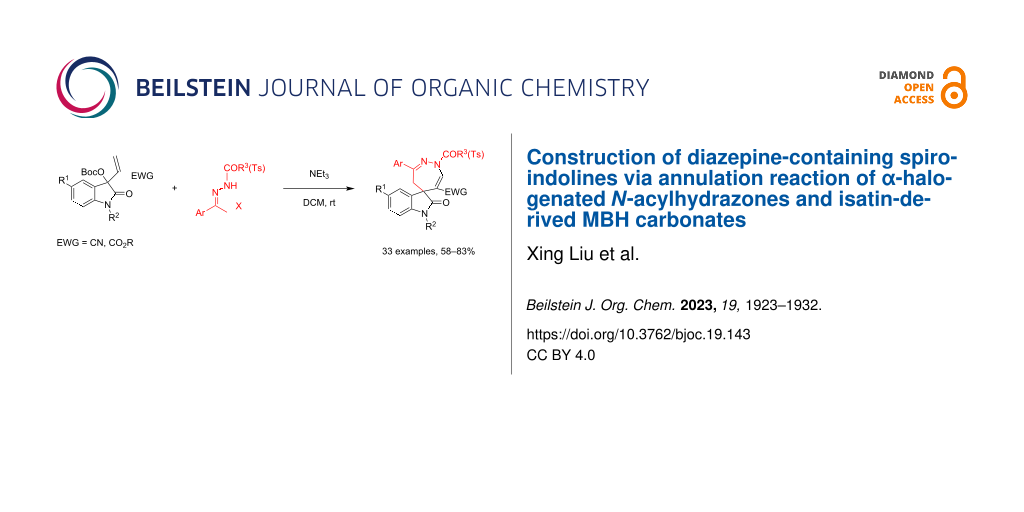
Abstract
A straightforward synthetic protocol for the efficient construction of diazepine-containing spiroindolines has been developed and proceeds through a by base-promoted annulation reaction of α-halogenated N-acylhydrazones and isatin-derived MBH carbonates. The reaction mechanism of this formal [4 + 3] annulation includes the in situ generated allylic ylide, nucleophilic substitution, Michael additon, and elimination processes. Additionally, the similar reaction with α-halogenated N-tosylhydrazones also afforded N-tosyl-substituted spiro[indoline-3,5'-[1,2]diazepine] in satisfactory yields. This protocol provides a convenient approach for the assembly of diverse highly functionalized spiro[indoline-3,5'-[1,2]diazepines] and also features a broad substrate scope, simple reaction conditions, and high molecular convergence.

Graphical Abstract
Introduction
Among the various N-containing heterocyclic compounds, 1,2-diazepine represents one of the important privileged structural motif, which is frequently found in natural products, bioactive molecules, and pharmaceuticals [1-4]. 1,2-Diazepine derivatives exhibit promising biological activities such as anticonvulsant, antibacterial, and antiproliferative effects [5-13]. Thus, the development of alternative synthetic methodologies for functionalized 1,2-diazepines has drawn extensive attention [14-21]. One of the most attractive strategies to synthesize the 1,2-diazepine motif represents the [4 + 3] cycloaddition reaction between activated azoalkenes and 1,3-dipolarophiles [22-27].
