/endo14/12 = 1.1, was negligible [12]. With the best fullerene catalyst 21, the presence of the fullerene made the diastereoselectivity ratio with exo/endo21/20 = 1.9 nearly doubled. The same was true for the enantioselectivity, which increased from 23% ee for control 20 to 55% ee for anion–π catalyst 21. These results were in support that stabilization of the intrinsically disfavored exo transition state VII on the polarizable surface of carbon allotropes can increase diastereo- and enantioselectivity of Diels–Alder reactions.
![[1860-5397-19-140-4]](/bjoc/content/figures/1860-5397-19-140-4.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Asymmetric anion–π catalysis of intrinsically disfavored exo-selective Diels–Alder reactions on fullerene catalyst 21, with notional endo and exo transition states VI and VII, respectively.
Figure 4: Asymmetric anion–π catalysis of intrinsically disfavored exo-selective Diels–Alder reactions on ful...
The direct formation of 1,3-nonadjacent stereocenters is a topic of concern in asymmetric catalysis [67]. To explore compatibility with anion–π catalysis, the addition of ethyl 2-cyano-2-phenylacetate (28) to 2-chloroacrylonitrile (29) was selected (Figure 5) [68]. In the presence of 5 mol % of the best fullerene catalyst 21, conversion into dicyanide 30 reached 72% within 5 days at ambient temperature [67]. While enantioselectivity was negligible, dr 5.3:1 was the best diastereoselectivity among all tested anion–π catalysts. This result was consistent with the stabilization of the anionic intermediates IX and X and the respective transition states on the polarized fullerene surface.
![[1860-5397-19-140-5]](/bjoc/content/figures/1860-5397-19-140-5.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Asymmetric anion–π catalysis to install remote stereogenic centers on fullerene catalyst 21, with notional transition states IX and X.
Figure 5: Asymmetric anion–π catalysis to install remote stereogenic centers on fullerene catalyst 21, with n...
Anion–π autocatalysis on fullerenes
The autocatalysis of epoxide-opening ether cyclization on π-acidic aromatic surfaces has been identified in 2018 as an emergent property of anion–π catalysis [69]. In this series, fullerene catalyst 31 was found to catalyze the cyclization of epoxide 32 into THF 33 (Figure 6). The rate enhancement for catalysis was 270, whereas autocatalysis accelerated the reaction by 1045 M−1. The origin of autocatalysis on π-acidic surfaces was clarified only this year because the involvement of two molecules of water in the decisive transition state XI complicated the situation [70].
![[1860-5397-19-140-6]](/bjoc/content/figures/1860-5397-19-140-6.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Primary anion–π autocatalysis on monofunctional fullerene 31, with catalytic and autocatalytic rate enhancements, representative cascade cyclizations with anti-Baldwin (XII) and Baldwin (XIII) selectivity in nature, and primary anion–π autocatalysis of cascade cyclization XIV on catalysts other than carbon allotropes.
Figure 6: Primary anion–π autocatalysis on monofunctional fullerene 31, with catalytic and autocatalytic rate...
This reaction was intriguing for reasons beyond autocatalysis. It is the only example so far where fullerenes act as anion–π catalyst without additional activating groups, usually a tethered base to inject a negative charge into the substrate directly on top of the catalytic π surface [69]. The tert-butyl esters in Bingel fullerene 31 serve only to improve solubility and are not expected to participate in catalysis. Moreover, epoxide opening polyether cyclizations are among the most impressive cascade reactions in nature [71-73]. Best known is the hypothetical cascade XII in the biosynthesis of brevetoxin B [74]. It affords eleven fused ethers by violating the Eschenmoser–Dunitz–Baldwin guidelines [75-78] in every step. Among the Baldwin compatible examples from nature, the cascade XIII leading to monensin A is arguably the most popular [79]. On anion–π catalysts other than carbon allotropes, up to four epoxides 34 have been cyclized into the monensin-like tetra-THF 35 as outlined in intermediate XIV [24,25]. Thus, this reaction can be considered as the anion–π catalysis counterpart of the steroid cyclizations catalyzed in nature with the charge inverted, conventional cation–π interactions [15].
Anion–π catalysis on fullerene dimers
The strength of anion–π interactions increases with face-to-face π stacking because the delocalized π electrons move within the stack away from the charge, which induces a macrodipole along the stack that supports the binding of the anion (Figure 1B) [61]. What works for anion–(π)n–π catalysis on π-stacked foldamers [61] and micelles [8] should apply to fullerenes as well. The unique polarizability of fullerene monomers like 1 is thought to induce strong anion–π interactions and thus account for the observed catalytic activity (Figure 7A) [12]. This polarizability should further increase in fullerene dimers like 36 [80]. Anions, anionic reactive intermediates and anionic transition states on top of fullerenes dimers should thus induce a larger macrodipole which should in turn strengthen their binding to the expanded π system and thus increase anion–π catalysis (Figure 1B).
![[1860-5397-19-140-7]](/bjoc/content/figures/1860-5397-19-140-7.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: (A) Macrodipoles induced by anionic transition states account for anion–π catalysis on fullerenes. (B) Fullerene dimers double polarizability of the π system and thus anion–π catalysis, with structure of fullerene dimer 37, intercalating activators and inactivators, monomeric fullerene and fullerene-NDI controls, and A/D ratios for products 6 and 7 normalized against the A/D0 ratio of the fullerene-free control 23, increasing A/D ratios indicate increasing anion-π catalysis.
Figure 7: (A) Macrodipoles induced by anionic transition states account for anion–π catalysis on fullerenes. ...
Fullerene dimer 37 was equipped with the tethered tertiary amine needed to deprotonate the substrate 4 and produce the conjugate bases I and II as the first reactive intermediates directly on the active π surface (Figure 2 and Figure 7) [80]. Calibrated against triethylamine 23, fullerene dimer 37 catalyzed enolate addition with an A/D37/23 = 12.5. This is more than twice the A/D22/23 = 4.6 of the respective monomer 22, which was already best among fullerene monomers. In computational simulations, the positive maximum of the MEP surface next to the amine base was the same for dimer 37 (+13.9 kJ mol−1) and monomer 22 (+13.8 kJ mol−1), supporting that the dramatic increase in activity did not originate from a change in intrinsic anion–π interactions. The computed polarizability of dimer 37 (α = 1499 a.u.) was almost twice as high as that of monomer 22 (α = 903 a.u.), confirming that powerful induced anion–π interactions account for the high catalytic activity.
Twenty equivalents of the π-acidic intercalator 38 increased the catalytic activity of fullerene dimer 37 by 20%. This increase was consistent with the formation of the π-stacked complex 39 with increased polarizability and electron deficiency. With the π-basic intercalator 40, the catalytic activity of fullerene dimer 37 decreased by 20%. This complementary decrease supported the formation of complex 41 with decreased electron deficiency. Control experiments with fullerene monomer 22 gave unchanged catalytic activity with intercalators 38 or 40, which suggested that complexes 42 and 43 do not form. The insensitivity of fullerene monomers 22 thus supported that 38 and 40 activate and inactivate fullerene dimers 37 by intercalation in-between the two fullerenes.
Replacement of the second fullerene in dimer 37 with a poorly polarizable naphthalenediimide (NDI) in 44 increased the catalytic activity of fullerene monomer 22 much less significantly. With less electron-deficient NDIs carrying two sulfide donors in the core, the catalytic activity of 45 dropped below that of fullerene monomer 22. Oxidation of the sulfide donors into sulfoxide acceptors increased the catalytic activity much less than expected, resting below that of unsubstituted NDIs despite stronger π acidity. Supported by experimental and computational data, this poor performance of dyad 46 was explained by lone-pair π interactions of the fullerene with the donating oxygens. These lone-pair π interactions were even more impactful on the sulfone level. Compared to the sulfoxides in 46, the catalytic activity of dyad 47 decreased rather than increased despite stronger π acidity.
Supported by computational predictions [80,81], record activities in anion–π catalysis with fullerene dimers called for higher oligomers. However, synthetic efforts were not fruitful, mostly due to poor solubility.
Anion–π catalysis on carbon nanotubes
With fullerenes confirmed as privileged scaffold for induced anion–π catalysis but higher oligomers inaccessible [12,67,80], the obvious next move was to switch to carbon nanotubes. Compared to the sixty free electrons rushing toward one side of a C60 fullerene 1 to create a large macrodipole in response to an anionic transition state, the number of electrons available to maximize polarizability already in SWCNTs 2 is much higher (Figure 1D) [9-11]. Continuing in this series to increase polarizability with carbon allotropes, MWCNTs 3 emerge as privileged scaffold for anion–π catalysis because polarizability is possible not only along the tubes but also between the nanotubes, like in π-stacked foldamers (Figure 1E).
Numerous reports on catalysis with carbon nanotubes have appeared [45,46,82-94]. They serve as catch-and-release scaffolds in different variations, and, less frequently, as (photo)redox partners. Although they might contribute to these activities, anion–π interactions have not been considered. Anion–π catalysis on carbon nanotubes has been introduced explicitly in 2019 [13]. Already in the presence of pristine SWCNTs 2, the ability of TEA 23 to catalyze enolate addition with 4 increased to A/D48/23 = 1.2 for a virtual catalytic complex 48 between the two (Figure 8). Covalent modification of SWCNTs with tertiary amines as in 49 further increased activity to A/D49/23 = 2.0. Suppression of this increase in a virtual complex 50 with a competitive inhibitor 51 further supported that the observed activity originates from anion–π catalysis.
![[1860-5397-19-140-8]](/bjoc/content/figures/1860-5397-19-140-8.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Structure and activity of covalently and non-covalently modified SWCNTs and MWCNTs, with A/D ratios for products 6 and 7 normalized against the A/D0 ratios of the CNT-free controls, increasing A/D ratios indicate increasing anion–π catalysis.
Figure 8: Structure and activity of covalently and non-covalently modified SWCNTs and MWCNTs, with A/D ratios...
With A/D52/23 = 1.3, pristine MWCNTs 3 failed to increase the activity of TEA 23 much more than SWCNTs 2. This is not surprising because virtual complexes 52 and 48 are not expected to exist to an appreciable extent. With covalent modification, MWCNTs 53 with A/D53/23 = 7.3 outperformed the corresponding SWCNTs 49 with A/D49/23 = 2.0 clearly. This significant increase in activity was consistent with the increase in polarizability from SWCNTs 2 to MWCNTs 3 (Figure 1). Compared to fullerene monomers and dimers, this activity cannot be overestimated because MWCNTs operate in suspension rather than solution, that is as formal heterogenous rather than homogenous catalysts.
Inhibition of the covalently modified MWCNT 53 with inhibitor 51 in complex 54 was supported by a drop from A/D53/23 = 7.3 to A/D54/23 = 4.8. Under identical conditions, the much more π-basic inhibitor 55 inhibited enolate addition by the MWCNT 53 almost completely, i.e., A/D56/23 = 1.9 for the formal catalyst-inhibitor complex 56. Increasing inhibition with the π basicity of the inhibitor was consistent with powerful anion–π interactions accessible on MWCNTs for efficient anion–π catalysis.
