Abstract
2,2’-Bis(4-dimethylaminophenyl)- and 2,2'-dicyclohexyl-1,1',3,3'-tetramethyl-2,2',3,3'-tetrahydro-2,2'-bibenzo[d]imidazole ((N-DMBI)2 and (Cyc-DMBI)2) are quite strong reductants with effective potentials of ca. −2 V vs ferrocenium/ferrocene, yet are relatively stable to air due to the coupling of redox and bond-breaking processes. Here, we examine their use in accomplishing electron transfer-induced bond-cleavage reactions, specifically dehalogenations. The dimers reduce halides that have reduction potentials less cathodic than ca. −2 V vs ferrocenium/ferrocene, especially under UV photoexcitation (using a 365 nm LED). In the case of benzyl halides, the products are bibenzyl derivatives, whereas aryl halides are reduced to the corresponding arenes. The potentials of the halides that can be reduced in this way, quantum-chemical calculations, and steady-state and transient absorption spectroscopy suggest that UV irradiation accelerates the reactions via cleavage of the dimers to the corresponding radical monomers.

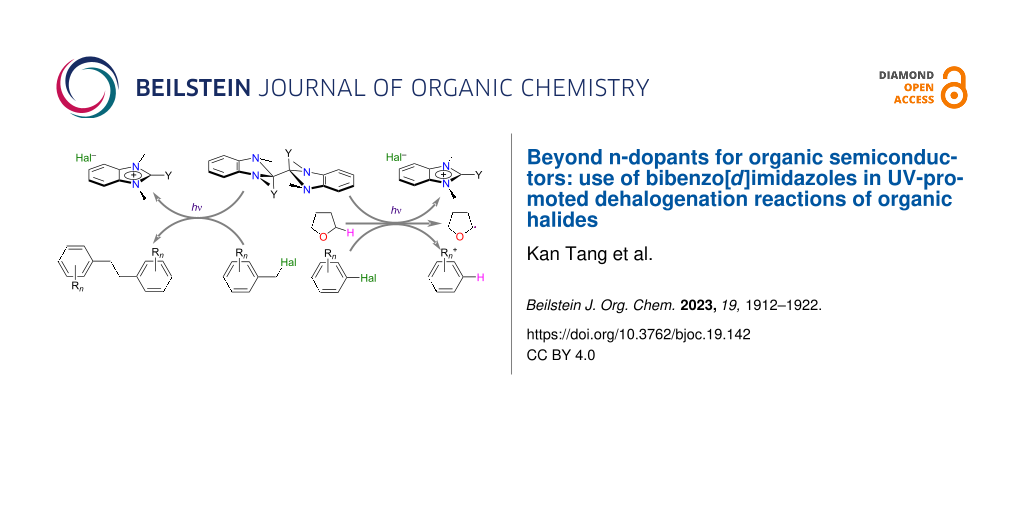
Graphical Abstract
Introduction
Reductive dehalogenation reactions of organic halides can be used in organic synthesis as a means of generating carbon-centered radical or anion intermediates and could have relevance to the treatment of waste halogenated polymers. While such reactions can be achieved using highly reducing metals, molecular reductants can potentially enable more selectivity, as required for the use of such reactions in the synthesis of molecules bearing various functional groups. In particular, Wanzlick dimers (C=C-bonded dimers of N-heterocyclic carbenes, Figure 1a, i) have been used by Murphy’s group and others for a variety of transformations, such as the formation of indolines from N-allyl-2-iodoanilines [1], indanones from 3-(2-halophenyl)propanoic esters [2,3], and 3-methyl-2,3-dihydrobenzofuran from 1-allyloxy-2-halobenzenes [4]. Related species have also been used to initiate the coupling of aryl halides and arenes [5]. However, even relatively easily reduced organic halides have sufficiently cathodic reduction potentials (e.g., ca. −1.6 V and −1.8 vs ferrocenium/ferrocene (FeCp2+/0) for diethyl bromomalonate [6] and 4-iodotoluene, see Table 2, respectively) that simple one- or two-electron donors capable of exergonic ground-state electron transfer to these substrates will be rather air sensitive, complicating their handling and use. In addition some molecular reductants can themselves react with the reactive intermediates; for example, the dehalogenation of alkyl halides, RX, by CoCp2 (Cp = η5-cyclopentadienyl; E = −1.3 V vs FeCp2+/0), gives CoCp2+ and X−, but the organic radicals R• react with another molecule of CoCp2 to afford CoCp(η4-C5H5R) [7]. In some cases, issues of reductant air sensitivity can be circumvented by the use of photocatalysts in concert with sacrificial weak reductants (Figure 1b) [8-11]. Another approach is to add ambient-stable precursors to reaction mixtures: for example, reducing Wanzlick dimers and related species (Figure 1a, ii) have been formed from precursors through in situ decarboxylation [12] or deprotonation [4,5], while other reducing species have been formed from in situ reactions of simple diols or diamines [13].
![[1860-5397-19-142-1]](/bjoc/content/figures/1860-5397-19-142-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a, b) Schematics of previous approaches to dehalogenation-based reactions using molecular reductants, along with representative structures of reductants and photocatalysts used, and (c) schematic for the present approach, along with structures of 1,1',3,3'-tetramethyl-2,2',3,3'-tetrahydro-2,2'-bibenzo[d]imidazoles, (Y-DMBI)2, and the corresponding monomer cations, specifically the first reported in the literature and those used in this work.
Figure 1: (a, b) Schematics of previous approaches to dehalogenation-based reactions using molecular reductan...
Another approach is to utilize dimers formed by highly reducing radicals, such the bibenzoimidazoles (Y-DMBI)2 (Figure 1c). (Me-DMBI)2 was first reported in 1984 and used as a reductant in studies of electrochemically generated reactive species [14-16]. More recently, several examples, including (Cyc-DMBI)2 (Y = cyclohexyl) and (N-DMBI)2 (Y = 4-dimethylaminophenyl) have been used as effective n-dopants for organic semiconductors [17-24] and redox mediators for the electrochemical depolymerization of poly(ethylene terephthalate) [25]. These dimers (D2 = (Y-DMBI)2) undergo reactions with organic semiconductors A to afford two monomeric Y-DMBI+ (D+) cations and two reduced semiconductors, A•−. The effective redox potentials, E(D+/0.5D2), are estimated to be ca. −2 V vs FeCp2+/0, yet the dimers are reasonably stable to air due to the kinetic barriers associated with the coupling of electron-transfer and bond-cleavage reactions [26]. Here we demonstrate that (N-DMBI)2 and (Cyc-DMBI)2 (Figure 1c) can be used to accomplish dehalogenation of benzyl, alkyl, and aryl halides (RX) and discuss the scope and possible mechanism of these reactions.