In addition, spirooxindole is also a privileged structural scaffold, which has been recognized as key structural unit in many bioactive natural products and pharmaceuticals with broad biological activities [28-30]. The development of elegant synthetic methodologies for the synthesis of spirooxindole derivatives continues to be a highly active subject in organic synthesis [31-35]. In recent years, the readily available isatin-derived Morita–Baylis–Hillman (MBH) carbonates have become one of the most powerful reagents for the construction of diverse spirooxindoles [36-43]. In the presence of Lewis bases, isatin-derived MBH carbonates usually undergo [3 + 2] and [3 + 3] cycloaddition reactions with a broad range of active C–C and C–N double bonds and 1,3-dipolarpohiles to give various five- or six-membered cyclic spirooxindoles [44-48]. However, the [4 + 3] cycloaddition reaction of isatin-derived MBH carbonates with active diene components has not been well developed probably due to the lack of suitable active diene compounds [49-53]. In this respect, Chen and co-workers were the first who reported the efficient synthesis of spiro[azepine-4,3'-indoline] derivatives via the [4 + 3] cycloaddition reaction of bromo-substituted isatin-derived MBH adducts and N-(o-chloromethyl)arylamides. In this efficient protocol, the reactive allylic phosphonium ylides and aza-o-quinone methides were generated in situ and sequentially underwent a [4 + 3] cycloaddition reaction (reaction 1 in Scheme 1) [54]. Recently, Chen and co-workers reported a chiral tertiary amine-catalyzed asymmetric γ-regioselective [4 + 3] annulation reaction of isatin-derived MBH carbonates and cyclic 2-benzylidenebenzo[b]thiophen-3-ylidene)benzenesulfonamides to give chiral azepane spirooxindoles with excellent stereoselectivity (reaction 2 in Scheme 1) [55,56]. Du and co-workers reported a DABCO-mediated [4 + 3] cycloaddition reaction between o-quinone methides and isatin-derived MBH carbonates to give functionalized benzo[b]oxepine derivatives in satisfactory yields and with good diastereoselectivity (reaction 3, Scheme 1) [57]. Very recently, we found that the base-catalyzed [4 + 3] cycloaddition reaction of isatin-derived MBH carbonates with various α,β-unsaturated N-arylaldimines affords spiro[indoline-3,5'-pyrrolo[3,4-b]azepine] derivatives (reaction 4 in Scheme 1) [58]. Inspired by these elegant synthetic protocols and in continuation of our aim to provide efficient synthetic protocols for diverse spirooxindoles [59-67], we herein wish to report the base-mediated formal [4 + 3] annulation reaction of isatin-derived MBH carbonates with α-halogenated N-acylhydrazones for the convenient synthesis of functionalized spiro[indoline-3,5'-[1,2]diazepine] derivatives (reaction 5 in Scheme 1).
![[1860-5397-19-143-i1]](/bjoc/content/inline/1860-5397-19-143-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative [4 + 3] cycloaddition reactions of MBH carbonates derived from isatins.
Scheme 1: Representative [4 + 3] cycloaddition reactions of MBH carbonates derived from isatins.
Results and Discussion
Initially, the reaction conditions were screened by employing α-chloro-N-benzoylhydrazone 1a and MBH nitrile of isatin 2a as standard reaction. The main experiments are briefly summarized in Table 1. At first, the reaction in DCM in the presence of common organic bases such as DMAP, DABCO, or DBU gave the expected spiro[indoline-3,5'-[1,2]diazepine] 3a in low to moderate yields (Table 1, entries 1–3). However, piperidine, TMG, and Na2CO3 failed to promote the annulation reaction (Table 1, entries 4–6). When triethylamine was employed as the base, the reaction gave the spiro compound 3a in 71% yield (Table 1, entry 7). The yields of product 3a remained nearly unchanged when the reaction time was either shortened to 12 h or prolonged to 36 h (Table 1, entries 8 and 9). Also, neither decreasing or increasing the reaction temperature did improve the yield of product 3a which was obtained in 26% yield at 0 °C and 52% yield in refluxing DCM, respectively (Table 1, entries 10 and 11). In the presence of triethylamine, the reaction in other solvents such as DCE, THF, and CHCl3 gave the product 3a in 61%, 46% and 56% yields, respectively (Table 1, entries 12–14). However, the reaction did not proceed in toluene and acetonitrile (Table 1, entries 15 and 16). When lowering the amount of triethylamine to one equivalent, the yield of spiro compound 3a decreased to 35% (Table 1, entry 17). At last, if the amount of α-halogenated acylhydrazone was reduced, the yield of product 3a also decreased to 57% yield (Table 1, entry 18). Thus, the best reaction conditions were carrying out the reaction in DCM at room temperature for 24 hours in the presence of an excess amount of triethylamine.
Table 1: Optimizing the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-143-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Solvent | Time (h) | Temp. (°C) | Yield of 3a (%)b |
| 1 | DMAP | DCM | 24 | rt | 45 |
| 2 | DABCO | DCM | 24 | rt | 33 |
| 3 | DBU | DCM | 24 | rt | 15 |
| 4 | piperidine | DCM | 12 | rt | 0 |
| 5 | TMG | DCM | 12 | rt | 0 |
| 6 | Na2CO3 | DCM | 12 | rt | 0 |
| 7 | Et3N | DCM | 24 | rt | 71 |
| 8 | Et3N | DCM | 12 | rt | 72 |
| 9 | Et3N | DCM | 36 | rt | 72 |
| 10 | Et3N | DCM | 12 | 0 | 26 |
| 11 | Et3N | DCM | 12 | reflux | 52 |
| 12 | Et3N | DCE | 12 | rt | 61 |
| 13 | Et3N | THF | 12 | rt | 42 |
| 14 | Et3N | CHCl3 | 12 | rt | 56 |
| 15 | Et3N | MeCN | 12 | rt | 0 |
| 16 | Et3N | PhMe | 12 | rt | 0 |
| 17 | Et3Nc | DCM | 12 | rt | 35 |
| 18 | Et3Nd | DCM | 12 | rt | 57 |
aReaction conditions: α-halogenated acylhydrazone 1a (0.2 mmol), MBH nitrile of isatin 2a (0.1 mmol), base (0.2 mmol), solvent (4.0 mL); bIsolated yields. cEt3N (0.1 mmol); dα-halogenated acylhydrazone (0.12 mmol).