Like in the fullerene series, elongation of the tether in 57 decreased catalytic activity to still important A/D57/23 = 5.0, presumably due to entropic reasons. The activity of inhibitor 51 remained detectable under standard conditions, thus supporting the formation of complex 58 with A/D58/23 = 3.6. Unlike the fullerene series, preorganization of the tight tether in MWCNT 59 with A/D59/23 = 4.7 did not give the best activity. This suggested that with the fully rigid and tight tether, small differences in local environment turned an advantageous match with the most convex surface of fullerene 21 into a slight mismatch with MWCNTs 59. However, activities remained high and inhibition with inhibitor 51 in formal complex 60 was with A/D60/23 = 3.3 preserved, also with this mildly mismatched tether.
The π-acidic NDI 61 with a Leonard-turned tertiary amine is a privileged small-molecule anion–π catalyst that catalyzes the enolate addition of malonate 4 with an activity that, however, does not reach that of the best carbon allotropes [95]. In the presence of pristine MWCNTs 3, the activity of the NDI catalyst 61 increased significantly, implying the formation of complex 62 with A/D62/61 = 7.1. While the reaction presumably still occurred on the NDI surface, this very high A/D value suggested that efficient face-to-face stacking connected the active NDI surfaces to the nanotube to increase intrinsic anion–π interactions with the highest polarizability.
For interfacing with CNTs, the π-basic pyrenes rather than the π-acidic NDIs are most popular [82,88,90,96-99]. Equipped with a tertiary amine, the catalytic activity of dyad 63 increased only slightly in the presence of MWCNTs 3, suggesting that the formed π-basic complex 64 is with A/D64/63 = 1.2 much less active than the π-acidic NDI complex 62 with A/D62/61 = 7.1, which is as expected for operational anion–π catalysis. With a longer tether, the already weak activity of pyrene 65 even decreased rather than increased in the presence of MWCNTs 3, with a formal complex 66 operating with A/D66/65 = 0.5. The activity of the fullerene catalyst 8 did not increase much in the presence of MWCNTs 3, giving formal complex 67 with A/D67/8 = 1.4. This was meaningful because the high activity of fullerene 8 with A/D8/9 = 2.6 (Figure 3) leaves little room to improve, and the interaction between convex aromatic surfaces is not favorable.
Taken together, anion–π catalysis on multiwalled carbon nanotubes (53, A/D = 7.3) is better than on single-walled carbon nanotubes (49, A/D = 2.0) and monomeric fullerenes (22, A/D = 4.6) but weaker than on fullerene dimers (37, A/D = 12.5). The true activity is presumably much higher because MWCNTs, and SWCNTs, are heterogenous catalysts that operate as suspensions with only a minor fraction of the total π surface accessible for function. Fullerene monomers and dimers, in contrast, are homogenous catalysts that are fully dissolved and thus fully accessible. Anion–π catalysis thus increases roughly with the polarizability of the carbon allotrope. The overall less convincing activities found with SWCNTs could originate from more favorable weakening of the exclusively in-plane polarization by multiple substrate/transition-state binding under the selected experimental conditions. Besides these overall minor reservations for SWCMTs, the found activities are among the best observed [3], confirming the promise of induced anion–π interactions on carbon allotropes for catalysis.
In contrast to these conclusions made with modified carbon nanotubes, epoxide-opening ether cyclization of 32 occurred on undermodified fullerene 31 (Figure 6) but not on pristine MWCNTs 3 (Figure 9). Modified carbon nanotubes exert their catalytic power because the tethered base injects the negative charge into the substrate right on top of the polarizable π surface of the tube. Inability of pristine MWCNTs 3 to open and cyclize epoxide 32 thus implied insufficient substrate binding to form substrate-catalyst complexes that can proceed to transition state XV (Figure 9A). To increase binding, substrates would have to be equipped with interfacers. For MWCNTs 3, interfacing with pyrene is most popular [82,88,90,96-99]. A pyrene interfacer was thus attached to substrate 32 [44]. Binding of the pyrene in the resulting substrate 68 to the MWCNT catalyst 3 should then afford a substrate-catalyst complex strong enough to access the interfaced transition state XVI and yield product 69. Already the presence of 9 wt % of MWCNT suspensions gave rate enhancements of 390. They increased with increasing MWCNT concentrations.
![[1860-5397-19-140-9]](/bjoc/content/figures/1860-5397-19-140-9.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: (A) Epoxide-opening ether cyclization on pristine carbon nanotubes occurs with (XVI) but not without (XV) interfacers such as pyrene in 68 or DAN in 73, catalyst inactivation with 70 in formal complex 71 and product 69 in formal complex 72 supports anion-π catalysis but not autocatalysis. (B) Electric-field-induced anion–π catalysis on MWCNTs 3 in electrochemical microfluidic reactors, with reactor design (C) and notional transition states for ether cyclization of 68 at positive (XVIII), zero (XVII) and negative voltage (XIX). Parts B and C of Figure 9 were adapted from [44] (© 2023 M. A. Gutiérrez López et al., published by the American Association for the Advancement of Science, distributed under the terms of the Creative Commons Attribution 4.0 International License, https://creativecommons.org/licenses/by/4.0).
Figure 9: (A) Epoxide-opening ether cyclization on pristine carbon nanotubes occurs with (XVI) but not withou...
In the presence of 25 mol % of inhibitor 70, rate enhancements dropped to 11. This decrease was consistent with the formation of the formal inhibitor-catalyst complex 71, where the π-basic DANs 70 stacked onto MWCNTs hinder the access of substrate 68, physically, electrostatically, or both. The addition of 25 mol % product 69 at the beginning of the reaction decelerated rather than accelerated the cyclization from rate enhancements of 390 down to 5. The formal product-catalyst complexes 72 were, however, weak enough not to interfere with full substrate conversion, that is turnover. They provided support for the occurrence of anion–π catalysis but not autocatalysis on MWCNTs. The disappearance of autocatalysis on MWCNTs provided consistent support that the polarization of the extended π system induced by substrate/transition-state binding hinders additional binding. An anionic transition state on the extended π surface thus not only induces its own stabilization by polarization, i.e., self-creates its own catalyst, but also self-protects against its destruction by multiple binding at sufficiently low concentrations (Figure 1).
Rate enhancements for the conversion of substrate 73 into product 74 in the presence of 9 wt % pristine MWCNTs 3 were not as high as with substrate 68. This result was consistent with pyrene as best interfacers for MWCNTs. For anion–π catalysis on NDI stacks, the reversed order was obtained, consistent with the preferred formation of DAN-NDI charge transfer complexes [8,100-106].
The efficient conversion of interfaced substrates 68 and 73 on pristine MWCNTs 3 was of particular interest with regard to catalysis initiated by oriented external electric fields (OEEFs, Figure 9B). The idea of OEEF catalysis has been around for a long time as a promising, bioinspired concept to revolutionize organic synthesis on the broadest sense [32-35]. Indeed, every chemical transformation can be seen as a directional displacement of point charges, electrons. Control over speed and direction of this charge translocation by OEEFs should allow to generally manipulate molecular transformation, from speed to selectivity and access to completely new reactions. Internal electric fields have been shown to account for much of the power of enzymes [41-43]. The translation of these lessons from nature into OEEF catalysis has so far been slow for a series of most demanding challenges [32-40]. It has been shown that anion–π catalysis with NDIs on ITO electrodes could solve some but not all of these challenges, and relevance for practice remained negligible [14].
Operational anion–π catalysis on MWCNTs fundamentally changed this situation. Integrated into electrochemical microfluidic reactors [107-110], this breakthrough promised to solve all problems obstructing the use of OEEF catalysis in a remarkably straightforward manner. Electrochemical microfluidic reactors should provide access to strong fields at voltages low enough to avoid electron transfer, offer high enough effective catalyst to substrate ratios and work without interfering electrolytes (Figure 9C). MWCNTs drop casted on graphite anodes [111] then should translate the applied OEEFs into strong local macrodipoles. These oriented macrodipoles, depending on the sign of the applied voltages, should then enable strong anion–π and cation–π interactions [112-115] and accelerate and direct the electron displacement during the reaction. Possible limitations at high concentrations in suspension from catalyst depolarization by multiple binding (Figure 1) naturally do not apply to permanent polarization by OEEFs (Figure 9B).
To elaborate on these great expectations, pristine MWCNTs 3 were drop casted on the graphite anodes of electrochemical microfluidic reactors [44]. The substrate was injected by a syringe pump, the product was collected at the other end of the microflow channel and analyzed by HPLC (Figure 9C). Without applied voltage, the reaction essentially did not occur under these heterogenous conditions. This inactivity indicated that transition state XVII, characterized by interfacing and induced MWCNT polarization by the transition state itself but not from OEEFs, is hardly accessible (Figure 9B). With increasing positive voltage applied, conversion increased in a pseudo-linear manner. At 6 V, the conversion after one passage through the system at a flow rate Qv = 25 µL min–1 increased to 31%. The emergence of conversion in response to applied voltage was consistent with operational OEEF catalysis, that is the stabilization of transition state XVIII by the local oriented macrodipole of MWCNTs that are polarized by the OEEF applied. Inversion of the applied voltage to –3 V removed the minimal activity present without voltage. This result was important because it was not only consistent with the existence of OEEF catalysis with a destabilized transition state XIX. It also disfavored contributions from SN1-type mechanisms and, most important, from electron transfer.
Anion–π catalysis on graphite
Beginning with spherical fullerenes, expansion of the aromatic surface of carbon allotropes leads to SWCNTs and MWCNTs. Further expansion by unrolling nanotubes into infinite sheets leads to graphene 75 as formal homolog of SWCNTs 2 and graphite 76 as formal homolog of MWCNTs 3 (Figure 10) [112,114-123]. Graphite is the oldest, most common carbon allotrope composed of sp2 hybridized atoms, complementary to diamond as the archetypal sp3 carbon allotrope. Conductive because of the delocalized electrons of the giant π system, graphite is commonly used as electrode. With this giant π system, graphite qualifies as potential anion–π and cation–π catalyst, depending on the orientation of the planes in the solid.
![[1860-5397-19-140-10]](/bjoc/content/figures/1860-5397-19-140-10.jpg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Electric-field-induced anion–π catalysis on MWCNTs 3 on graphite 76 in electrochemical microfluidic reactors, with reactor design (A) and notional transition states for ether cyclization of 68 at positive (XX) and zero voltage (XXI) on graphite 76. Figure 10 was adapted from [44] (© 2023 M. A. Gutiérrez López et al., published by the American Association for the Advancement of Science, distributed under the terms of the Creative Commons Attribution 4.0 International License, https://creativecommons.org/licenses/by/4.0).
Figure 10: Electric-field-induced anion–π catalysis on MWCNTs 3 on graphite 76 in electrochemical microfluidic...
Also in electromicrofluidic reactors, graphite electrodes are used. It was thus tempting to try OEEF-induced anion–π catalysis in the absence of MWCNTs, directly on graphite. However, without MWCNTs drop casted on the graphite electrode, the conductivity of the reactor was much lower [44]. This meant that already small currents produce high voltage. Despite this undesirable situation, conversion of 68 was observed to increase with increasing voltage. This observation was particularly intriguing because electric-field-induced anion–π catalysis on graphite might be relevant with regard to early steps in the origin of life [124,125].
Conclusion
Anion–π catalysis on carbon allotropes originates from the observation that access to strong anion–π interactions by increasing the intrinsic π acidity of aromatic surfaces is not realistic. Any permanent withdrawal of electron density destabilizes the aromatic system before anion–π interactions would become really attractive for catalysis [3]. This observation called for a shift of attention from intrinsic to induced anion–π interactions. Polarizability of the aromatic system has been predicted since the beginning to provide access to really strong anion–π interactions [61]. For anion–π catalysis, the shift from intrinsic to induced anion–π interactions suggests that the anionic transition state will induce the formation of its own catalyst. Close to a polarizable π surface, such an anionic transition state will drive all movable electrons away. This induced charge relocation will generate strong macrodipoles which are oriented to stabilize the same anionic transition state that induces their formation (Figure 1).