Results and Discussion
Reaction of (Y-DMBI)2 with benzyl bromide
We began our investigations of dehalogenation reactions using benzyl bromide (BnBr, 1a), which has a reduction peak potential (Epc) of −1.6 V vs FeCp2+/0, as the substrate (RX), anhydrous THF (without stabilizer) as the solvent, and (N-DMBI)2 or (Cyc-DMBI)2 as a stoichiometric reductant (0.5 equiv, assuming one electron is needed for each substrate molecule and that each dimer molecule contributes two electrons). Reaction outcomes were analyzed by GC–MS (see Supporting Information File 1 for details); products were identified based on the observed m/z values and comparison of retention times with authentic samples, while conversions and yields were based on calibration curves established using authentic samples. Table 1 summarizes the conversion of BnBr and product yields at various reaction times, with or without dimers, and with or without irradiation at a nominal wavelength of 365 nm (see Supporting Information File 1 for experimental details and see [27] for the spectrum of the excitation source used), which is anticipated to selectively excite the dimeric reductants rather than the substrate (see below and Supporting Information File 1, Figure S4 for further details). In the dark at low concentrations of substrate and reductant low conversions are observed after 1 h (Table 1, entry 1), with the dehalogenation products being a mixture of toluene and bibenzyl, i.e., RH and R2 products, which were identified by the masses observed through GC–MS and through comparison of the GC–MS retention times with authentic samples. Higher concentrations and reaction times (entries 7–9 in Tabe 1) lead to larger extents of conversion (up to 61% at 18 h) and favor formation of bibenzyl over toluene (up to 52% yield at 18 h). [BnBr]•– presumably cleaves to afford Bn•, which can react further to form Bn2 (see following section) or can form toluene through reaction with THF, which is known to have a reasonably weak α-CH bond and act as a H• donor towards many radicals [28-31].
Table 1: Debromination of benzyl bromide (1a).
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-142-i5.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Time [h] | UV | [BnBr] [mM]a | Reductant | Conversion [%]b | BnH yield [%]b | Bn2 yield [%]b |
| 1 | 1 | no | 3.0 | (N-DMBI)2 | 20 | 13 | 10 |
| 2 | 0.5 | yes | 3.0 | (N-DMBI)2 | 90 | 8.6 | 82 |
| 3 | 1 | yes | 3.0 | (N-DMBI)2 | 100 | 9 | 92 |
| 4 | 1 | no | 3.0 | (Cyc-DMBI)2 | 10 | 5 | 5 |
| 5 | 0.5 | yes | 3.0 | (Cyc-DMBI)2 | 70 | 4 | 65 |
| 6 | 1 | yes | 3.0 | (Cyc-DMBI)2 | 100 | 4 | 95 |
| 7 | 1 | no | 18.7 | (N-DMBI)2 | 29 | 7.5 | 22 |
| 8 | 2 | no | 18.7 | (N-DMBI)2 | 39 | 9 | 30 |
| 9 | 18 | no | 18.7 | (N-DMBI)2 | 61 | 7 | 52 |
| 10 | 1 | yes | 18.7 | (N-DMBI)2 | 70 | 6 | 66 |
| 11 | 2 | yes | 18.7 | (N-DMBI)2 | 80 | 6 | 74 |
| 12 | 2 | yes | 18.7 | – | 6 | 6 | –c |
| 13 | 4 | yes | 18.7 | (N-DMBI)2 | 92 | 6 | 86 |
| 14 | 18 | yes | 18.7 | (N-DMBI)2 | 100 | 8 | 93 |
| 15 | 18 | yes | 18.7 | – | 61 | 58 | –c |
a3.0 and 18.7 mM BnBr concentrations used, BnBr in quantities of 12 and 75 µmol (2 and 13 mg), respectively, corresponding to dimer quantities of 6.0 and 37.5 µmol, respectively (3.2 and 20 mg for (N-DMBI)2; 2.8 and 17 mg for (Cyc-DMBI)2. bConversions and yields were determined by GC–MS as described in Supporting Information File 1. cNone detected.
The reaction is substantially accelerated by UV excitation at 365 nm; quantitative conversion of benzyl bromide at low initial concentration (3 mM) can be achieved within 1 h using both (N-DMBI)2 or (Cyc-DMBI)2 and UV (Table 1, entries 3 and 6), while even at higher substrate concentrations (18.7 mM) 80% and near-quantitative (>90%) conversions can be obtained using (N-DMBI)2 within 2 h and 4 h, respectively (Table 1, entries 11 and 13). We also investigated the impact of photoexcitation in the absence of the dimeric reductant; however, extents of conversion are much lower for a given reaction time (compare entries 12 and 15 to 11 and 14 in Table 1) and the sole detected product is toluene rather than bibenzyl.
Furthermore, in one of the cases of complete conversion (Table 1, entry 14), 1H NMR spectroscopy indicated that the reductant-based side product is a salt of the monomeric cation N-DMBI+ (Supporting Information File 1, Figure S15). Thus, the overall reaction is consistent with:
![[1860-5397-19-142-i3]](/bjoc/content/inline/1860-5397-19-142-i3.svg?max-width=590&scale=1.18182)
Scope of reaction (Y-DMBI)2 with other benzyl, alkyl, and aryl halides
Table 2 summarizes the conversions and product yields for the reactions of (N-DMBI)2 or (Cyc-DMBI)2 with several other benzyl halides (1b–e), an alkyl halide 2, and several aryl halides (3a–f). Again GC–MS was used to identify and quantify the products; the necessary authentic samples were mostly commercially available, but the R2 products from 1b and 1c were not, although well-known in the literature (for example, see ref. [32]), and were synthesized as described in Supporting Information File 1. More complete data are shown in Supporting Information File 1, Tables S1 and S2. As in the case of 1a, conversions and yields under UV irradiation in the absence of reductant are low on a 2 h timescale (≤10%) and the main products are those in which the halide is replaced by a hydrogen atom. The more easily reduced benzyl halides examined (1b and 1c) are dehalogenated by (N-DMBI)2 in the dark, and with (N-DMBI)2 and UV irradiation are quantitatively dehalogenated in 2–6 h with the corresponding substituted bibenzyls being the dominant products. These reaction conditions represent an alternative metal-free approach to the conventional synthesis of bibenzyls through the reaction of Grignard or organolithium reagents with benzyl halides, or to the use of highly active metal reagents [33-36] or metal-containing catalysts [32]. We note that another all-organic reductive dimerization of benzyl halides using 2,3,5,6-tetrakis(tetramethylguanidino)pyridine has recently been reported [37]. The less readily reduced halides examined here (1d,e, and 2) are only sluggishly converted, even when using both (N-DMBI)2 (or (Cyc-DMBI)2) and light. Moreover, in the cases of 1d and 1e there are significant mismatches between conversion and the yields of the corresponding RH and R2 species, indicating that additional products must be formed. Indeed, in the case of 4-methylbenzyl chloride GC–MS shows a product with a mass consistent with the formation of 4-methylbenzyl-substituted THF (see Supporting Information File 1, Figure S2) and a somewhat better R2 yield is obtained in toluene (see Table S2). For 1-bromooctadecane (2), around half the product obtained using (N-DMBI)2 and UV is RH, i.e., octadecane. The remaining unidentified product may be R2 (C36H74), but this product is not easily detected by GC–MS.