With the optimized reaction conditions in hands, we next examined the scope of the reaction by employing various functionalized substrates and the results are summarized in Scheme 2. As it can be seen, the expected dihydrospiro[indoline-3,5'-[1,2]diazepines] 3a–m were obtained in reasonable to good yields. Both, α-chloro- and α-bromo-N-acylhydrazones could be successfully used in the reaction and gave similar results. Also, hydrazones with different benzoyl-protecting groups were well tolerated in the reaction. In general, α-bromo-N-acetylhydrazones gave higher yields than the corresponding α-bromo-N-benzoylhydrazones. Also, substituents present in the MBH nitriles of isatins showed marginal effects on the yields. The obtained spiro[indoline-3,5'-[1,2]diazepines] 3a–m were fully characterized by various spectroscopic methods. Because there are one C=C bond and one C=N bond in the molecules 3a–m, no diastereoisomers are obtained. Therefore, the 1H NMR spectra gave simple absorptions for the characteristic groups in the molecules.
![[1860-5397-19-143-i2]](/bjoc/content/inline/1860-5397-19-143-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of spiro[indoline-3,5'-[1,2]diazepines] 3a–m. Conditions: α-halogenated acylhydrazone (0.2 mmol), MBH nitrile of isatin (0.1 mmol), Et3N (0.2 mmol), DCM (4.0 mL), rt, 24 h; athe α-chloro-N-acylhydrazone was used, for other compounds α-bromo-N-acylhydrazones were used. Yields refer to isolated compounds.
Scheme 2: Synthesis of spiro[indoline-3,5'-[1,2]diazepines] 3a–m. Conditions: α-halogenated acylhydrazone (0....
For further developing the scope of the [4 + 3] cycloaddition reaction, MBH esters of isatins 4 were also employed in the reaction, but it was found that the reaction proceeded sluggishly in the presence of triethylamine (Scheme 3). However, the expected annulation reaction proceeded smoothly in dichloromethane within 24 hours in the presence of DABCO as base, affording the corresponding spiro[indoline-3,5'-[1,2]diazepine]-6'-carboxylates 5a–g in 63–77% yields (Scheme 3). The substituents on both substrates also showed little effect on the yields. The chemical structures were fully characterized by HRMS, IR, 1H and 13C NMR spectra.
![[1860-5397-19-143-i3]](/bjoc/content/inline/1860-5397-19-143-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of spiro[indoline-3,5'-[1,2]diazepines] 5a–g. Conditions: α-halogenated acylhydrazone (0.2 mmol), MBH ester of isatin (0.1 mmol), DABCO (0.2 mmol), DCM (4.0 mL), rt, 24 h; aα-chloro-N-acylhydrazone was used, for other compounds α-bromo-N-acylhydrazone was used. Yields refer to isolated compounds.
Scheme 3: Synthesis of spiro[indoline-3,5'-[1,2]diazepines] 5a–g. Conditions: α-halogenated acylhydrazone (0....
For demonstrating the synthetic value of this protocol, α-halogenated p-toluenesulfonylhydrazones 6 were also used in the reaction and the results are summarized in Scheme 4. As it can be seen, the triethylamine-mediated [4 + 3] cycloaddition could be accomplished at room temperature in two hours. These results clearly showed that the α-halogenated p-toluenesulfonylhydrazones 6 have a much higher reactivity compared to that of α-halogenated benzoylsulfonylhydrazones 1 in this reaction. The desired spiro[indoline-3,5'-[1,2]diazepines] 7a–n were obtained in satisfactory yields of 58–83% and the substituents in MBH nitriles of isatins showed only marginal effects on the yields. The chemical structures of the spiro compounds 7a–n were established by various spectroscopy methods. In addition, the single crystal structure of compound 7a was also determined by X-ray diffraction (Figure 1). As can be seen from Figure 1, both the C–C and C–N double bonds are part of the cyclic 1,2-diazepine ring and the methylene unit is connected to the 3-positon of the oxindole moiety.