The shift of attention from intrinsic to induced anion–π interactions thus called for aromatic systems of highest polarizability, that is carbon allotropes [9-11]. This account recapitulates how anion–π catalysis on carbon allotropes was explored first on fullerene monomers, then fullerene dimers, SWCNTs and MWCNTs. Studies mainly focusing on enolate addition chemistry showed that selectivity generally increases with the polarizability of the carbon allotrope. Other reactions like asymmetric anion Diels-Alder reactions, the construction of 1,3-nonadjacent stereocenters and bioinspired ether cyclizations have been realized as well.
The emerging combination with oriented external electric fields changes the mechanism of anion–π catalysis on carbon allotropes [44]. Rather than an anionic transition state creating its own catalyst, the OEEF polarizes the carbon allotrope in advance. The resulting macrodipoles then should enable strong anion–π and cation–π interactions depending on their orientation, and accelerate and direct electron displacement during the reaction. This translation of the external field into local oriented macrodipoles solves one important challenge that has delayed progress with OEEF-induced catalysis. Namely, the fields predicted to accelerate and direct the flow of electrons during a reaction are much larger than the voltage needed to turn-on electron transfer and redox chemistry. This dilemma is overcome by carbon allotropes. They translate voltages weak enough to avoid electron transfer into oriented local molecular macrodipoles that are strong enough to access significant anion–π and cation–π interactions and thus accelerate electrons movement during a reaction.
The rich collection of additional problems holding back progress with OEEF-induced catalysis could be addressed with electrochemical microfluidic reactors. Most important are high effective catalyst to substrate ratios, high fields from small voltages, and no need to add electrolytes. These electrochemical microfluidic reactors have been constructed for a completely different purpose, that is practical access to organic redox chemistry [107-110]. Our results suggest that there might be more to win before the electrons jump. Being general and easy to use, the introduced supramolecular organocatalytic systems promise to lift all involved topics to a new level of significance. Essentially every reaction consists of the movement of electrons, from nucleophile to electrophile. To accelerate and direct this charge displacement, any electron-rich motif in transition states and reactive intermediates should be stabilizable by induced anion–π interactions with MWCNTs that are polarized by an electric field. Inversion of the applied voltage should allow to stabilize the respective electron-poor motifs by induced cation–π interactions, and the combination with co-catalysts interfaced on MWCNTs should provide general access to asymmetric catalysis. OEEF-induced anion–π and cation–π catalysis on carbon allotropes, affordable, clean and general, thus have the potential to non-covalently electrify organic synthesis in the broadest sense.
References
-
Zhao, Y.; Domoto, Y.; Orentas, E.; Beuchat, C.; Emery, D.; Mareda, J.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2013, 52, 9940–9943. doi:10.1002/anie.201305356
Return to citation in text: [1] -
Zhao, Y.; Benz, S.; Sakai, N.; Matile, S. Chem. Sci. 2015, 6, 6219–6223. doi:10.1039/c5sc02563j
Return to citation in text: [1] [2] [3] [4] -
Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Eur. J. 2022, 28, e202103303. doi:10.1002/chem.202103303
Return to citation in text: [1] [2] -
Miros, F. N.; Zhao, Y.; Sargsyan, G.; Pupier, M.; Besnard, C.; Beuchat, C.; Mareda, J.; Sakai, N.; Matile, S. Chem. – Eur. J. 2016, 22, 2648–2657. doi:10.1002/chem.201504008
Return to citation in text: [1] -
Frontera, A.; Quiñonero, D.; Garau, C.; Costa, A.; Ballester, P.; Deyà, P. M. J. Phys. Chem. A 2006, 110, 9307–9309. doi:10.1021/jp062176e
Return to citation in text: [1] -
Keshri, S. K.; Ishizuka, T.; Kojima, T.; Matsushita, Y.; Takeuchi, M. J. Am. Chem. Soc. 2021, 143, 3238–3244. doi:10.1021/jacs.0c13389
Return to citation in text: [1] -
Tan, M.-L.; Ángeles Gutiérrez López, M.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2023, 62, e202310393. doi:10.1002/anie.202310393
Return to citation in text: [1] [2] [3] -
Sabirov, D. S. RSC Adv. 2014, 4, 44996–45028. doi:10.1039/c4ra06116k
Return to citation in text: [1] [2] [3] -
Zhang, Y.; Wang, D.; Wang, W. Comput. Theor. Chem. 2018, 1128, 56–59. doi:10.1016/j.comptc.2018.02.011
Return to citation in text: [1] [2] [3] -
Sabirov, D. S.; Tukhbatullina, A. A. Nanomaterials 2022, 12, 4404. doi:10.3390/nano12244404
Return to citation in text: [1] [2] [3] -
López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Bornhof, A.-B.; Vázquez‐Nakagawa, M.; Rodríguez‐Pérez, L.; Ángeles Herranz, M.; Sakai, N.; Martín, N.; Matile, S.; López‐Andarias, J. Angew. Chem., Int. Ed. 2019, 58, 16097–16100. doi:10.1002/anie.201909540
Return to citation in text: [1] [2] [3] -
Akamatsu, M.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2017, 139, 6558–6561. doi:10.1021/jacs.7b02421
Return to citation in text: [1] [2] -
Kennedy, C. R.; Lin, S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2016, 55, 12596–12624. doi:10.1002/anie.201600547
Return to citation in text: [1] [2] -
Kutateladze, D. A.; Strassfeld, D. A.; Jacobsen, E. N. J. Am. Chem. Soc. 2020, 142, 6951–6956. doi:10.1021/jacs.0c02665
Return to citation in text: [1] -
Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2021, 60, 20650–20655. doi:10.1002/anie.202106509
Return to citation in text: [1] -
Maynard, J. R. J.; Galmés, B.; Stergiou, A. D.; Symes, M. D.; Frontera, A.; Goldup, S. M. Angew. Chem., Int. Ed. 2022, 61, e202115961. doi:10.1002/anie.202115961
Return to citation in text: [1] [2] -
Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Asian J. 2021, 16, 3599–3603. doi:10.1002/asia.202100920
Return to citation in text: [1] -
Giese, M.; Albrecht, M.; Rissanen, K. Chem. Commun. 2016, 52, 1778–1795. doi:10.1039/c5cc09072e
Return to citation in text: [1] -
Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature 2017, 543, 637–646. doi:10.1038/nature21701
Return to citation in text: [1] -
Guo, S.-Y.; Guo, Q.-H.; Tong, S.; Wang, M.-X. Angew. Chem., Int. Ed. 2020, 59, 8078–8083. doi:10.1002/anie.201915839
Return to citation in text: [1] -
Cotelle, Y.; Lebrun, V.; Sakai, N.; Ward, T. R.; Matile, S. ACS Cent. Sci. 2016, 2, 388–393. doi:10.1021/acscentsci.6b00097
Return to citation in text: [1] -
Paraja, M.; Matile, S. Angew. Chem., Int. Ed. 2020, 59, 6273–6277. doi:10.1002/anie.202000579
Return to citation in text: [1] [2] -
Paraja, M.; Hao, X.; Matile, S. Angew. Chem., Int. Ed. 2020, 59, 15093–15097. doi:10.1002/anie.202000681
Return to citation in text: [1] [2] -
Gini, A.; Paraja, M.; Galmés, B.; Besnard, C.; Poblador-Bahamonde, A. I.; Sakai, N.; Frontera, A.; Matile, S. Chem. Sci. 2020, 11, 7086–7091. doi:10.1039/d0sc02551h
Return to citation in text: [1] -
Chen, H.; Frontera, A.; Ángeles Gutiérrez López, M.; Sakai, N.; Matile, S. Helv. Chim. Acta 2022, 105, e202200119. doi:10.1002/hlca.202200119
Return to citation in text: [1] -
Chen, H.; Li, T.-R.; Sakai, N.; Besnard, C.; Guénée, L.; Pupier, M.; Viger-Gravel, J.; Tiefenbacher, K.; Matile, S. Chem. Sci. 2022, 13, 10273–10280. doi:10.1039/d2sc03991e
Return to citation in text: [1] -
Hao, X.; Li, T.-R.; Chen, H.; Gini, A.; Zhang, X.; Rosset, S.; Mazet, C.; Tiefenbacher, K.; Matile, S. Chem. – Eur. J. 2021, 27, 12215–12223. doi:10.1002/chem.202101548
Return to citation in text: [1] -
Humeniuk, H. V.; Gini, A.; Hao, X.; Coelho, F.; Sakai, N.; Matile, S. JACS Au 2021, 1, 1588–1593. doi:10.1021/jacsau.1c00345
Return to citation in text: [1] -
Akamatsu, M.; Yamanaga, K.; Tanaka, K.; Kanehara, Y.; Sumita, M.; Sakai, K.; Sakai, H. Langmuir 2023, 39, 5833–5839. doi:10.1021/acs.langmuir.3c00127
Return to citation in text: [1] -
Shaik, S.; Danovich, D.; Joy, J.; Wang, Z.; Stuyver, T. J. Am. Chem. Soc. 2020, 142, 12551–12562. doi:10.1021/jacs.0c05128
Return to citation in text: [1] [2] [3] -