Table 2: Dehalogenation of various benzyl, alkyl, and aryl halides using (N-DMBI)2 (or, in parentheses Cyc-DMBI)2.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-142-i6.svg?max-width=637&scale=1.0)
|
||||
| RX | Epc [V]b | Conversion [%]c | RH yield [%]c | R2 yield [%]c |
| 1a | −1.6 | 92 | 6 | 86 |
| 1b | −1.5 | 80 | 2 | 75 |
| 1c | −1.6 | 100 | 14 | 86 |
| 1d | −2.3 | 39 | 7 | 5 |
| 1e | −2.4 | 73 (60) | 3 (3) | 3 (32) |
| 2 | −2.1 | 39 | 19 | –d |
| 3ae | −1.8 | 98 | 98 | –f |
| 3be | −1.6 | 99 | 99 | –f |
| 3c | −1.6 | 55 (99) | 41 (60) | –f (–f) |
| 3d | −2.4 | 5 (5) | 5 (5) | –f |
| 3e | −2.5 | 4 (2) | 0 (2) | –f |
| 3f | −2.4 | 1 (<1) | 1 (<1) | –f |
aThe reaction scale in each case was 75 µmol (9.5–25 mg) RX and 37.5 µmol dimer (20 and 17 mg for (N-DMBI)2 and (Cyc-DMBI)2, respectively). bPeak reduction potential vs FeCp2+/0 in THF/0.1 M Bu4NPF6 (see Supporting Information File 1, Figures S7 and S8). cConversions and yields were determined by GC–MS as described in Supporting Information File 1. dLikely not detectable by GC–MS. e2 h reaction time. fNone detected.
In sharp contrast to the case of benzyl derivatives, where the use of dimer reductants primarily affords R2 products, biaryls are not observed as dehalogenation products of aryl halides by dimeric reductants and/or light. This may be attributable to the lower stability and greater reactivity of aryl radicals relative to that of their benzyl counterparts. Indeed, aryl radicals are known to abstract H• from THF [30,31] and presumably do so in the present reactions before any further reactions can occur. Resonance-stabilized benzyl radicals, on the other hand, are sufficiently long-lived to react further to afford dimeric bibenzyl derivatives (especially at higher concentrations or when photoirradiation is used, presumably affording higher steady-state [R•] concentrations). In principle, this is possible by either dimerization of 2R• or by a second reduction of R• to R− (as invoked in the reductive cyclization of (2-halophenyl)propanoic esters [2]) which then acts as a nucleophile towards a second molecule of RX. However, addition of Me3SiCl to a photoirradiated BnBr/(N-DMBI)2 reaction mixture did not lead to any detectable BnSiMe3, thus supporting a radical dimerization pathway (see Supporting Information File 1, Table S3).
As in the case of sp2 R–X systems, only small extents of dehalogenation for R = aryl are observed in the absence of reductants. For the more easily reduced aryl halides (3a–c), moderate to high extents of conversion are obtained in the dark using dimeric reductants, while higher extents, in some cases near-quantitative in 2 h, are obtained using UV and reductants. In the case of 3a and 3b the conversion and yields of the RH compounds are in good agreement, while discrepancies in the case of 3c indicate additional side reactions.
The observation that some of the organic halides tested in Table 2 are cleanly dehalogenated and others essentially unreactive suggested the possibility of selective dehalogenation of compounds containing different halogenated functionalities. Specifically, we examined 2-iodobenzyl chloride, in which the C–I and C–Cl bond strengths are expected to be fairly similar (C–X bond dissociation enthalpies of 280 and 310 kJ mol−1 have been reported for BnCl and PhI, respectively [38]), but the aryl–I bond is likely to be better coupled to the reduction process (given the reduction potentials in Table 2). As shown in Scheme 1, we found 2-iodobenzyl chloride is cleanly converted to benzyl chloride under the standard conditions used in Table 2 (see Supporting Information File 1, Table S4 for more details). This selectivity is not typically achievable using electropositive metals and may be of use in more elaborate chemical transformations.
![[1860-5397-19-142-i1]](/bjoc/content/inline/1860-5397-19-142-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Selective deiodination of 2-iodobenzyl chloride.
Scheme 1: Selective deiodination of 2-iodobenzyl chloride.
Mechanism of dark reactions
Doping of organic semiconductors by (Y-DMBI)2 dimers [18,39] or by various dimers formed by 18-electron sandwich compounds [18,40,41], as well as redox reactions of other dimers formed by organic radicals [42,43], are known to proceed by two distinct pathways. When the dimer (D2) is relatively weakly bound, its dissociation to D• can be the initial step (“cleavage-first”), which is then followed by fast electron-transfer (ET) reactions (Scheme 2). On the other hand, in the “electron-transfer first” mechanism, the first step is an ET reaction, resulting in the formation of D2•+, which subsequently cleaves to form D+ and D•, which is much more readily oxidized than the dimer itself and thus participates in a second fast ET (Scheme 2). In the present case, knowledge of the operative mechanism(s) is important to understand what substrates might be cleavable on what timescales in the dark; in particular, if the cleavage-first mechanism is viable, substrates with Ered as cathodic as, roughly, E(D+/D•), should react, whereas more strongly bonded dimers might not react with the most challenging of these substrates due to the non-viability of the cleavage-first mechanism and prohibitively endergonic initial steps for the ET-first mechanism.