![[1860-5397-19-143-i4]](/bjoc/content/inline/1860-5397-19-143-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of dihydrospiro[indoline-3,5'-[1,2]diazepines] 7a–n. Conditions: α-halogenated N-tosylhydrazone (0.2 mmol), MBH nitrile of isatin (0.2 mmol), Et3N (0.4 mmol), DCM (4.0 mL), rt, 3 h; aα-chloro-N-tosylhydrazone was used, for other compounds α-bromo-N-tosylhydrazone was used. Yields refer to isolated compounds.
Scheme 4: Synthesis of dihydrospiro[indoline-3,5'-[1,2]diazepines] 7a–n. Conditions: α-halogenated N-tosylhyd...
![[1860-5397-19-143-1]](/bjoc/content/figures/1860-5397-19-143-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Single crystal structure of the spiro compound 7a.
Figure 1: Single crystal structure of the spiro compound 7a.
On the basis of the current results and previous works [54-61], a reaction mechanism for the formation of the spiro[indoline-3,5'-[1,2]diazepines] has been proposed and is depicted in Scheme 5. At first, MBH carbonates of isatin 2 is attacked at the α-position by the Lewis base to give the ammonium salt A with elimination of carbon dioxide and a tert-butoxide ion. Secondly, the ammonium salt A is deprotonated by the in situ generated tert-butoxide ion to give the allylic ylide B. Thirdly, the intermediate C is formed by the nucleophilic substitution of a halide ion in substrate 1 by the allylic ylide B. Then, Michael addition of the amino group to the C=C bond results in the cyclic intermediate D. Finally, the spiro[indoline-3,5'-[1,2]diazepine] 3 is produced by the elimination of a proton and the Lewis base. Obviously, the spiro compounds 5 and 7 are formed by a similar reaction mechanism.
![[1860-5397-19-143-i5]](/bjoc/content/inline/1860-5397-19-143-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Additionally, the method was applied to a gram-scale reaction of α-halogenated p-toluenesulfonylhydrazone 6c and MBH nitrile of isatin 2c under the standard conditions (Scheme 6). The expected spiro product 7c was successfully obtained in 70% yield, which clearly demonstrated that this base-promoted annulation reaction is applicable for the large-scale synthesis of diazepine-containing spiroindolines.
![[1860-5397-19-143-i6]](/bjoc/content/inline/1860-5397-19-143-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Gram-scale synthesis of compound 7c.
Scheme 6: Gram-scale synthesis of compound 7c.
Conclusion
In summary, we have developed a synthetic protocol for the base-mediated annulation reaction of α-halogenated acylhydrazones with isatin-derived MBH carbonates. The reaction provides a straightforward synthetic route for the efficient construction of novel spiro[indoline-3,5'-[1,2]diazepine] derivatives in satisfactory yields. The advantages of this reaction include the use of readily available reagents, mild conditions, satisfactory yields, broad substrate scope, high molecular convergence, and atomic economy. The synthetic applications of this annulation reaction in heterocyclic chemistry might be significant.
Experimental
General procedure for the preparation of dihydrospiro[indoline-3,5'-[1,2]diazepines] 3a–l: A 10 mL reaction tube was charged with α-halogenated acylhydrazone (0.2 mmol), MBH nitrile of isatin (0.1 mmol), triethylamine (0.2 mmol) and dichloromethane (4.0 mL) and the mixture was stirred at room temperature for 24 hours. After removing the solvent by rotatory evaporation at reduced pressure, the residue was subjected to column chromatography with ethyl acetate, dichloromethane and petroleum ether 1:3:7 (v/v/v) to give pure product for analysis.