Ciampi, S.; Darwish, N.; Aitken, H. M.; Díez-Pérez, I.; Coote, M. L. Chem. Soc. Rev. 2018, 47, 5146–5164. doi:10.1039/c8cs00352a
Return to citation in text: [1] [2] [3] -
Kareem, S.; Vali, S. R.; Reddy, B. V. S. Eur. J. Org. Chem. 2023, e202300103. doi:10.1002/ejoc.202300103
Return to citation in text: [1] [2] [3] -
Shaik, S.; Ramanan, R.; Danovich, D.; Mandal, D. Chem. Soc. Rev. 2018, 47, 5125–5145. doi:10.1039/c8cs00354h
Return to citation in text: [1] [2] [3] -
Delley, M. F.; Nichols, E. M.; Mayer, J. M. J. Am. Chem. Soc. 2021, 143, 10778–10792. doi:10.1021/jacs.1c05419
Return to citation in text: [1] [2] -
Yu, L.-J.; Coote, M. L. J. Phys. Chem. A 2019, 123, 582–589. doi:10.1021/acs.jpca.8b11579
Return to citation in text: [1] [2] -
Zhang, B.; Schaack, C.; Prindle, C. R.; Vo, E. A.; Aziz, M.; Steigerwald, M. L.; Berkelbach, T. C.; Nuckolls, C.; Venkataraman, L. Chem. Sci. 2023, 14, 1769–1774. doi:10.1039/d2sc06411a
Return to citation in text: [1] [2] -
Gorin, C. F.; Beh, E. S.; Bui, Q. M.; Dick, G. R.; Kanan, M. W. J. Am. Chem. Soc. 2013, 135, 11257–11265. doi:10.1021/ja404394z
Return to citation in text: [1] [2] -
Blyth, M. T.; Noble, B. B.; Russell, I. C.; Coote, M. L. J. Am. Chem. Soc. 2020, 142, 606–613. doi:10.1021/jacs.9b12186
Return to citation in text: [1] [2] -
Fried, S. D.; Boxer, S. G. Annu. Rev. Biochem. 2017, 86, 387–415. doi:10.1146/annurev-biochem-061516-044432
Return to citation in text: [1] [2] -
Vaissier Welborn, V.; Head-Gordon, T. Chem. Rev. 2019, 119, 6613–6630. doi:10.1021/acs.chemrev.8b00399
Return to citation in text: [1] [2] -
Warshel, A.; Sharma, P. K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M. H. M. Chem. Rev. 2006, 106, 3210–3235. doi:10.1021/cr0503106
Return to citation in text: [1] [2] -
Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Campisciano, V.; Gruttadauria, M.; Giacalone, F. ChemCatChem 2019, 11, 90–133. doi:10.1002/cctc.201801414
Return to citation in text: [1] [2] -
Garrido, M.; Gualandi, L.; Di Noja, S.; Filippini, G.; Bosi, S.; Prato, M. Chem. Commun. 2020, 56, 12698–12716. doi:10.1039/d0cc05316c
Return to citation in text: [1] [2] -
Toganoh, M.; Matsuo, Y.; Nakamura, E. J. Organomet. Chem. 2003, 683, 295–300. doi:10.1016/s0022-328x(03)00465-0
Return to citation in text: [1] -
Vidal, S.; Marco-Martínez, J.; Filippone, S.; Martín, N. Chem. Commun. 2017, 53, 4842–4844. doi:10.1039/c7cc01267e
Return to citation in text: [1] -
Sun, Y.-B.; Cao, C.-Y.; Yang, S.-L.; Huang, P.-P.; Wang, C.-R.; Song, W.-G. Chem. Commun. 2014, 50, 10307–10310. doi:10.1039/c4cc04891a
Return to citation in text: [1] -
Sun, Y.; Cao, C.; Huang, P.; Yang, S.; Song, W. RSC Adv. 2015, 5, 86082–86087. doi:10.1039/c5ra16011a
Return to citation in text: [1] -
Nierengarten, J.-F. Chem. Commun. 2017, 53, 11855–11868. doi:10.1039/c7cc07479d
Return to citation in text: [1] -
Yamada, M. ChemPlusChem 2023, 88, e202300062. doi:10.1002/cplu.202300062
Return to citation in text: [1] -
Li, C.-Z.; Chueh, C.-C.; Yip, H.-L.; Ding, F.; Li, X.; Jen, A. K.-Y. Adv. Mater. (Weinheim, Ger.) 2013, 25, 2457–2461. doi:10.1002/adma.201204543
Return to citation in text: [1] -
Sun, X.; Chen, W.; Liang, L.; Hu, W.; Wang, H.; Pang, Z.; Ye, Y.; Hu, X.; Wang, Q.; Kong, X.; Jin, Y.; Lei, M. Chem. Mater. 2016, 28, 8726–8731. doi:10.1021/acs.chemmater.6b04056
Return to citation in text: [1] -
Sun, X.; Ji, L. Y.; Chen, W. W.; Guo, X.; Wang, H. H.; Lei, M.; Wang, Q.; Li, Y. F. J. Mater. Chem. A 2017, 5, 20720–20728. doi:10.1039/c7ta06335k
Return to citation in text: [1] -
Yamada, M.; Sahara, K.; Koizumi, M.; Maeda, Y.; Suzuki, M. Chem. – Eur. J. 2023, 29, e202300877. doi:10.1002/chem.202300877
Return to citation in text: [1] -
Sun, S.; Liu, Z.; Colombo, F.; Gao, R.; Yu, Y.; Qiu, Y.; Su, J.; Gan, L. Angew. Chem., Int. Ed. 2022, 61, e202212090. doi:10.1002/anie.202212090
Return to citation in text: [1] -
Kobuke, Y.; Yoshida, J.-i. Tetrahedron Lett. 1978, 19, 367–370. doi:10.1016/s0040-4039(01)85127-3
Return to citation in text: [1] -
Sakai, N.; Sordé, N.; Matile, S. Molecules 2001, 6, 845–851. doi:10.3390/61100845
Return to citation in text: [1] -
Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187
Return to citation in text: [1] [2] -
Bornhof, A.-B.; Bauzá, A.; Aster, A.; Pupier, M.; Frontera, A.; Vauthey, E.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 4884–4892. doi:10.1021/jacs.8b00809
Return to citation in text: [1] [2] [3] [4] -
Liu, L.; Cotelle, Y.; Bornhof, A.-B.; Besnard, C.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2017, 56, 13066–13069. doi:10.1002/anie.201707730
Return to citation in text: [1] -
Hoffmann, R.; Woodward, R. B. J. Am. Chem. Soc. 1965, 87, 4388–4389. doi:10.1021/ja00947a033
Return to citation in text: [1] -
Suzuki, T.; Watanabe, S.; Kobayashi, S.; Tanino, K. Org. Lett. 2017, 19, 922–925. doi:10.1021/acs.orglett.7b00085
Return to citation in text: [1] -
Okamura, H.; Shimizu, H.; Nakamura, Y.; Iwagawa, T.; Nakatani, M. Tetrahedron Lett. 2000, 41, 4147–4150. doi:10.1016/s0040-4039(00)00555-4
Return to citation in text: [1] -
Chatelet, B.; Dufaud, V.; Dutasta, J.-P.; Martinez, A. J. Org. Chem. 2014, 79, 8684–8688. doi:10.1021/jo501457d
Return to citation in text: [1] -
Zhang, X.; Liu, L.; López-Andarias, J.; Wang, C.; Sakai, N.; Matile, S. Helv. Chim. Acta 2018, 101, e1700288. doi:10.1002/hlca.201700288
Return to citation in text: [1] [2] [3] -
Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2006, 128, 3928–3930. doi:10.1021/ja060312n
Return to citation in text: [1] -
Zhang, X.; Hao, X.; Liu, L.; Pham, A.-T.; López-Andarias, J.; Frontera, A.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 17867–17871. doi:10.1021/jacs.8b11788
Return to citation in text: [1] [2] -
Gutiérrez López, M. Á.; Tan, M.-L.; Frontera, A.; Matile, S. JACS Au 2023, 3, 1039–1051. doi:10.1021/jacsau.2c00656
Return to citation in text: [1] -
Sittihan, S.; Jamison, T. F. J. Am. Chem. Soc. 2019, 141, 11239–11244. doi:10.1021/jacs.9b04696
Return to citation in text: [1] -
Liu, H.; Lin, S.; Jacobsen, K. M.; Poulsen, T. B. Angew. Chem., Int. Ed. 2019, 58, 13630–13642. doi:10.1002/anie.201812982
Return to citation in text: [1] -
Li, F.-X.; Ren, S.-J.; Li, P.-F.; Yang, P.; Qu, J. Angew. Chem., Int. Ed. 2020, 59, 18473–18478. doi:10.1002/anie.202007980
Return to citation in text: [1] -
Nakanishi, K. Toxicon 1985, 23, 473–479. doi:10.1016/0041-0101(85)90031-5
Return to citation in text: [1] -
Tenud, L.; Farooq, S.; Seibl, J.; Eschenmoser, A. Helv. Chim. Acta 1970, 53, 2059–2069. doi:10.1002/hlca.19700530816
Return to citation in text: [1] -
Bürgi, H. B.; Dunitz, J. D.; Lehn, J. M.; Wipff, G. Tetrahedron 1974, 30, 1563–1572. doi:10.1016/s0040-4020(01)90678-7
Return to citation in text: [1] -
Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734–736. doi:10.1039/c39760000734
Return to citation in text: [1] -
Gilmore, K.; Mohamed, R. K.; Alabugin, I. V. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2016, 6, 487–514. doi:10.1002/wcms.1261
Return to citation in text: [1] -
Cane, D. E.; Celmer, W. D.; Westley, J. W. J. Am. Chem. Soc. 1983, 105, 3594–3600. doi:10.1021/ja00349a040
Return to citation in text: [1] -
López‐Andarias, J.; Bauzá, A.; Sakai, N.; Frontera, A.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 10883–10887. doi:10.1002/anie.201804092
Return to citation in text: [1] [2] [3] [4] -
Panneerselvam, M.; Akash, H.; Patnaik, A. Phys. Chem. Chem. Phys. 2023, 25, 10647–10660. doi:10.1039/d2cp06017e
Return to citation in text: [1] -
Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chem. Rev. 2006, 106, 1105–1136. doi:10.1021/cr050569o
Return to citation in text: [1] [2] [3] -
Liu, D.; Lungerich, D.; Nakamuro, T.; Harano, K.; Nakamura, E. Micron 2022, 160, 103316. doi:10.1016/j.micron.2022.103316
Return to citation in text: [1] -
Blanco, M.; Nieto-Ortega, B.; de Juan, A.; Vera-Hidalgo, M.; López-Moreno, A.; Casado, S.; González, L. R.; Sawada, H.; González-Calbet, J. M.; Pérez, E. M. Nat. Commun. 2018, 9, 2671. doi:10.1038/s41467-018-05183-8
Return to citation in text: [1] -
Kitanosono, T.; Xu, P.; Kobayashi, S. Science 2018, 362, 311–315. doi:10.1126/science.aap7883
Return to citation in text: [1] -
Gholinejad, M.; Naghshbandi, Z.; Nájera, C. ChemCatChem 2019, 11, 1792–1823. doi:10.1002/cctc.201802101
Return to citation in text: [1] -