![[1860-5397-19-142-i2]](/bjoc/content/inline/1860-5397-19-142-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction mechanisms for the reactions of dimeric reductants (D2) such as (Y-DMBI)2 derivatives with acceptors (A) such as organic semiconductors or, in this work, organic halides that react further (the relative rates of steps are indicated for cases where the A reduction potential falls between D2 and D• oxidation potentials).
Scheme 2: Reaction mechanisms for the reactions of dimeric reductants (D2) such as (Y-DMBI)2 derivatives with...
We investigated the dark reactions of the two dimers with BnBr in THF, using GC–MS to determine RX conversion and R2 and RH yields at various time intervals, as shown in the example of Figure 2a. We repeated these experiments for a variety of different initial reductant and RX concentrations, [D2]0 and [RX]0 (see Supporting Information File 1, Figure S3), and plotted the initial rate normalized for dimer concentrations, (d[RX]/dt)0/[D2]0, vs [RX]0 (Figure 2b). In the case of D2 = (Cyc-DMBI)2, a linear plot with approximately zero intercept is obtained, consistent with a reaction first order in [RX] and [D2], as expected if the reaction proceeds via an initial rate-determining ET. On the other hand, in the case of D2 = (N-DMBI)2, the linear plot has a distinctly non-zero intercept, a behavior which conforms with a rate law consisting of the sum of two terms, one first order in [D2] and one first order in both [D2] and [RX]
![[1860-5397-19-142-i4]](/bjoc/content/inline/1860-5397-19-142-i4.svg?max-width=590&scale=1.18182)
and is similar to doping behavior we have recently seen for some weakly bonded dimers where both “cleavage-first” and “ET-first” mechanisms are competitive. In the case of (Cyc-DMBI)2, the second-order rate constant, kET, is estimated as 6.0 × 10−3 M−1 s−1, whereas for (N-DMBI)2, kcl = 4.7 × 10−5 s−1 and kET = 1.0 × 10−2 M−1s−1. The difference in kET values is qualitatively consistent with the peak oxidation potentials, Epa(D2•+/D2), of the two dimers; values of −0.06 and −0.13 V vs FeCp2+/0 are found for (Cyc-DMBI)2 and (N-DMBI)2, respectively [18,44], indicating that ET from the former to benzyl bromide is more endergonic than from the latter. The rate constant for (Cyc-DMBI)2-to-BnBr ET is also much smaller than that previously determined for the doping of 6,13-bis(triisopropylsilylethynyl)pentacene (TIPS-pentacene, E1/20/− = −1.45 V, kET ≈ 0.15 M−1s−1) by (Cyc-DMBI)2 [18], consistent with the differences in the reduction potentials between BnBr and TIPS-pentacene. The observation of the “cleavage-first” mechanism for (N-DMBI)2 and not for (Cyc-DMBI)2 is consistent with DFT estimates of bond dissociation energies for these two dimers (ΔUdiss = 163 and 210 kJ mol−1, respectively [18,44]) and with their reactivity towards TIPS-pentacene [18,39].
![[1860-5397-19-142-2]](/bjoc/content/figures/1860-5397-19-142-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) A representative temporal evolution of % conversion (blue squares), % toluene yield (red diamonds), and % bibenzyl yield (black circles) during the dark dehalogenation reaction of benzyl bromide (BnBr) using (N-DMBI)2 in THF (these data were acquired using 3 mM BnBr and 1.5 mM of (N-DMBI)2. (b) Plot of [D2]0-normalized initial reaction rate (d([RX]/dt)0/[D2]0) as a function of the initial benzyl bromide concentration ([RX]0) obtained from several experiments of the type shown in part (a) for different [D2] and [RX] for both D2 = (Cyc-DMBI)2 and (N-DMBI)2. For these slow reactions, the “initial” rates were estimated from the change of substrate concentration over the first 30 min reaction time.
Figure 2: (a) A representative temporal evolution of % conversion (blue squares), % toluene yield (red diamon...
Impact of photoexcitation
The absorptivities, ε, of (N-DMBI)2 and (Cyc-DMBI)2 at 365 nm in THF are ca. 420 and 43 M−1 cm−1, respectively. Figure 3a and b show the absorption spectra of the two dimeric reductants in THF (see Supporting Information File 1, Figure S6 for data in toluene). (N-DMBI)2 shows a strong absorption feature with a maximum at 304 nm (εmax = 28000 M−1 cm−1) and a weak shoulder at ca. 400 nm (ε400 ≈ 150 M−1 cm−1), whereas (Cyc-DMBI)2 exhibits only a strong feature with a maximum at 327 nm (εmax = 13000 M−1 cm−1). TD-DFT calculations (M06/6-31G(d,p), isolated molecules) qualitatively reproduce the different behavior of the two dimers: for (Cyc-DMBI)2 the S0→S1 transition is calculated to be strong (oscillator strength, f = 0.27). The natural transition orbitals (NTOs, see Supporting Information File 1, including Figure S10, for more information) indicate that this excitation is largely confined to the bibenzoimidazole portion of the molecule. In the case of (N-DMBI)2 the lowest reasonably strong (f = 0.21) transition is S0→S6 and has bibenzoimidazole-dominated NTOs (Figure 3c) similar to those for the S0→S1 transition of (Cyc-DMBI)2, but is seen at higher energy. The strongest of the weaker lower energy transitions are S0→S4 (f = 0.018), which is calculated to lie lower in energy than the S0→S1 transition of (Cyc-DMBI)2 and presumably corresponds to the shoulder observed in the experimental spectrum of (N-DMBI)2, and S0→S5, which lies very close to S0→S6. For both of these transitions, the NTOs indicate considerable charge transfer from the bibenzoimidazole portion of the molecules to the C6H4NMe2 Y substituents (see Figure 3d and Supporting Information File 1, Figure S11).
![[1860-5397-19-142-3]](/bjoc/content/figures/1860-5397-19-142-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Top: UV–vis absorption spectra for the two dimeric reductants in THF emphasizing (a) the different positions of the relatively strong absorption peaks and (b) the presence of a low-energy shoulder in the spectrum of (N-DMBI)2. Bottom: TD-DFT (M06/6-31G(d, p)) natural transition orbitals for selected (N-DMBI)2 transitions that are thought to be primarily responsible for (c) the main absorption peak and (d) the weak shoulder seen experimentally.
Figure 3: Top: UV–vis absorption spectra for the two dimeric reductants in THF emphasizing (a) the different ...