1'-Benzoyl-1-benzyl-5-methyl-2-oxo-3'-phenyl-1',4'-dihydrospiro[indoline-3,5'-[1,2]diazepine]-6'-carbonitrile (3a): yellow solid, 0.370 g, 71%; mp 186–187 °C; 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H, ArH), 7.75–7.72 (m, 2H, ArH), 7.57–7.53 (m, 1H, ArH), 7.47–7.43 (m, 2H, ArH), 7.35–7.31 (m, 2H, ArH), 7.30–7.27 (m, 5H, ArH), 7.26–7.23 (m, 1H, ArH), 7.22–7.18 (m, 2H, ArH), 7.05–7.02 (m, 1H, ArH), 6.96 (s, 1H, ArH), 6.73 (d, J = 8.0 Hz, 1H, ArH), 4.95–4.90 (m, 2H, CH2), 3.47 (d, J = 14.0 Hz, 1H, CH), 3.24 (d, J = 14.0 Hz, 1H, CH), 2.16 (s, 3H, CH3) ppm; 13C NMR (100 MHz, CDCl3) δ 173.8, 170.9, 159.6, 139.6, 138.4, 136.2, 135.2, 133.3, 133.2, 131.8, 130.7, 130.3, 130.0, 129.2, 128.9, 128.4, 127.9, 127.8, 127.4, 127.3, 125.5, 117.4, 109.9, 95.0, 52.3, 44.4, 37.0, 20.9 ppm; IR (KBr) ν: 2960, 2936, 2870, 2211, 1717, 1626, 1498, 1445, 1367, 1268, 1193, 1112, 1090, 1009, 903, 868, 815 cm−1; HRMS–ESI TOF (m/z): [M + Na]+ calcd for C34H26N4O2Na, 545.1956; found, 545.1948.
General procedure for the preparation of dihydrospiro[indoline-3,5'-[1,2]diazepines] 5a–i: A 10 mL reaction tube was charged with α-halogenated acylhydrazone (0.2 mmol), MBH ester of isatin (0.1 mmol), DABCO (0.2 mmol, 0.0224 g), and dichloromethane (4.0 mL) and the mixture was stirred at room temperature for 24 hours. After removing the solvent by rotatory evaporation at reduced pressure, the residue was subjected to column chromatography with ethyl acetate, dichloromethane and petroleum ether 1:3:7 (v/v/v) to give pure product for analysis.
Methyl 1'-benzoyl-1-benzyl-5-chloro-2-oxo-3'-phenyl-1',4'-dihydrospiro[indoline-3,5'-[1,2]diazepine]-6'-carboxylate (5a): yellow solid, 0.391 g, 68%; mp 189–191 °C; 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H, ArH), 7.73–7.71 (m, 2H, ArH), 7.56–7.53 (m, 1H, ArH), 7.48–7.42 (m, 4H, ArH), 7.38–7.35 (m, 2H, ArH), 7.32–7.31 (m, 1H, ArH), 7.29–7.27 (m, 1H, ArH), 7.14 (t, J = 8.0 Hz, 2H, ArH), 7.09–7.05 (m, 3H, ArH), 6.86–6.85 (m, 1H, ArH), 6.70 (d, J = 8.4 Hz, 1H, ArH), 5.02 (s, 2H, CH2), 3.65 (s, 3H, OCH3), 3.49 (d, J = 13.6 Hz, 1H, CH), 3.10 (d, J = 13.6 Hz, 1H, CH) ppm; 13C NMR (100 MHz, CDCl3) δ 176.3, 171.3, 165.8, 160.6, 141.1, 137.3, 135.9, 135.5, 133.8, 131.6, 130.6, 129.7, 128.9, 128.5, 128.4, 127.9, 127.8, 127.5, 127.0, 124.4, 111.6, 110.4, 52.2, 51.1, 44.4, 36.8 ppm; IR (KBr) ν: 2924, 2853, 1721, 1608, 1484, 1456, 1430, 1340, 1170, 812 cm−1; HRMS–ESI TOF (m/z): [M + H]+ calcd for C34H27ClN3O4, 576.1685; found, 576.1683.
General procedure for the preparation of dihydrospiro[indoline-3,5'-[1,2]diazepines] 7a–l: A 10 mL reaction tube was charged with α-halogenated N-tosylhydrazone (0.2 mmol), MBH ester of isatin (0.2 mmol), triethylamine (0.4 mmol, 0.0364 g), and dichloromethane (4.0 mL) and the mixture was stirred at room temperature for three hours. After removing the solvent by rotatory evaporation at reduced pressure, the residue was subjected to column chromatography with ethyl acetate, dichloromethane, and petroleum ether 1:3.7 (v/v/v) to give pure product for analysis.