Chen, Z.; Guan, Z.; Li, M.; Yang, Q.; Li, C. Angew. Chem., Int. Ed. 2011, 50, 4913–4917. doi:10.1002/anie.201006870
Return to citation in text: [1] -
Xing, L.; Xie, J.-H.; Chen, Y.-S.; Wang, L.-X.; Zhou, Q.-L. Adv. Synth. Catal. 2008, 350, 1013–1016. doi:10.1002/adsc.200700617
Return to citation in text: [1] [2] [3] -
Li, H.; Zhong, M.; Li, C.; Ren, Y.; Chen, J.; Yang, Q. ChemCatChem 2019, 11, 3952–3958. doi:10.1002/cctc.201900311
Return to citation in text: [1] -
Zhang, L.; Zhang, W.; Serp, P.; Sun, W.-H.; Durand, J. ChemCatChem 2014, 6, 1310–1316. doi:10.1002/cctc.201301063
Return to citation in text: [1] [2] [3] -
Ding, Q.; Yu, Y.; Huang, F.; Zhang, L.; Zheng, J.-G.; Xu, M.; Baell, J. B.; Huang, H. Chem. – Eur. J. 2020, 26, 4592–4598. doi:10.1002/chem.201905468
Return to citation in text: [1] -
Chronopoulos, D. D.; Kokotos, C. G.; Karousis, N.; Kokotos, G.; Tagmatarchis, N. Nanoscale 2015, 7, 2750–2757. doi:10.1039/c4nr06543c
Return to citation in text: [1] -
Hajipour, A. R.; Khorsandi, Z. ChemistrySelect 2017, 2, 8976–8982. doi:10.1002/slct.201700847
Return to citation in text: [1] -
Mercadante, A.; Campisciano, V.; Morena, A.; Valentino, L.; La Parola, V.; Aprile, C.; Gruttadauria, M.; Giacalone, F. Eur. J. Org. Chem. 2022, e202200497. doi:10.1002/ejoc.202200497
Return to citation in text: [1] -
Cotelle, Y.; Benz, S.; Avestro, A.-J.; Ward, T. R.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2016, 55, 4275–4279. doi:10.1002/anie.201600831
Return to citation in text: [1] -
Li, F.; Zhang, B.; Li, X.; Jiang, Y.; Chen, L.; Li, Y.; Sun, L. Angew. Chem., Int. Ed. 2011, 50, 12276–12279. doi:10.1002/anie.201105044
Return to citation in text: [1] [2] -
Das, A.; Stahl, S. S. Angew. Chem., Int. Ed. 2017, 56, 8892–8897. doi:10.1002/anie.201704921
Return to citation in text: [1] [2] -
Glanzer, S.; Sax, A. F. Mol. Phys. 2013, 111, 2427–2438. doi:10.1080/00268976.2013.831499
Return to citation in text: [1] [2] -
Garrido, M.; Volland, M. K.; Münich, P. W.; Rodríguez-Pérez, L.; Calbo, J.; Ortí, E.; Herranz, M. Á.; Martín, N.; Guldi, D. M. J. Am. Chem. Soc. 2020, 142, 1895–1903. doi:10.1021/jacs.9b10772
Return to citation in text: [1] [2] -
Hagihara, S.; Tanaka, H.; Matile, S. J. Am. Chem. Soc. 2008, 130, 5656–5657. doi:10.1021/ja801094p
Return to citation in text: [1] -
Talukdar, P.; Bollot, G.; Mareda, J.; Sakai, N.; Matile, S. Chem. – Eur. J. 2005, 11, 6525–6532. doi:10.1002/chem.200500516
Return to citation in text: [1] -
Gabriel, G. J.; Iverson, B. L. J. Am. Chem. Soc. 2002, 124, 15174–15175. doi:10.1021/ja0275358
Return to citation in text: [1] -
Cougnon, F. B. L.; Sanders, J. K. M. Acc. Chem. Res. 2012, 45, 2211–2221. doi:10.1021/ar200240m
Return to citation in text: [1] -
Iijima, T.; Vignon, S. A.; Tseng, H.-R.; Jarrosson, T.; Sanders, J. K. M.; Marchioni, F.; Venturi, M.; Apostoli, E.; Balzani, V.; Stoddart, J. F. Chem. – Eur. J. 2004, 10, 6375–6392. doi:10.1002/chem.200400651
Return to citation in text: [1] -
Ikkanda, B. A.; Samuel, S. A.; Iverson, B. L. J. Org. Chem. 2014, 79, 2029–2037. doi:10.1021/jo402704z
Return to citation in text: [1] -
Mukhopadhyay, P.; Iwashita, Y.; Shirakawa, M.; Kawano, S.-i.; Fujita, N.; Shinkai, S. Angew. Chem., Int. Ed. 2006, 45, 1592–1595. doi:10.1002/anie.200503158
Return to citation in text: [1] -
Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497
Return to citation in text: [1] [2] -
Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412
Return to citation in text: [1] [2] -
Folgueiras‐Amador, A. A.; Philipps, K.; Guilbaud, S.; Poelakker, J.; Wirth, T. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. doi:10.1002/anie.201709717
Return to citation in text: [1] [2] -
Gnaim, S.; Bauer, A.; Zhang, H.-J.; Chen, L.; Gannett, C.; Malapit, C. A.; Hill, D. E.; Vogt, D.; Tang, T.; Daley, R. A.; Hao, W.; Zeng, R.; Quertenmont, M.; Beck, W. D.; Kandahari, E.; Vantourout, J. C.; Echeverria, P.-G.; Abruna, H. D.; Blackmond, D. G.; Minteer, S. D.; Reisman, S. E.; Sigman, M. S.; Baran, P. S. Nature 2022, 605, 687–695. doi:10.1038/s41586-022-04595-3
Return to citation in text: [1] [2] -
Gabriel, G.; Gómez-Martínez, R.; Villa, R. Physiol. Meas. 2008, 29, S203–S212. doi:10.1088/0967-3334/29/6/s18
Return to citation in text: [1] -
Foroutan-Nejad, C.; Marek, R. Phys. Chem. Chem. Phys. 2014, 16, 2508–2514. doi:10.1039/c3cp52671b
Return to citation in text: [1] [2] -
Novák, M.; Foroutan-Nejad, C.; Marek, R. J. Chem. Theory Comput. 2016, 12, 3788–3795. doi:10.1021/acs.jctc.6b00586
Return to citation in text: [1] -
Farajpour, E.; Sohrabi, B.; Beheshtian, J. Phys. Chem. Chem. Phys. 2016, 18, 7293–7299. doi:10.1039/c5cp07710a
Return to citation in text: [1] [2] -
Chen, J.; Li, J.; Liu, X.; He, Z.; Shi, G. Phys. Chem. Chem. Phys. 2023, 25, 13260–13264. doi:10.1039/d3cp00986f
Return to citation in text: [1] [2] -
Hu, H.; Xin, J. H.; Hu, H.; Wang, X.; Kong, Y. Appl. Catal., A 2015, 492, 1–9. doi:10.1016/j.apcata.2014.11.041
Return to citation in text: [1] -
Zhou, K.; Xu, Z. Phys. Rev. Res. 2020, 2, 042034. doi:10.1103/physrevresearch.2.042034
Return to citation in text: [1] -
Chen, L.; Liu, S.; Xu, Z.; Yang, X. J. Phys. Chem. Lett. 2019, 10, 5735–5741. doi:10.1021/acs.jpclett.9b02074
Return to citation in text: [1] -
Dai, L. Acc. Chem. Res. 2013, 46, 31–42. doi:10.1021/ar300122m
Return to citation in text: [1] -
Acocella, M. R.; Mauro, M.; Guerra, G. ChemSusChem 2014, 7, 3279–3283. doi:10.1002/cssc.201402770
Return to citation in text: [1] -
Bian, S.; Scott, A. M.; Cao, Y.; Liang, Y.; Osuna, S.; Houk, K. N.; Braunschweig, A. B. J. Am. Chem. Soc. 2013, 135, 9240–9243. doi:10.1021/ja4042077
Return to citation in text: [1] -
Sun, P. Z.; Xiong, W. Q.; Bera, A.; Timokhin, I.; Wu, Z. F.; Mishchenko, A.; Sellers, M. C.; Liu, B. L.; Cheng, H. M.; Janzen, E.; Edgar, J. H.; Grigorieva, I. V.; Yuan, S. J.; Geim, A. K. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2300481120. doi:10.1073/pnas.2300481120
Return to citation in text: [1] -
Su, D. S.; Qi, W.; Wen, G. Carbon and Graphite for Catalysis. Industrial Carbon and Graphite Materials; Wiley-VCH: Weinheim, Germany, 2021; Vol. I, pp 457–490. doi:10.1002/9783527674046.ch8
Return to citation in text: [1] -
Liu, Z.; Wu, L.-F.; Kufner, C. L.; Sasselov, D. D.; Fischer, W. W.; Sutherland, J. D. Nat. Chem. 2021, 13, 1126–1132. doi:10.1038/s41557-021-00789-w
Return to citation in text: [1] -
Muchowska, K. B.; Varma, S. J.; Moran, J. Chem. Rev. 2020, 120, 7708–7744. doi:10.1021/acs.chemrev.0c00191
Return to citation in text: [1]
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 62. | Liu, L.; Cotelle, Y.; Bornhof, A.-B.; Besnard, C.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2017, 56, 13066–13069. doi:10.1002/anie.201707730 |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 63. | Hoffmann, R.; Woodward, R. B. J. Am. Chem. Soc. 1965, 87, 4388–4389. doi:10.1021/ja00947a033 |
| 124. | Liu, Z.; Wu, L.-F.; Kufner, C. L.; Sasselov, D. D.; Fischer, W. W.; Sutherland, J. D. Nat. Chem. 2021, 13, 1126–1132. doi:10.1038/s41557-021-00789-w |
| 125. | Muchowska, K. B.; Varma, S. J.; Moran, J. Chem. Rev. 2020, 120, 7708–7744. doi:10.1021/acs.chemrev.0c00191 |
| 112. | Foroutan-Nejad, C.; Marek, R. Phys. Chem. Chem. Phys. 2014, 16, 2508–2514. doi:10.1039/c3cp52671b |
| 114. | Farajpour, E.; Sohrabi, B.; Beheshtian, J. Phys. Chem. Chem. Phys. 2016, 18, 7293–7299. doi:10.1039/c5cp07710a |
| 115. | Chen, J.; Li, J.; Liu, X.; He, Z.; Shi, G. Phys. Chem. Chem. Phys. 2023, 25, 13260–13264. doi:10.1039/d3cp00986f |
| 116. | Hu, H.; Xin, J. H.; Hu, H.; Wang, X.; Kong, Y. Appl. Catal., A 2015, 492, 1–9. doi:10.1016/j.apcata.2014.11.041 |
| 117. | Zhou, K.; Xu, Z. Phys. Rev. Res. 2020, 2, 042034. doi:10.1103/physrevresearch.2.042034 |
| 118. | Chen, L.; Liu, S.; Xu, Z.; Yang, X. J. Phys. Chem. Lett. 2019, 10, 5735–5741. doi:10.1021/acs.jpclett.9b02074 |
| 119. | Dai, L. Acc. Chem. Res. 2013, 46, 31–42. doi:10.1021/ar300122m |
| 120. | Acocella, M. R.; Mauro, M.; Guerra, G. ChemSusChem 2014, 7, 3279–3283. doi:10.1002/cssc.201402770 |
| 121. | Bian, S.; Scott, A. M.; Cao, Y.; Liang, Y.; Osuna, S.; Houk, K. N.; Braunschweig, A. B. J. Am. Chem. Soc. 2013, 135, 9240–9243. doi:10.1021/ja4042077 |