Most of the above-mentioned substrates exhibit little or no absorption at 365 nm (see Supporting Information File 1, Figure S4 for comparison of spectra of the reductants and BnBr); thus, although slight overlap between the tail of substrate absorption and the tail of the output of the nominally 365 nm LED may be responsible for the reactivity seen in the absence of dimeric reductants, the stronger absorption of the two dimeric reductants examined suggests photo-acceleration of the reactions involves the excited states of the dimers. In principle excitation of the dimers might result in cleavage of the dimers to the corresponding radical monomers, D•, which can then reduce RX, or to ET from dimer excited states, D2*, to RX. The results in Table 2 support the former effect: if photoinduced cleavage occurs we would expect the scope of RX cleavage to be more-or-less limited by the reducing strength of D• (E(D+/D•) = ca. −2.4 V for both species used here), whereas the singlet excited state of the dimer, D2*, should be more reducing, allowing more challenging substrates to be reduced (E(D2•+/D2*) can be estimated as ca. −3.4 and −2.8 V for (Cyc-DMBI)2 and (Cyc-DMBI)2, respectively, using values of E(D2•+/D2) and estimated absorption onsets). Furthermore, the NTOs for the strong absorptions of both dimers and the weak low-energy absorption of (N-DMBI)2 both involve some depopulations of the bibenzoimidazole-based HOMO, which, as well as π-character also has significant C–σ-bonding character associated with the inter-monomer bond, suggesting that excitation should weaken the bond (in a similar way to oxidation associated with removal of an electron from the same orbital). To investigate this possibility in more detail we recorded transient absorption spectra of the dimers following excitation at 350 nm. Importantly, the samples did not degrade during the experiment, indicating that the species observed eventually reform the dimers. The transient absorption spectra of (N-DMBI)2 (Figure 4a) show significant spectral evolution within the first 1 ps following photoexcitation at 350 nm in MeCN. The (negative) bleach feature at 450 nm and photoinduced absorption (PIA) peaking at 775 nm transform into a broad PIA spanning the visible wavelengths with a notable isosbestic point at 600 nm. There is a secondary evolution occurring with a time constant of approximately 60 ps resulting in a long-lived feature existing beyond the 5 ns window of the ultrafast experiment. In toluene (Figure 4b), the spectral features at early times are different; however, the kinetics at 654 nm are the same between solvents suggesting that the initial features dominating the spectra at 250 fs in MeCN are present in toluene.
![[1860-5397-19-142-4]](/bjoc/content/figures/1860-5397-19-142-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Transient absorption spectra of (a) (N-DMBI)2 in MeCN, (b) (N-DMBI)2 in toluene, and (c) (Cyc-DMBI)2 in toluene with excitation at 350 nm in the absence of oxygen. (d) Comparison of temporal evolution of the absorption at 654 nm for both dimers in toluene. (e) Spectra for the two monomeric radicals obtained from M06/6-31G(d,p) TD-DFT calculations.
Figure 4: Transient absorption spectra of (a) (N-DMBI)2 in MeCN, (b) (N-DMBI)2 in toluene, and (c) (Cyc-DMBI)2...
The (Cyc-DMBI)2 exhibits faster decay kinetics (Figure 4c) than (N-DMBI)2 under the same experimental conditions. The spectral shape is similar to the late time spectra of the (N-DMBI)2 with a broad PIA spanning the visible wavelengths, which may suggest that the initial evolution observed in the (N-DMBI)2 occurs within our instrument response. In toluene, the signal amplitude at 5 ns for (Cyc-DMBI)2 is significantly smaller than that of the (N-DMBI)2 and 96% of the signal amplitude at 654 nm decays with a 1 ps lifetime. The isosbestic point in Figure 4a is consistent with a clean transformation such as monomer formation. The corresponding monomeric radicals are calculated using TD-DFT (M06/6-31G(d,p)) (Figure 4e) to have absorptions at similar energies to those seen in the longer-time spectra with Cyc-DMBI• having a blue-shifted absorption relative to N-DMBI•, consistent with the observed features arising from the monomeric radicals. Intersystem crossing (ISC) from the singlet to triplet excited state is also a possibility and TD-DFT calculations suggest that the dimer T1 states absorb at similar wavelengths to the monomer radicals (see Supporting Information File 1, Figure S12). However, ISC is typically much slower than 1 ps in organic molecules, especially so for those lacking heavy atoms [45].
Conclusion
Dimeric reductants of the (Y-DMBI)2 type can accomplish the dehalogenation of a variety of organic halides with peak reduction potentials less cathodic than ca. −2.0 V vs FeCp2+/0, affording essentially quantitative conversions in a few hours under 365 nm illumination. In the case of benzyl halides the primary products are bibenzyls, some of which may be challenging to synthesize by other methods, whereas aryl halides afford dehalogenated arenes. One example also demonstrates that the approach may have utility for selectively dehalogenating compounds containing different halogens. The photoacceleration of these reactions appears to occur through photocleavage of the dimers to the more strongly reducing monomers, which may help inform the use of these and related reductants in achieving other chemical transformations.
Supporting Information
| Supporting Information File 1: Synthetic and other experimental procedures, additional data, additional TD-DFT results, and NMR spectra of compounds. | ||
| Format: PDF | Size: 3.6 MB | Download |
Funding
This work was authored in part by the National Renewable Energy Laboratory (NREL), operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. This work was primarily supported by the Laboratory Directed Research and Development (LDRD) Program at NREL. The DFT and TD-DFT calculations, performed at the University of Kentucky, were supported by the Center for Soft PhotoElectroChemical Systems (SPECS), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award No. DE-SC0023411. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.