1-Benzyl-2-oxo-3'-(p-tolyl)-1'-tosyl-1',4'-dihydrospiro[indoline-3,5'-[1,2]diazepine]-6'-carbonitrile (7a): white solid, 64%; mp 226–230 °C; 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H, C=CH-N), 7.97 (d, J = 8.0 Hz, 2H, ArH), 7.42 (d, J = 8.0 Hz, 2H, ArH), 7.29 (d, J = 8.4 Hz, 2H, ArH), 7.28–7.26 (s, 1H, ArH), 7.26–7.24 (m, 2H, ArH), 7.22–7.20 (m, 1H, ArH), 7.19–7.16 (m, 2H, ArH), 7.09 (s, 1H, ArH), 7.06 (d, J = 4.8 Hz, 2H, ArH), 6.98 (t, J = 7.6 Hz, 1H, ArH), 6.78 (d, J = 7.6 Hz, 1H, ArH), 4.88 (d, J = 15.6 Hz, 1H, CH), 4.79 (d, J = 15.6 Hz, 1H, CH), 3.29 (d, J = 10.0 Hz, 1H, CH), 3.20 (d, J = 10.0 Hz, 1H, CH), 2.49 (s, 3H, CH3), 2.34 (s, 3H, CH3) ppm; 13C {1H} NMR (100 MHz, CDCl3) δ 173.7, 160.5, 145.9, 141.8, 141.1, 139.4, 135.1, 133.5, 132.7, 129.9, 129.9, 129.5, 129.2, 129.0, 128.8, 127.8, 127.3, 127.2, 124.5, 123.5, 117.3, 109.9, 91.9, 52.1, 44.3, 37.3, 21.8, 21.3 ppm; IR (KBr) ν: 3057, 3055, 2928, 2217, 1716, 1613, 1488, 1467, 1449, 1369, 1297, 1259, 1189, 1175, 1090, 1021, 999, 869, 804, 764, 754, 696, 667, 657, 639 cm−1; HRMS–ESI TOF (m/z): [M + Na]+ calcd for C34H28ClN4O3SNa, 595.1774; found, 595.1765.
The crystallographic data of compound 7a (CCDC 2280223) have been deposited at the Cambridge Crystallographic Database Centre.
Supporting Information
| Supporting Information File 1: Characterization data and 1H, 13C NMR, and HRMS spectra for all new compounds. | ||
| Format: PDF | Size: 7.6 MB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Ogawa, H.; Yamashita, H.; Kondo, K.; Yamamura, Y.; Miyamoto, H.; Kan, K.; Kitano, K.; Tanaka, M.; Nakaya, K.; Nakamura, S.; Mori, T.; Tominaga, M.; Yabuuchi, Y. J. Med. Chem. 1996, 39, 3547–3555. doi:10.1021/jm960133o
Return to citation in text: [1] -
Tabata, H.; Nakagomi, J.; Morizono, D.; Oshitari, T.; Takahashi, H.; Natsugari, H. Angew. Chem., Int. Ed. 2011, 50, 3075–3079. doi:10.1002/anie.201007772
Return to citation in text: [1] -
Michelini, S.; Cassano, G. B.; Frare, F.; Perugi, G. Pharmacopsychiatry 2007, 29, 127–134. doi:10.1055/s-2007-979558
Return to citation in text: [1] -
Chakraborty, S.; Shah, N. H.; Fishbein, J. C.; Hosmane, R. S. Bioorg. Med. Chem. Lett. 2011, 21, 756–759. doi:10.1016/j.bmcl.2010.11.109
Return to citation in text: [1] -
Wiethe, R. W.; Stewart, E. L.; Drewry, D. H.; Gray, D. W.; Mehbob, A.; Hoekstra, W. J. Bioorg. Med. Chem. Lett. 2006, 16, 3777–3779. doi:10.1016/j.bmcl.2006.04.055
Return to citation in text: [1] -
Łuszczki, J. J. Pharmacol. Rep. 2009, 61, 197–216. doi:10.1016/s1734-1140(09)70024-6
Return to citation in text: [1] -
Iwamoto, F. M.; Kreisl, T. N.; Kim, L.; Duic, J. P.; Butman, J. A.; Albert, P. S.; Fine, H. A. Cancer 2010, 116, 1776–1782. doi:10.1002/cncr.24957
Return to citation in text: [1] -
Murthy, K. S. K.; Knaus, E. E. Drug Dev. Res. 1999, 46, 155–162. doi:10.1002/(sici)1098-2299(199902)46:2<155::aid-ddr9>3.0.co;2-w
Return to citation in text: [1] -
Isono, K. J. Antibiot. 1988, 41, 1711–1739. doi:10.7164/antibiotics.41.1711
Return to citation in text: [1] -
Kim, H.; Kim, M.; Lee, J.; Yu, H.; Hah, J.-M. Bioorg. Med. Chem. 2011, 19, 6760–6767. doi:10.1016/j.bmc.2011.09.042
Return to citation in text: [1] -