| 122. | Sun, P. Z.; Xiong, W. Q.; Bera, A.; Timokhin, I.; Wu, Z. F.; Mishchenko, A.; Sellers, M. C.; Liu, B. L.; Cheng, H. M.; Janzen, E.; Edgar, J. H.; Grigorieva, I. V.; Yuan, S. J.; Geim, A. K. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2300481120. doi:10.1073/pnas.2300481120 |
| 123. | Su, D. S.; Qi, W.; Wen, G. Carbon and Graphite for Catalysis. Industrial Carbon and Graphite Materials; Wiley-VCH: Weinheim, Germany, 2021; Vol. I, pp 457–490. doi:10.1002/9783527674046.ch8 |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 70. | Gutiérrez López, M. Á.; Tan, M.-L.; Frontera, A.; Matile, S. JACS Au 2023, 3, 1039–1051. doi:10.1021/jacsau.2c00656 |
| 69. | Zhang, X.; Hao, X.; Liu, L.; Pham, A.-T.; López-Andarias, J.; Frontera, A.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 17867–17871. doi:10.1021/jacs.8b11788 |
| 67. | Zhang, X.; Liu, L.; López-Andarias, J.; Wang, C.; Sakai, N.; Matile, S. Helv. Chim. Acta 2018, 101, e1700288. doi:10.1002/hlca.201700288 |
| 107. | Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497 |
| 108. | Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412 |
| 109. | Folgueiras‐Amador, A. A.; Philipps, K.; Guilbaud, S.; Poelakker, J.; Wirth, T. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. doi:10.1002/anie.201709717 |
| 110. | Gnaim, S.; Bauer, A.; Zhang, H.-J.; Chen, L.; Gannett, C.; Malapit, C. A.; Hill, D. E.; Vogt, D.; Tang, T.; Daley, R. A.; Hao, W.; Zeng, R.; Quertenmont, M.; Beck, W. D.; Kandahari, E.; Vantourout, J. C.; Echeverria, P.-G.; Abruna, H. D.; Blackmond, D. G.; Minteer, S. D.; Reisman, S. E.; Sigman, M. S.; Baran, P. S. Nature 2022, 605, 687–695. doi:10.1038/s41586-022-04595-3 |
| 69. | Zhang, X.; Hao, X.; Liu, L.; Pham, A.-T.; López-Andarias, J.; Frontera, A.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 17867–17871. doi:10.1021/jacs.8b11788 |
| 67. | Zhang, X.; Liu, L.; López-Andarias, J.; Wang, C.; Sakai, N.; Matile, S. Helv. Chim. Acta 2018, 101, e1700288. doi:10.1002/hlca.201700288 |
| 9. | Sabirov, D. S. RSC Adv. 2014, 4, 44996–45028. doi:10.1039/c4ra06116k |
| 10. | Zhang, Y.; Wang, D.; Wang, W. Comput. Theor. Chem. 2018, 1128, 56–59. doi:10.1016/j.comptc.2018.02.011 |
| 11. | Sabirov, D. S.; Tukhbatullina, A. A. Nanomaterials 2022, 12, 4404. doi:10.3390/nano12244404 |
| 68. | Wang, Y.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2006, 128, 3928–3930. doi:10.1021/ja060312n |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 64. | Suzuki, T.; Watanabe, S.; Kobayashi, S.; Tanino, K. Org. Lett. 2017, 19, 922–925. doi:10.1021/acs.orglett.7b00085 |
| 65. | Okamura, H.; Shimizu, H.; Nakamura, Y.; Iwagawa, T.; Nakatani, M. Tetrahedron Lett. 2000, 41, 4147–4150. doi:10.1016/s0040-4039(00)00555-4 |
| 66. | Chatelet, B.; Dufaud, V.; Dutasta, J.-P.; Martinez, A. J. Org. Chem. 2014, 79, 8684–8688. doi:10.1021/jo501457d |
| 3. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 61. | Bornhof, A.-B.; Bauzá, A.; Aster, A.; Pupier, M.; Frontera, A.; Vauthey, E.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 4884–4892. doi:10.1021/jacs.8b00809 |
| 71. | Sittihan, S.; Jamison, T. F. J. Am. Chem. Soc. 2019, 141, 11239–11244. doi:10.1021/jacs.9b04696 |
| 72. | Liu, H.; Lin, S.; Jacobsen, K. M.; Poulsen, T. B. Angew. Chem., Int. Ed. 2019, 58, 13630–13642. doi:10.1002/anie.201812982 |
| 73. | Li, F.-X.; Ren, S.-J.; Li, P.-F.; Yang, P.; Qu, J. Angew. Chem., Int. Ed. 2020, 59, 18473–18478. doi:10.1002/anie.202007980 |
| 75. | Tenud, L.; Farooq, S.; Seibl, J.; Eschenmoser, A. Helv. Chim. Acta 1970, 53, 2059–2069. doi:10.1002/hlca.19700530816 |
| 76. | Bürgi, H. B.; Dunitz, J. D.; Lehn, J. M.; Wipff, G. Tetrahedron 1974, 30, 1563–1572. doi:10.1016/s0040-4020(01)90678-7 |
| 77. | Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734–736. doi:10.1039/c39760000734 |
| 78. | Gilmore, K.; Mohamed, R. K.; Alabugin, I. V. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2016, 6, 487–514. doi:10.1002/wcms.1261 |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 80. | López‐Andarias, J.; Bauzá, A.; Sakai, N.; Frontera, A.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 10883–10887. doi:10.1002/anie.201804092 |
| 61. | Bornhof, A.-B.; Bauzá, A.; Aster, A.; Pupier, M.; Frontera, A.; Vauthey, E.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 4884–4892. doi:10.1021/jacs.8b00809 |
| 8. | Tan, M.-L.; Ángeles Gutiérrez López, M.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2023, 62, e202310393. doi:10.1002/anie.202310393 |
| 15. | Kennedy, C. R.; Lin, S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2016, 55, 12596–12624. doi:10.1002/anie.201600547 |
| 61. | Bornhof, A.-B.; Bauzá, A.; Aster, A.; Pupier, M.; Frontera, A.; Vauthey, E.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 4884–4892. doi:10.1021/jacs.8b00809 |
| 79. | Cane, D. E.; Celmer, W. D.; Westley, J. W. J. Am. Chem. Soc. 1983, 105, 3594–3600. doi:10.1021/ja00349a040 |
| 24. | Paraja, M.; Matile, S. Angew. Chem., Int. Ed. 2020, 59, 6273–6277. doi:10.1002/anie.202000579 |
| 25. | Paraja, M.; Hao, X.; Matile, S. Angew. Chem., Int. Ed. 2020, 59, 15093–15097. doi:10.1002/anie.202000681 |
| 80. | López‐Andarias, J.; Bauzá, A.; Sakai, N.; Frontera, A.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 10883–10887. doi:10.1002/anie.201804092 |
| 81. | Panneerselvam, M.; Akash, H.; Patnaik, A. Phys. Chem. Chem. Phys. 2023, 25, 10647–10660. doi:10.1039/d2cp06017e |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 67. | Zhang, X.; Liu, L.; López-Andarias, J.; Wang, C.; Sakai, N.; Matile, S. Helv. Chim. Acta 2018, 101, e1700288. doi:10.1002/hlca.201700288 |
| 80. | López‐Andarias, J.; Bauzá, A.; Sakai, N.; Frontera, A.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 10883–10887. doi:10.1002/anie.201804092 |
| 80. | López‐Andarias, J.; Bauzá, A.; Sakai, N.; Frontera, A.; Matile, S. Angew. Chem., Int. Ed. 2018, 57, 10883–10887. doi:10.1002/anie.201804092 |
| 1. | Zhao, Y.; Domoto, Y.; Orentas, E.; Beuchat, C.; Emery, D.; Mareda, J.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2013, 52, 9940–9943. doi:10.1002/anie.201305356 |
| 6. | Frontera, A.; Quiñonero, D.; Garau, C.; Costa, A.; Ballester, P.; Deyà, P. M. J. Phys. Chem. A 2006, 110, 9307–9309. doi:10.1021/jp062176e |
| 15. | Kennedy, C. R.; Lin, S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2016, 55, 12596–12624. doi:10.1002/anie.201600547 |
| 16. | Kutateladze, D. A.; Strassfeld, D. A.; Jacobsen, E. N. J. Am. Chem. Soc. 2020, 142, 6951–6956. doi:10.1021/jacs.0c02665 |
| 82. | Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chem. Rev. 2006, 106, 1105–1136. doi:10.1021/cr050569o |
| 88. | Xing, L.; Xie, J.-H.; Chen, Y.-S.; Wang, L.-X.; Zhou, Q.-L. Adv. Synth. Catal. 2008, 350, 1013–1016. doi:10.1002/adsc.200700617 |
| 90. | Zhang, L.; Zhang, W.; Serp, P.; Sun, W.-H.; Durand, J. ChemCatChem 2014, 6, 1310–1316. doi:10.1002/cctc.201301063 |
| 96. | Li, F.; Zhang, B.; Li, X.; Jiang, Y.; Chen, L.; Li, Y.; Sun, L. Angew. Chem., Int. Ed. 2011, 50, 12276–12279. doi:10.1002/anie.201105044 |
| 97. | Das, A.; Stahl, S. S. Angew. Chem., Int. Ed. 2017, 56, 8892–8897. doi:10.1002/anie.201704921 |
| 98. | Glanzer, S.; Sax, A. F. Mol. Phys. 2013, 111, 2427–2438. doi:10.1080/00268976.2013.831499 |
| 99. | Garrido, M.; Volland, M. K.; Münich, P. W.; Rodríguez-Pérez, L.; Calbo, J.; Ortí, E.; Herranz, M. Á.; Martín, N.; Guldi, D. M. J. Am. Chem. Soc. 2020, 142, 1895–1903. doi:10.1021/jacs.9b10772 |
| 3. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 3. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 4. | Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Eur. J. 2022, 28, e202103303. doi:10.1002/chem.202103303 |
| 17. | Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Angew. Chem., Int. Ed. 2021, 60, 20650–20655. doi:10.1002/anie.202106509 |
| 18. | Maynard, J. R. J.; Galmés, B.; Stergiou, A. D.; Symes, M. D.; Frontera, A.; Goldup, S. M. Angew. Chem., Int. Ed. 2022, 61, e202115961. doi:10.1002/anie.202115961 |
| 19. | Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Asian J. 2021, 16, 3599–3603. doi:10.1002/asia.202100920 |
| 20. | Giese, M.; Albrecht, M.; Rissanen, K. Chem. Commun. 2016, 52, 1778–1795. doi:10.1039/c5cc09072e |
| 21. | Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature 2017, 543, 637–646. doi:10.1038/nature21701 |