References
-
Murphy, J. A.; Khan, T. A.; Zhou, S.-z.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038
Return to citation in text: [1] -
Murphy, J. A.; Zhou, S.-z.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554
Return to citation in text: [1] [2] -
Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.-z.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g
Return to citation in text: [1] -
Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378
Return to citation in text: [1] [2] -
Zhou, S.; Anderson, G. M.; Mondal, B.; Doni, E.; Ironmonger, V.; Kranz, M.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2014, 5, 476–482. doi:10.1039/c3sc52315b
Return to citation in text: [1] [2] -
Huang, M.; Tang, J.; Kim, J. K.; Gong, M.; Zhang, J.; Li, Y.; Wu, Y. Org. Biomol. Chem. 2022, 20, 6459–6463. doi:10.1039/d2ob00835a
Return to citation in text: [1] -
Herberich, G. E.; Bauer, E.; Schwarzer, J. J. Organomet. Chem. 1969, 17, 445–452. doi:10.1016/s0022-328x(00)88230-3
Return to citation in text: [1] -
Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582
Return to citation in text: [1] -
Furst, L.; Matsuura, B. S.; Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. Org. Lett. 2010, 12, 3104–3107. doi:10.1021/ol101146f
Return to citation in text: [1] -
Ghosh, I.; Ghosh, T.; Bardagi, J. I.; König, B. Science 2014, 346, 725–728. doi:10.1126/science.1258232
Return to citation in text: [1] -
Constantin, T.; Zanini, M.; Regni, A.; Sheikh, N. S.; Juliá, F.; Leonori, D. Science 2020, 367, 1021–1026. doi:10.1126/science.aba2419
Return to citation in text: [1] -
Tintori, G.; Fall, A.; Assani, N.; Zhao, Y.; Bergé-Lefranc, D.; Redon, S.; Vanelle, P.; Broggi, J. Org. Chem. Front. 2021, 8, 1197–1205. doi:10.1039/d0qo01488e
Return to citation in text: [1] -
Zhou, S.; Doni, E.; Anderson, G. M.; Kane, R. G.; MacDougall, S. W.; Ironmonger, V. M.; Tuttle, T.; Murphy, J. A. J. Am. Chem. Soc. 2014, 136, 17818–17826. doi:10.1021/ja5101036
Return to citation in text: [1] -
Ludvík, J.; Volke, J.; Pragst, F. J. Electroanal. Chem. Interfacial Electrochem. 1986, 215, 179–190. doi:10.1016/0022-0728(86)87014-0
Return to citation in text: [1] -
Ludvík, J.; Pragst, F.; Volke, J. J. Electroanal. Chem. Interfacial Electrochem. 1984, 180, 141–156. doi:10.1016/0368-1874(84)83576-5
Return to citation in text: [1] -
Pragst, F.; Niazymbetov, M. J. Electroanal. Chem. Interfacial Electrochem. 1986, 197, 245–264. doi:10.1016/0022-0728(86)80153-x
Return to citation in text: [1] -
Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668
Return to citation in text: [1] -
Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Naab, B. D.; Gu, X.; Kurosawa, T.; To, J. W. F.; Salleo, A.; Bao, Z. Adv. Electron. Mater. 2016, 2, 1600004. doi:10.1002/aelm.201600004
Return to citation in text: [1] -
Yuan, D.; Huang, D.; Zhang, C.; Zou, Y.; Di, C.-a.; Zhu, X.; Zhu, D. ACS Appl. Mater. Interfaces 2017, 9, 28795–28801. doi:10.1021/acsami.7b07282
Return to citation in text: [1] -
Schwarze, M.; Gaul, C.; Scholz, R.; Bussolotti, F.; Hofacker, A.; Schellhammer, K. S.; Nell, B.; Naab, B. D.; Bao, Z.; Spoltore, D.; Vandewal, K.; Widmer, J.; Kera, S.; Ueno, N.; Ortmann, F.; Leo, K. Nat. Mater. 2019, 18, 242–248. doi:10.1038/s41563-018-0277-0
Return to citation in text: [1] -
Al Kurdi, K.; Gregory, S. A.; Jhulki, S.; Conte, M.; Barlow, S.; Yee, S. K.; Marder, S. R. Mater. Adv. 2020, 1, 1829–1834. doi:10.1039/d0ma00406e
Return to citation in text: [1] -
Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817
Return to citation in text: [1] -
Lungwitz, D.; Joy, S.; Mansour, A. E.; Opitz, A.; Karunasena, C.; Li, H.; Panjwani, N. A.; Moudgil, K.; Tang, K.; Behrends, J.; Barlow, S.; Marder, S. R.; Brédas, J.-L.; Graham, K.; Koch, N.; Kahn, A. J. Phys. Chem. Lett. 2023, 14, 5633–5640. doi:10.1021/acs.jpclett.3c01022
Return to citation in text: [1] -
Pham, P. H.; Barlow, S.; Marder, S. R.; Luca, O. R. Chem Catal. 2023, 3, 100675. doi:10.1016/j.checat.2023.100675
Return to citation in text: [1] -
Mohapatra, S. K.; Marder, S. R.; Barlow, S. Acc. Chem. Res. 2022, 55, 319–332. doi:10.1021/acs.accounts.1c00612
Return to citation in text: [1] -
https://www.hepatochem.com/product/hck1012-xx-011/ (accessed Sept 27, 2023).
Return to citation in text: [1] -
Newcomb, M.; Park, S. U. J. Am. Chem. Soc. 1986, 108, 4132–4134. doi:10.1021/ja00274a046
Return to citation in text: [1] -
Bietti, M.; Martella, R.; Salamone, M. Org. Lett. 2011, 13, 6110–6113. doi:10.1021/ol202561z
Return to citation in text: [1] -
Jing, L.; Nash, J. J.; Kenttämaa, H. I. J. Am. Chem. Soc. 2008, 130, 17697–17709. doi:10.1021/ja801707p
Return to citation in text: [1] [2] -
Higginson, B.; Sanjosé-Orduna, J.; Gu, Y.; Martin, R. Synlett 2021, 32, 1633–1636. doi:10.1055/a-1457-2399
Return to citation in text: [1] [2] -
Yu, D.; To, W.-P.; Tong, G. S. M.; Wu, L.-L.; Chan, K.-T.; Du, L.; Phillips, D. L.; Liu, Y.; Che, C.-M. Chem. Sci. 2020, 11, 6370–6382. doi:10.1039/d0sc01340d