Assimon, V. A.; Tang, Y.; Vargas, J. D.; Lee, G. J.; Wu, Z. Y.; Lou, K.; Yao, B.; Menon, M.-K.; Pios, A.; Perez, K. C.; Madriaga, A.; Buchowiecki, P. K.; Rolfe, M.; Shawver, L.; Jiao, X.; Le Moigne, R.; Zhou, H.-J.; Anderson, D. J. ACS Chem. Biol. 2019, 14, 236–244. doi:10.1021/acschembio.8b00904
Return to citation in text: [1] -
Snieckus, V.; Streith, J. Acc. Chem. Res. 1981, 14, 348–355. doi:10.1021/ar00071a004
Return to citation in text: [1] -
Wang, M.; Huang, Z.; Xu, J.; Chi, Y. R. J. Am. Chem. Soc. 2014, 136, 1214–1217. doi:10.1021/ja411110f
Return to citation in text: [1] -
Guo, C.; Sahoo, B.; Daniliuc, C. G.; Glorius, F. J. Am. Chem. Soc. 2014, 136, 17402–17405. doi:10.1021/ja510737n
Return to citation in text: [1] -
Wang, L.; Li, S.; Blümel, M.; Philipps, A. R.; Wang, A.; Puttreddy, R.; Rissanen, K.; Enders, D. Angew. Chem., Int. Ed. 2016, 55, 11110–11114. doi:10.1002/anie.201604819
Return to citation in text: [1] -
Mojikhalifeh, S.; Hasaninejad, A. Org. Chem. Front. 2018, 5, 1516–1521. doi:10.1039/c8qo00210j
Return to citation in text: [1] -
Belyy, A. Y.; Levina, A. A.; Platonov, D. N.; Salikov, R. F.; Medvedev, M. G.; Tomilov, Y. V. Eur. J. Org. Chem. 2019, 4133–4138. doi:10.1002/ejoc.201801861
Return to citation in text: [1] -
Mohammed, K. S.; Elbeily, E. E.; El‐Taweel, F. M.; Fadda, A. A. J. Heterocycl. Chem. 2019, 56, 493–500. doi:10.1002/jhet.3425
Return to citation in text: [1] -
Asamdi, M.; Shaikh, M. M.; Chauhan, P. M.; Chikhalia, K. H. Tetrahedron 2018, 74, 3719–3727. doi:10.1016/j.tet.2018.05.051
Return to citation in text: [1] -
Attanasi, O. A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F. R.; Santeusanio, S. Eur. J. Org. Chem. 2009, 3109–3127. doi:10.1002/ejoc.200900243
Return to citation in text: [1] -
Zhang, L.; Xu, Y.; Zhang, X.; Zhang, X.; Fan, X. Org. Chem. Front. 2020, 7, 2284–2290. doi:10.1039/d0qo00657b
Return to citation in text: [1] -
Matsuya, Y.; Ohsawa, N.; Nemoto, H. J. Am. Chem. Soc. 2006, 128, 13072–13073. doi:10.1021/ja065277z
Return to citation in text: [1] -
Zhu, S.-Y.; Zhang, Y.; Wang, W.; Hui, X.-P. Org. Lett. 2017, 19, 5380–5383. doi:10.1021/acs.orglett.7b02657
Return to citation in text: [1] -
Deng, Y.; Pei, C.; Arman, H.; Dong, K.; Xu, X.; Doyle, M. P. Org. Lett. 2016, 18, 5884–5887. doi:10.1021/acs.orglett.6b02965
Return to citation in text: [1] -
Yuan, C.; Zhou, L.; Xia, M.; Sun, Z.; Wang, D.; Guo, H. Org. Lett. 2016, 18, 5644–5647. doi:10.1021/acs.orglett.6b02885
Return to citation in text: [1] -
Li, T.; Yang, Z.; Song, Z.; Chauvin, R.; Cui, X. Org. Lett. 2020, 22, 4078–4082. doi:10.1021/acs.orglett.0c01139
Return to citation in text: [1] -
Ding, Y.-L.; Zhao, Y.-L.; Niu, S.-S.; Wu, P.; Cheng, Y. J. Org. Chem. 2020, 85, 612–621. doi:10.1021/acs.joc.9b02693
Return to citation in text: [1] -
Yu, B.; Yu, Z.; Qi, P.-P.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 95, 35–40. doi:10.1016/j.ejmech.2015.03.020
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342
Return to citation in text: [1] -
Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. J. Med. Chem. 2019, 62, 448–466. doi:10.1021/acs.jmedchem.8b00909
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808