| 22. | Guo, S.-Y.; Guo, Q.-H.; Tong, S.; Wang, M.-X. Angew. Chem., Int. Ed. 2020, 59, 8078–8083. doi:10.1002/anie.201915839 |
| 23. | Cotelle, Y.; Lebrun, V.; Sakai, N.; Ward, T. R.; Matile, S. ACS Cent. Sci. 2016, 2, 388–393. doi:10.1021/acscentsci.6b00097 |
| 24. | Paraja, M.; Matile, S. Angew. Chem., Int. Ed. 2020, 59, 6273–6277. doi:10.1002/anie.202000579 |
| 25. | Paraja, M.; Hao, X.; Matile, S. Angew. Chem., Int. Ed. 2020, 59, 15093–15097. doi:10.1002/anie.202000681 |
| 26. | Gini, A.; Paraja, M.; Galmés, B.; Besnard, C.; Poblador-Bahamonde, A. I.; Sakai, N.; Frontera, A.; Matile, S. Chem. Sci. 2020, 11, 7086–7091. doi:10.1039/d0sc02551h |
| 27. | Chen, H.; Frontera, A.; Ángeles Gutiérrez López, M.; Sakai, N.; Matile, S. Helv. Chim. Acta 2022, 105, e202200119. doi:10.1002/hlca.202200119 |
| 28. | Chen, H.; Li, T.-R.; Sakai, N.; Besnard, C.; Guénée, L.; Pupier, M.; Viger-Gravel, J.; Tiefenbacher, K.; Matile, S. Chem. Sci. 2022, 13, 10273–10280. doi:10.1039/d2sc03991e |
| 29. | Hao, X.; Li, T.-R.; Chen, H.; Gini, A.; Zhang, X.; Rosset, S.; Mazet, C.; Tiefenbacher, K.; Matile, S. Chem. – Eur. J. 2021, 27, 12215–12223. doi:10.1002/chem.202101548 |
| 30. | Humeniuk, H. V.; Gini, A.; Hao, X.; Coelho, F.; Sakai, N.; Matile, S. JACS Au 2021, 1, 1588–1593. doi:10.1021/jacsau.1c00345 |
| 31. | Akamatsu, M.; Yamanaga, K.; Tanaka, K.; Kanehara, Y.; Sumita, M.; Sakai, K.; Sakai, H. Langmuir 2023, 39, 5833–5839. doi:10.1021/acs.langmuir.3c00127 |
| 3. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 4. | Luo, N.; Ao, Y.-F.; Wang, D.-X.; Wang, Q.-Q. Chem. – Eur. J. 2022, 28, e202103303. doi:10.1002/chem.202103303 |
| 5. | Miros, F. N.; Zhao, Y.; Sargsyan, G.; Pupier, M.; Besnard, C.; Beuchat, C.; Mareda, J.; Sakai, N.; Matile, S. Chem. – Eur. J. 2016, 22, 2648–2657. doi:10.1002/chem.201504008 |
| 13. | Bornhof, A.-B.; Vázquez‐Nakagawa, M.; Rodríguez‐Pérez, L.; Ángeles Herranz, M.; Sakai, N.; Martín, N.; Matile, S.; López‐Andarias, J. Angew. Chem., Int. Ed. 2019, 58, 16097–16100. doi:10.1002/anie.201909540 |
| 82. | Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chem. Rev. 2006, 106, 1105–1136. doi:10.1021/cr050569o |
| 88. | Xing, L.; Xie, J.-H.; Chen, Y.-S.; Wang, L.-X.; Zhou, Q.-L. Adv. Synth. Catal. 2008, 350, 1013–1016. doi:10.1002/adsc.200700617 |
| 90. | Zhang, L.; Zhang, W.; Serp, P.; Sun, W.-H.; Durand, J. ChemCatChem 2014, 6, 1310–1316. doi:10.1002/cctc.201301063 |
| 96. | Li, F.; Zhang, B.; Li, X.; Jiang, Y.; Chen, L.; Li, Y.; Sun, L. Angew. Chem., Int. Ed. 2011, 50, 12276–12279. doi:10.1002/anie.201105044 |
| 97. | Das, A.; Stahl, S. S. Angew. Chem., Int. Ed. 2017, 56, 8892–8897. doi:10.1002/anie.201704921 |
| 98. | Glanzer, S.; Sax, A. F. Mol. Phys. 2013, 111, 2427–2438. doi:10.1080/00268976.2013.831499 |
| 99. | Garrido, M.; Volland, M. K.; Münich, P. W.; Rodríguez-Pérez, L.; Calbo, J.; Ortí, E.; Herranz, M. Á.; Martín, N.; Guldi, D. M. J. Am. Chem. Soc. 2020, 142, 1895–1903. doi:10.1021/jacs.9b10772 |
| 2. | Zhao, Y.; Benz, S.; Sakai, N.; Matile, S. Chem. Sci. 2015, 6, 6219–6223. doi:10.1039/c5sc02563j |
| 14. | Akamatsu, M.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2017, 139, 6558–6561. doi:10.1021/jacs.7b02421 |
| 3. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 13. | Bornhof, A.-B.; Vázquez‐Nakagawa, M.; Rodríguez‐Pérez, L.; Ángeles Herranz, M.; Sakai, N.; Martín, N.; Matile, S.; López‐Andarias, J. Angew. Chem., Int. Ed. 2019, 58, 16097–16100. doi:10.1002/anie.201909540 |
| 9. | Sabirov, D. S. RSC Adv. 2014, 4, 44996–45028. doi:10.1039/c4ra06116k |
| 10. | Zhang, Y.; Wang, D.; Wang, W. Comput. Theor. Chem. 2018, 1128, 56–59. doi:10.1016/j.comptc.2018.02.011 |
| 11. | Sabirov, D. S.; Tukhbatullina, A. A. Nanomaterials 2022, 12, 4404. doi:10.3390/nano12244404 |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 95. | Cotelle, Y.; Benz, S.; Avestro, A.-J.; Ward, T. R.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2016, 55, 4275–4279. doi:10.1002/anie.201600831 |
| 8. | Tan, M.-L.; Ángeles Gutiérrez López, M.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2023, 62, e202310393. doi:10.1002/anie.202310393 |
| 9. | Sabirov, D. S. RSC Adv. 2014, 4, 44996–45028. doi:10.1039/c4ra06116k |
| 10. | Zhang, Y.; Wang, D.; Wang, W. Comput. Theor. Chem. 2018, 1128, 56–59. doi:10.1016/j.comptc.2018.02.011 |
| 11. | Sabirov, D. S.; Tukhbatullina, A. A. Nanomaterials 2022, 12, 4404. doi:10.3390/nano12244404 |
| 7. | Keshri, S. K.; Ishizuka, T.; Kojima, T.; Matsushita, Y.; Takeuchi, M. J. Am. Chem. Soc. 2021, 143, 3238–3244. doi:10.1021/jacs.0c13389 |
| 13. | Bornhof, A.-B.; Vázquez‐Nakagawa, M.; Rodríguez‐Pérez, L.; Ángeles Herranz, M.; Sakai, N.; Martín, N.; Matile, S.; López‐Andarias, J. Angew. Chem., Int. Ed. 2019, 58, 16097–16100. doi:10.1002/anie.201909540 |
| 45. | Campisciano, V.; Gruttadauria, M.; Giacalone, F. ChemCatChem 2019, 11, 90–133. doi:10.1002/cctc.201801414 |
| 46. | Garrido, M.; Gualandi, L.; Di Noja, S.; Filippini, G.; Bosi, S.; Prato, M. Chem. Commun. 2020, 56, 12698–12716. doi:10.1039/d0cc05316c |
| 82. | Tasis, D.; Tagmatarchis, N.; Bianco, A.; Prato, M. Chem. Rev. 2006, 106, 1105–1136. doi:10.1021/cr050569o |
| 83. | Liu, D.; Lungerich, D.; Nakamuro, T.; Harano, K.; Nakamura, E. Micron 2022, 160, 103316. doi:10.1016/j.micron.2022.103316 |
| 84. | Blanco, M.; Nieto-Ortega, B.; de Juan, A.; Vera-Hidalgo, M.; López-Moreno, A.; Casado, S.; González, L. R.; Sawada, H.; González-Calbet, J. M.; Pérez, E. M. Nat. Commun. 2018, 9, 2671. doi:10.1038/s41467-018-05183-8 |
| 85. | Kitanosono, T.; Xu, P.; Kobayashi, S. Science 2018, 362, 311–315. doi:10.1126/science.aap7883 |
| 86. | Gholinejad, M.; Naghshbandi, Z.; Nájera, C. ChemCatChem 2019, 11, 1792–1823. doi:10.1002/cctc.201802101 |
| 87. | Chen, Z.; Guan, Z.; Li, M.; Yang, Q.; Li, C. Angew. Chem., Int. Ed. 2011, 50, 4913–4917. doi:10.1002/anie.201006870 |
| 88. | Xing, L.; Xie, J.-H.; Chen, Y.-S.; Wang, L.-X.; Zhou, Q.-L. Adv. Synth. Catal. 2008, 350, 1013–1016. doi:10.1002/adsc.200700617 |
| 89. | Li, H.; Zhong, M.; Li, C.; Ren, Y.; Chen, J.; Yang, Q. ChemCatChem 2019, 11, 3952–3958. doi:10.1002/cctc.201900311 |
| 90. | Zhang, L.; Zhang, W.; Serp, P.; Sun, W.-H.; Durand, J. ChemCatChem 2014, 6, 1310–1316. doi:10.1002/cctc.201301063 |
| 91. | Ding, Q.; Yu, Y.; Huang, F.; Zhang, L.; Zheng, J.-G.; Xu, M.; Baell, J. B.; Huang, H. Chem. – Eur. J. 2020, 26, 4592–4598. doi:10.1002/chem.201905468 |
| 92. | Chronopoulos, D. D.; Kokotos, C. G.; Karousis, N.; Kokotos, G.; Tagmatarchis, N. Nanoscale 2015, 7, 2750–2757. doi:10.1039/c4nr06543c |
| 93. | Hajipour, A. R.; Khorsandi, Z. ChemistrySelect 2017, 2, 8976–8982. doi:10.1002/slct.201700847 |
| 94. | Mercadante, A.; Campisciano, V.; Morena, A.; Valentino, L.; La Parola, V.; Aprile, C.; Gruttadauria, M.; Giacalone, F. Eur. J. Org. Chem. 2022, e202200497. doi:10.1002/ejoc.202200497 |
| 45. | Campisciano, V.; Gruttadauria, M.; Giacalone, F. ChemCatChem 2019, 11, 90–133. doi:10.1002/cctc.201801414 |
| 46. | Garrido, M.; Gualandi, L.; Di Noja, S.; Filippini, G.; Bosi, S.; Prato, M. Chem. Commun. 2020, 56, 12698–12716. doi:10.1039/d0cc05316c |
| 47. | Toganoh, M.; Matsuo, Y.; Nakamura, E. J. Organomet. Chem. 2003, 683, 295–300. doi:10.1016/s0022-328x(03)00465-0 |
| 48. | Vidal, S.; Marco-Martínez, J.; Filippone, S.; Martín, N. Chem. Commun. 2017, 53, 4842–4844. doi:10.1039/c7cc01267e |
| 49. | Sun, Y.-B.; Cao, C.-Y.; Yang, S.-L.; Huang, P.-P.; Wang, C.-R.; Song, W.-G. Chem. Commun. 2014, 50, 10307–10310. doi:10.1039/c4cc04891a |
| 50. | Sun, Y.; Cao, C.; Huang, P.; Yang, S.; Song, W. RSC Adv. 2015, 5, 86082–86087. doi:10.1039/c5ra16011a |
| 51. | Nierengarten, J.-F. Chem. Commun. 2017, 53, 11855–11868. doi:10.1039/c7cc07479d |
| 32. | Shaik, S.; Danovich, D.; Joy, J.; Wang, Z.; Stuyver, T. J. Am. Chem. Soc. 2020, 142, 12551–12562. doi:10.1021/jacs.0c05128 |
| 33. | Ciampi, S.; Darwish, N.; Aitken, H. M.; Díez-Pérez, I.; Coote, M. L. Chem. Soc. Rev. 2018, 47, 5146–5164. doi:10.1039/c8cs00352a |
| 34. | Kareem, S.; Vali, S. R.; Reddy, B. V. S. Eur. J. Org. Chem. 2023, e202300103. doi:10.1002/ejoc.202300103 |
| 35. | Shaik, S.; Ramanan, R.; Danovich, D.; Mandal, D. Chem. Soc. Rev. 2018, 47, 5125–5145. doi:10.1039/c8cs00354h |