Return to citation in text: [1] [2] -
Inaba, S.; Matsumoto, H.; Rieke, R. D. J. Org. Chem. 1984, 49, 2093–2098. doi:10.1021/jo00186a003
Return to citation in text: [1] -
Burns, T. P.; Rieke, R. D. J. Org. Chem. 1987, 52, 3674–3680. doi:10.1021/jo00392a033
Return to citation in text: [1] -
Kim, S.-H.; Rieke, R. D. J. Org. Chem. 2000, 65, 2322–2330. doi:10.1021/jo991478s
Return to citation in text: [1] -
Suh, Y.; Lee, J.-s.; Kim, S.-H.; Rieke, R. D. J. Organomet. Chem. 2003, 684, 20–36. doi:10.1016/s0022-328x(03)00500-x
Return to citation in text: [1] -
Wiesner, S.; Walter, P.; Wagner, A.; Kaifer, E.; Himmel, H.-J. Eur. J. Org. Chem. 2016, 5045–5054. doi:10.1002/ejoc.201600978
Return to citation in text: [1] -
Blanksby, S. J.; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255–263. doi:10.1021/ar020230d
Return to citation in text: [1] -
Mohapatra, S. K.; Al Kurdi, K.; Jhulki, S.; Bogdanov, G.; Bacsa, J.; Conte, M.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Beilstein J. Org. Chem. 2023, 19, 1651–1663. doi:10.3762/bjoc.19.121
Return to citation in text: [1] [2] -
Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591
Return to citation in text: [1] -
Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a
Return to citation in text: [1] -
Bennett, R. W.; Wharry, D. L.; Koch, T. H. J. Am. Chem. Soc. 1980, 102, 2345–2349. doi:10.1021/ja00527a036
Return to citation in text: [1] -
Colter, A. K.; Lai, C. C.; Parsons, A. G.; Ramsey, N. B.; Saito, G. Can. J. Chem. 1985, 63, 445–451. doi:10.1139/v85-073
Return to citation in text: [1] -
Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020
Return to citation in text: [1] [2] -
Imran, M.; Zhang, X.; Wang, Z.; Chen, X.; Zhao, J.; Barbon, A.; Voronkova, V. K. Phys. Chem. Chem. Phys. 2021, 23, 15835–15868. doi:10.1039/d1cp01937f
Return to citation in text: [1]
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 44. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 39. | Mohapatra, S. K.; Al Kurdi, K.; Jhulki, S.; Bogdanov, G.; Bacsa, J.; Conte, M.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Beilstein J. Org. Chem. 2023, 19, 1651–1663. doi:10.3762/bjoc.19.121 |
| 1. | Murphy, J. A.; Khan, T. A.; Zhou, S.-z.; Thomson, D. W.; Mahesh, M. Angew. Chem., Int. Ed. 2005, 44, 1356–1360. doi:10.1002/anie.200462038 |
| 6. | Huang, M.; Tang, J.; Kim, J. K.; Gong, M.; Zhang, J.; Li, Y.; Wu, Y. Org. Biomol. Chem. 2022, 20, 6459–6463. doi:10.1039/d2ob00835a |
| 5. | Zhou, S.; Anderson, G. M.; Mondal, B.; Doni, E.; Ironmonger, V.; Kranz, M.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2014, 5, 476–482. doi:10.1039/c3sc52315b |
| 28. | Newcomb, M.; Park, S. U. J. Am. Chem. Soc. 1986, 108, 4132–4134. doi:10.1021/ja00274a046 |
| 29. | Bietti, M.; Martella, R.; Salamone, M. Org. Lett. 2011, 13, 6110–6113. doi:10.1021/ol202561z |
| 30. | Jing, L.; Nash, J. J.; Kenttämaa, H. I. J. Am. Chem. Soc. 2008, 130, 17697–17709. doi:10.1021/ja801707p |
| 31. | Higginson, B.; Sanjosé-Orduna, J.; Gu, Y.; Martin, R. Synlett 2021, 32, 1633–1636. doi:10.1055/a-1457-2399 |
| 4. | Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378 |
| 25. | Pham, P. H.; Barlow, S.; Marder, S. R.; Luca, O. R. Chem Catal. 2023, 3, 100675. doi:10.1016/j.checat.2023.100675 |
| 2. | Murphy, J. A.; Zhou, S.-z.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 3. | Murphy, J. A.; Garnier, J.; Park, S. R.; Schoenebeck, F.; Zhou, S.-z.; Turner, A. T. Org. Lett. 2008, 10, 1227–1230. doi:10.1021/ol800134g |
| 26. | Mohapatra, S. K.; Marder, S. R.; Barlow, S. Acc. Chem. Res. 2022, 55, 319–332. doi:10.1021/acs.accounts.1c00612 |
| 4. | Hanson, S. S.; Doni, E.; Traboulsee, K. T.; Coulthard, G.; Murphy, J. A.; Dyker, C. A. Angew. Chem., Int. Ed. 2015, 54, 11236–11239. doi:10.1002/anie.201505378 |
| 5. | Zhou, S.; Anderson, G. M.; Mondal, B.; Doni, E.; Ironmonger, V.; Kranz, M.; Tuttle, T.; Murphy, J. A. Chem. Sci. 2014, 5, 476–482. doi:10.1039/c3sc52315b |
| 14. | Ludvík, J.; Volke, J.; Pragst, F. J. Electroanal. Chem. Interfacial Electrochem. 1986, 215, 179–190. doi:10.1016/0022-0728(86)87014-0 |
| 15. | Ludvík, J.; Pragst, F.; Volke, J. J. Electroanal. Chem. Interfacial Electrochem. 1984, 180, 141–156. doi:10.1016/0368-1874(84)83576-5 |
| 16. | Pragst, F.; Niazymbetov, M. J. Electroanal. Chem. Interfacial Electrochem. 1986, 197, 245–264. doi:10.1016/0022-0728(86)80153-x |
| 12. | Tintori, G.; Fall, A.; Assani, N.; Zhao, Y.; Bergé-Lefranc, D.; Redon, S.; Vanelle, P.; Broggi, J. Org. Chem. Front. 2021, 8, 1197–1205. doi:10.1039/d0qo01488e |
| 17. | Naab, B. D.; Zhang, S.; Vandewal, K.; Salleo, A.; Barlow, S.; Marder, S. R.; Bao, Z. Adv. Mater. (Weinheim, Ger.) 2014, 26, 4268–4272. doi:10.1002/adma.201400668 |
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 19. | Naab, B. D.; Gu, X.; Kurosawa, T.; To, J. W. F.; Salleo, A.; Bao, Z. Adv. Electron. Mater. 2016, 2, 1600004. doi:10.1002/aelm.201600004 |
| 20. | Yuan, D.; Huang, D.; Zhang, C.; Zou, Y.; Di, C.-a.; Zhu, X.; Zhu, D. ACS Appl. Mater. Interfaces 2017, 9, 28795–28801. doi:10.1021/acsami.7b07282 |
| 21. | Schwarze, M.; Gaul, C.; Scholz, R.; Bussolotti, F.; Hofacker, A.; Schellhammer, K. S.; Nell, B.; Naab, B. D.; Bao, Z.; Spoltore, D.; Vandewal, K.; Widmer, J.; Kera, S.; Ueno, N.; Ortmann, F.; Leo, K. Nat. Mater. 2019, 18, 242–248. doi:10.1038/s41563-018-0277-0 |
| 22. | Al Kurdi, K.; Gregory, S. A.; Jhulki, S.; Conte, M.; Barlow, S.; Yee, S. K.; Marder, S. R. Mater. Adv. 2020, 1, 1829–1834. doi:10.1039/d0ma00406e |
| 23. | Un, H.-I.; Gregory, S. A.; Mohapatra, S. K.; Xiong, M.; Longhi, E.; Lu, Y.; Rigin, S.; Jhulki, S.; Yang, C.-Y.; Timofeeva, T. V.; Wang, J.-Y.; Yee, S. K.; Barlow, S.; Marder, S. R.; Pei, J. Adv. Energy Mater. 2019, 9, 1900817. doi:10.1002/aenm.201900817 |
| 24. | Lungwitz, D.; Joy, S.; Mansour, A. E.; Opitz, A.; Karunasena, C.; Li, H.; Panjwani, N. A.; Moudgil, K.; Tang, K.; Behrends, J.; Barlow, S.; Marder, S. R.; Brédas, J.-L.; Graham, K.; Koch, N.; Kahn, A. J. Phys. Chem. Lett. 2023, 14, 5633–5640. doi:10.1021/acs.jpclett.3c01022 |
| 8. | Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582 |
| 9. | Furst, L.; Matsuura, B. S.; Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. Org. Lett. 2010, 12, 3104–3107. doi:10.1021/ol101146f |
| 10. | Ghosh, I.; Ghosh, T.; Bardagi, J. I.; König, B. Science 2014, 346, 725–728. doi:10.1126/science.1258232 |
| 11. | Constantin, T.; Zanini, M.; Regni, A.; Sheikh, N. S.; Juliá, F.; Leonori, D. Science 2020, 367, 1021–1026. doi:10.1126/science.aba2419 |
| 45. | Imran, M.; Zhang, X.; Wang, Z.; Chen, X.; Zhao, J.; Barbon, A.; Voronkova, V. K. Phys. Chem. Chem. Phys. 2021, 23, 15835–15868. doi:10.1039/d1cp01937f |
| 7. | Herberich, G. E.; Bauer, E.; Schwarzer, J. J. Organomet. Chem. 1969, 17, 445–452. doi:10.1016/s0022-328x(00)88230-3 |
| 13. | Zhou, S.; Doni, E.; Anderson, G. M.; Kane, R. G.; MacDougall, S. W.; Ironmonger, V. M.; Tuttle, T.; Murphy, J. A. J. Am. Chem. Soc. 2014, 136, 17818–17826. doi:10.1021/ja5101036 |
| 32. | Yu, D.; To, W.-P.; Tong, G. S. M.; Wu, L.-L.; Chan, K.-T.; Du, L.; Phillips, D. L.; Liu, Y.; Che, C.-M. Chem. Sci. 2020, 11, 6370–6382. doi:10.1039/d0sc01340d |
| 32. | Yu, D.; To, W.-P.; Tong, G. S. M.; Wu, L.-L.; Chan, K.-T.; Du, L.; Phillips, D. L.; Liu, Y.; Che, C.-M. Chem. Sci. 2020, 11, 6370–6382. doi:10.1039/d0sc01340d |
| 33. | Inaba, S.; Matsumoto, H.; Rieke, R. D. J. Org. Chem. 1984, 49, 2093–2098. doi:10.1021/jo00186a003 |
| 34. | Burns, T. P.; Rieke, R. D. J. Org. Chem. 1987, 52, 3674–3680. doi:10.1021/jo00392a033 |
| 35. | Kim, S.-H.; Rieke, R. D. J. Org. Chem. 2000, 65, 2322–2330. doi:10.1021/jo991478s |
| 36. | Suh, Y.; Lee, J.-s.; Kim, S.-H.; Rieke, R. D. J. Organomet. Chem. 2003, 684, 20–36. doi:10.1016/s0022-328x(03)00500-x |
| 42. | Bennett, R. W.; Wharry, D. L.; Koch, T. H. J. Am. Chem. Soc. 1980, 102, 2345–2349. doi:10.1021/ja00527a036 |
| 43. | Colter, A. K.; Lai, C. C.; Parsons, A. G.; Ramsey, N. B.; Saito, G. Can. J. Chem. 1985, 63, 445–451. doi:10.1139/v85-073 |
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 44. | Jhulki, S.; Un, H.-I.; Ding, Y.-F.; Risko, C.; Mohapatra, S. K.; Pei, J.; Barlow, S.; Marder, S. R. Chem 2021, 7, 1050–1065. doi:10.1016/j.chempr.2021.01.020 |
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 39. | Mohapatra, S. K.; Al Kurdi, K.; Jhulki, S.; Bogdanov, G.; Bacsa, J.; Conte, M.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Beilstein J. Org. Chem. 2023, 19, 1651–1663. doi:10.3762/bjoc.19.121 |
| 18. | Zhang, S.; Naab, B. D.; Jucov, E. V.; Parkin, S.; Evans, E. G. B.; Millhauser, G. L.; Timofeeva, T. V.; Risko, C.; Brédas, J.-L.; Bao, Z.; Barlow, S.; Marder, S. R. Chem. – Eur. J. 2015, 21, 10878–10885. doi:10.1002/chem.201500611 |
| 40. | Guo, S.; Mohapatra, S. K.; Romanov, A.; Timofeeva, T. V.; Hardcastle, K. I.; Yesudas, K.; Risko, C.; Brédas, J.-L.; Marder, S. R.; Barlow, S. Chem. – Eur. J. 2012, 18, 14760–14772. doi:10.1002/chem.201202591 |
| 41. | Longhi, E.; Risko, C.; Bacsa, J.; Khrustalev, V.; Rigin, S.; Moudgil, K.; Timofeeva, T. V.; Marder, S. R.; Barlow, S. Dalton Trans. 2021, 50, 13020–13030. doi:10.1039/d1dt02155a |
| 2. | Murphy, J. A.; Zhou, S.-z.; Thomson, D. W.; Schoenebeck, F.; Mahesh, M.; Park, S. R.; Tuttle, T.; Berlouis, L. E. A. Angew. Chem., Int. Ed. 2007, 46, 5178–5183. doi:10.1002/anie.200700554 |
| 38. | Blanksby, S. J.; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255–263. doi:10.1021/ar020230d |
| 37. | Wiesner, S.; Walter, P.; Wagner, A.; Kaifer, E.; Himmel, H.-J. Eur. J. Org. Chem. 2016, 5045–5054. doi:10.1002/ejoc.201600978 |
| 30. | Jing, L.; Nash, J. J.; Kenttämaa, H. I. J. Am. Chem. Soc. 2008, 130, 17697–17709. doi:10.1021/ja801707p |
| 31. | Higginson, B.; Sanjosé-Orduna, J.; Gu, Y.; Martin, R. Synlett 2021, 32, 1633–1636. doi:10.1055/a-1457-2399 |
© 2023 Tang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.