| 36. | Delley, M. F.; Nichols, E. M.; Mayer, J. M. J. Am. Chem. Soc. 2021, 143, 10778–10792. doi:10.1021/jacs.1c05419 |
| 37. | Yu, L.-J.; Coote, M. L. J. Phys. Chem. A 2019, 123, 582–589. doi:10.1021/acs.jpca.8b11579 |
| 38. | Zhang, B.; Schaack, C.; Prindle, C. R.; Vo, E. A.; Aziz, M.; Steigerwald, M. L.; Berkelbach, T. C.; Nuckolls, C.; Venkataraman, L. Chem. Sci. 2023, 14, 1769–1774. doi:10.1039/d2sc06411a |
| 39. | Gorin, C. F.; Beh, E. S.; Bui, Q. M.; Dick, G. R.; Kanan, M. W. J. Am. Chem. Soc. 2013, 135, 11257–11265. doi:10.1021/ja404394z |
| 40. | Blyth, M. T.; Noble, B. B.; Russell, I. C.; Coote, M. L. J. Am. Chem. Soc. 2020, 142, 606–613. doi:10.1021/jacs.9b12186 |
| 41. | Fried, S. D.; Boxer, S. G. Annu. Rev. Biochem. 2017, 86, 387–415. doi:10.1146/annurev-biochem-061516-044432 |
| 42. | Vaissier Welborn, V.; Head-Gordon, T. Chem. Rev. 2019, 119, 6613–6630. doi:10.1021/acs.chemrev.8b00399 |
| 43. | Warshel, A.; Sharma, P. K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M. H. M. Chem. Rev. 2006, 106, 3210–3235. doi:10.1021/cr0503106 |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 8. | Tan, M.-L.; Ángeles Gutiérrez López, M.; Sakai, N.; Matile, S. Angew. Chem., Int. Ed. 2023, 62, e202310393. doi:10.1002/anie.202310393 |
| 100. | Hagihara, S.; Tanaka, H.; Matile, S. J. Am. Chem. Soc. 2008, 130, 5656–5657. doi:10.1021/ja801094p |
| 101. | Talukdar, P.; Bollot, G.; Mareda, J.; Sakai, N.; Matile, S. Chem. – Eur. J. 2005, 11, 6525–6532. doi:10.1002/chem.200500516 |
| 102. | Gabriel, G. J.; Iverson, B. L. J. Am. Chem. Soc. 2002, 124, 15174–15175. doi:10.1021/ja0275358 |
| 103. | Cougnon, F. B. L.; Sanders, J. K. M. Acc. Chem. Res. 2012, 45, 2211–2221. doi:10.1021/ar200240m |
| 104. | Iijima, T.; Vignon, S. A.; Tseng, H.-R.; Jarrosson, T.; Sanders, J. K. M.; Marchioni, F.; Venturi, M.; Apostoli, E.; Balzani, V.; Stoddart, J. F. Chem. – Eur. J. 2004, 10, 6375–6392. doi:10.1002/chem.200400651 |
| 105. | Ikkanda, B. A.; Samuel, S. A.; Iverson, B. L. J. Org. Chem. 2014, 79, 2029–2037. doi:10.1021/jo402704z |
| 106. | Mukhopadhyay, P.; Iwashita, Y.; Shirakawa, M.; Kawano, S.-i.; Fujita, N.; Shinkai, S. Angew. Chem., Int. Ed. 2006, 45, 1592–1595. doi:10.1002/anie.200503158 |
| 32. | Shaik, S.; Danovich, D.; Joy, J.; Wang, Z.; Stuyver, T. J. Am. Chem. Soc. 2020, 142, 12551–12562. doi:10.1021/jacs.0c05128 |
| 33. | Ciampi, S.; Darwish, N.; Aitken, H. M.; Díez-Pérez, I.; Coote, M. L. Chem. Soc. Rev. 2018, 47, 5146–5164. doi:10.1039/c8cs00352a |
| 34. | Kareem, S.; Vali, S. R.; Reddy, B. V. S. Eur. J. Org. Chem. 2023, e202300103. doi:10.1002/ejoc.202300103 |
| 35. | Shaik, S.; Ramanan, R.; Danovich, D.; Mandal, D. Chem. Soc. Rev. 2018, 47, 5125–5145. doi:10.1039/c8cs00354h |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 44. | Gutiérrez López, M. Á.; Ali, R.; Tan, M.-L.; Sakai, N.; Wirth, T.; Matile, S. Sci. Adv. 2023, 9, eadj5502. doi:10.1126/sciadv.adj5502 |
| 2. | Zhao, Y.; Benz, S.; Sakai, N.; Matile, S. Chem. Sci. 2015, 6, 6219–6223. doi:10.1039/c5sc02563j |
| 3. | Zhao, Y.; Cotelle, Y.; Liu, L.; López-Andarias, J.; Bornhof, A.-B.; Akamatsu, M.; Sakai, N.; Matile, S. Acc. Chem. Res. 2018, 51, 2255–2263. doi:10.1021/acs.accounts.8b00223 |
| 18. | Maynard, J. R. J.; Galmés, B.; Stergiou, A. D.; Symes, M. D.; Frontera, A.; Goldup, S. M. Angew. Chem., Int. Ed. 2022, 61, e202115961. doi:10.1002/anie.202115961 |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 60. | Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187 |
| 111. | Gabriel, G.; Gómez-Martínez, R.; Villa, R. Physiol. Meas. 2008, 29, S203–S212. doi:10.1088/0967-3334/29/6/s18 |
| 2. | Zhao, Y.; Benz, S.; Sakai, N.; Matile, S. Chem. Sci. 2015, 6, 6219–6223. doi:10.1039/c5sc02563j |
| 61. | Bornhof, A.-B.; Bauzá, A.; Aster, A.; Pupier, M.; Frontera, A.; Vauthey, E.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2018, 140, 4884–4892. doi:10.1021/jacs.8b00809 |
| 112. | Foroutan-Nejad, C.; Marek, R. Phys. Chem. Chem. Phys. 2014, 16, 2508–2514. doi:10.1039/c3cp52671b |
| 113. | Novák, M.; Foroutan-Nejad, C.; Marek, R. J. Chem. Theory Comput. 2016, 12, 3788–3795. doi:10.1021/acs.jctc.6b00586 |
| 114. | Farajpour, E.; Sohrabi, B.; Beheshtian, J. Phys. Chem. Chem. Phys. 2016, 18, 7293–7299. doi:10.1039/c5cp07710a |
| 115. | Chen, J.; Li, J.; Liu, X.; He, Z.; Shi, G. Phys. Chem. Chem. Phys. 2023, 25, 13260–13264. doi:10.1039/d3cp00986f |
| 2. | Zhao, Y.; Benz, S.; Sakai, N.; Matile, S. Chem. Sci. 2015, 6, 6219–6223. doi:10.1039/c5sc02563j |
| 14. | Akamatsu, M.; Sakai, N.; Matile, S. J. Am. Chem. Soc. 2017, 139, 6558–6561. doi:10.1021/jacs.7b02421 |
| 58. | Kobuke, Y.; Yoshida, J.-i. Tetrahedron Lett. 1978, 19, 367–370. doi:10.1016/s0040-4039(01)85127-3 |
| 59. | Sakai, N.; Sordé, N.; Matile, S. Molecules 2001, 6, 845–851. doi:10.3390/61100845 |
| 60. | Lubkoll, J.; Wennemers, H. Angew. Chem., Int. Ed. 2007, 46, 6841–6844. doi:10.1002/anie.200702187 |
| 107. | Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497 |
| 108. | Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412 |
| 109. | Folgueiras‐Amador, A. A.; Philipps, K.; Guilbaud, S.; Poelakker, J.; Wirth, T. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. doi:10.1002/anie.201709717 |
| 110. | Gnaim, S.; Bauer, A.; Zhang, H.-J.; Chen, L.; Gannett, C.; Malapit, C. A.; Hill, D. E.; Vogt, D.; Tang, T.; Daley, R. A.; Hao, W.; Zeng, R.; Quertenmont, M.; Beck, W. D.; Kandahari, E.; Vantourout, J. C.; Echeverria, P.-G.; Abruna, H. D.; Blackmond, D. G.; Minteer, S. D.; Reisman, S. E.; Sigman, M. S.; Baran, P. S. Nature 2022, 605, 687–695. doi:10.1038/s41586-022-04595-3 |
| 52. | Yamada, M. ChemPlusChem 2023, 88, e202300062. doi:10.1002/cplu.202300062 |
| 53. | Li, C.-Z.; Chueh, C.-C.; Yip, H.-L.; Ding, F.; Li, X.; Jen, A. K.-Y. Adv. Mater. (Weinheim, Ger.) 2013, 25, 2457–2461. doi:10.1002/adma.201204543 |
| 54. | Sun, X.; Chen, W.; Liang, L.; Hu, W.; Wang, H.; Pang, Z.; Ye, Y.; Hu, X.; Wang, Q.; Kong, X.; Jin, Y.; Lei, M. Chem. Mater. 2016, 28, 8726–8731. doi:10.1021/acs.chemmater.6b04056 |
| 55. | Sun, X.; Ji, L. Y.; Chen, W. W.; Guo, X.; Wang, H. H.; Lei, M.; Wang, Q.; Li, Y. F. J. Mater. Chem. A 2017, 5, 20720–20728. doi:10.1039/c7ta06335k |
| 56. | Yamada, M.; Sahara, K.; Koizumi, M.; Maeda, Y.; Suzuki, M. Chem. – Eur. J. 2023, 29, e202300877. doi:10.1002/chem.202300877 |
| 57. | Sun, S.; Liu, Z.; Colombo, F.; Gao, R.; Yu, Y.; Qiu, Y.; Su, J.; Gan, L. Angew. Chem., Int. Ed. 2022, 61, e202212090. doi:10.1002/anie.202212090 |
| 41. | Fried, S. D.; Boxer, S. G. Annu. Rev. Biochem. 2017, 86, 387–415. doi:10.1146/annurev-biochem-061516-044432 |
| 42. | Vaissier Welborn, V.; Head-Gordon, T. Chem. Rev. 2019, 119, 6613–6630. doi:10.1021/acs.chemrev.8b00399 |
| 43. | Warshel, A.; Sharma, P. K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M. H. M. Chem. Rev. 2006, 106, 3210–3235. doi:10.1021/cr0503106 |
| 12. | López-Andarias, J.; Frontera, A.; Matile, S. J. Am. Chem. Soc. 2017, 139, 13296–13299. doi:10.1021/jacs.7b08113 |
| 32. | Shaik, S.; Danovich, D.; Joy, J.; Wang, Z.; Stuyver, T. J. Am. Chem. Soc. 2020, 142, 12551–12562. doi:10.1021/jacs.0c05128 |
| 33. | Ciampi, S.; Darwish, N.; Aitken, H. M.; Díez-Pérez, I.; Coote, M. L. Chem. Soc. Rev. 2018, 47, 5146–5164. doi:10.1039/c8cs00352a |
| 34. | Kareem, S.; Vali, S. R.; Reddy, B. V. S. Eur. J. Org. Chem. 2023, e202300103. doi:10.1002/ejoc.202300103 |
| 35. | Shaik, S.; Ramanan, R.; Danovich, D.; Mandal, D. Chem. Soc. Rev. 2018, 47, 5125–5145. doi:10.1039/c8cs00354h |
| 36. | Delley, M. F.; Nichols, E. M.; Mayer, J. M. J. Am. Chem. Soc. 2021, 143, 10778–10792. doi:10.1021/jacs.1c05419 |
| 37. | Yu, L.-J.; Coote, M. L. J. Phys. Chem. A 2019, 123, 582–589. doi:10.1021/acs.jpca.8b11579 |
| 38. | Zhang, B.; Schaack, C.; Prindle, C. R.; Vo, E. A.; Aziz, M.; Steigerwald, M. L.; Berkelbach, T. C.; Nuckolls, C.; Venkataraman, L. Chem. Sci. 2023, 14, 1769–1774. doi:10.1039/d2sc06411a |
| 39. | Gorin, C. F.; Beh, E. S.; Bui, Q. M.; Dick, G. R.; Kanan, M. W. J. Am. Chem. Soc. 2013, 135, 11257–11265. doi:10.1021/ja404394z |
| 40. | Blyth, M. T.; Noble, B. B.; Russell, I. C.; Coote, M. L. J. Am. Chem. Soc. 2020, 142, 606–613. doi:10.1021/jacs.9b12186 |
© 2023 Gutiérrez López et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.