Return to citation in text: [1] -
Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180
Return to citation in text: [1] -
Wang, Y.; Cobo, A. A.; Franz, A. K. Org. Chem. Front. 2021, 8, 4315–4348. doi:10.1039/d1qo00220a
Return to citation in text: [1] -
Saeed, R.; Sakla, A. P.; Shankaraiah, N. Org. Biomol. Chem. 2021, 19, 7768–7791. doi:10.1039/d1ob01176f
Return to citation in text: [1] -
Chen, Z.-C.; Chen, Z.; Du, W.; Chen, Y.-C. Chem. Rec. 2020, 20, 541–555. doi:10.1002/tcr.201900058
Return to citation in text: [1] -
Shanmugam, P.; Viswambharan, B.; Madhavan, S. Org. Lett. 2007, 9, 4095–4098. doi:10.1021/ol701533d
Return to citation in text: [1] -
Gomes, J. C.; Sirvent, J.; Moyano, A.; Rodrigues, M. T., Jr.; Coelho, F. Org. Lett. 2013, 15, 5838–5841. doi:10.1021/ol4029034
Return to citation in text: [1] -
Warghude, P. K.; Sabale, A. S.; Bhat, R. G. Org. Biomol. Chem. 2020, 18, 1794–1799. doi:10.1039/d0ob00007h
Return to citation in text: [1] -
Wang, Z.-H.; Lei, C.-W.; Zhang, X.-Y.; You, Y.; Zhao, J.-Q.; Yuan, W.-C. Org. Chem. Front. 2019, 6, 3342–3347. doi:10.1039/c9qo00890j
Return to citation in text: [1] -
Zhou, T.; Xia, T.; Liu, Z.; Liu, L.; Zhang, J. Adv. Synth. Catal. 2018, 360, 4475–4479. doi:10.1002/adsc.201801152
Return to citation in text: [1] -
He, X.-H.; Fu, X.-J.; Zhan, G.; Zhang, N.; Li, X.; Zhu, H.-P.; Peng, C.; He, G.; Han, B. Org. Chem. Front. 2022, 9, 1048–1055. doi:10.1039/d1qo01785c
Return to citation in text: [1] -
Zhang, L.; Liu, H.; Qiao, G.; Hou, Z.; Liu, Y.; Xiao, Y.; Guo, H. J. Am. Chem. Soc. 2015, 137, 4316–4319. doi:10.1021/jacs.5b01138
Return to citation in text: [1] -
Zhong, F.; Chen, G.-Y.; Han, X.; Yao, W.; Lu, Y. Org. Lett. 2012, 14, 3764–3767. doi:10.1021/ol301647g
Return to citation in text: [1] -
Deng, H.-P.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2012, 354, 783–789. doi:10.1002/adsc.201101012
Return to citation in text: [1] -
Wang, Y.; Liu, L.; Zhang, T.; Zhong, N.-J.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2012, 77, 4143–4147. doi:10.1021/jo3002535
Return to citation in text: [1] -
Hu, F.-L.; Wei, Y.; Shi, M. Chem. Commun. 2014, 50, 8912–8914. doi:10.1039/c4cc03479a
Return to citation in text: [1] -
Liu, Y.-L.; Wang, X.; Zhao, Y.-L.; Zhu, F.; Zeng, X.-P.; Chen, L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Angew. Chem., Int. Ed. 2013, 52, 13735–13739. doi:10.1002/anie.201307250
Return to citation in text: [1] -
Wei, F.; Huang, H.-Y.; Zhong, N.-J.; Gu, C.-L.; Wang, D.; Liu, L. Org. Lett. 2015, 17, 1688–1691. doi:10.1021/acs.orglett.5b00456
Return to citation in text: [1] -
Liu, J.; Wang, L.; Wang, X.; Xu, L.; Hao, Z.; Xiao, J. Org. Biomol. Chem. 2016, 14, 11510–11517. doi:10.1039/c6ob01953f
Return to citation in text: [1] -
Wani, I. A.; Bhattacharyya, A.; Sayyad, M.; Ghorai, M. K. Org. Biomol. Chem. 2018, 16, 2910–2922. doi:10.1039/c8ob00228b
Return to citation in text: [1] -
Gelis, C.; Levitre, G.; Merad, J.; Retailleau, P.; Neuville, L.; Masson, G. Angew. Chem., Int. Ed. 2018, 57, 12121–12125. doi:10.1002/anie.201807069
Return to citation in text: [1] -
Qiu, Z.-W.; Li, B. Q.; Liu, H.-F.; Zhu, Z.-Q.; Pan, H.-P.; Feng, N.; Ma, A.-J.; Peng, J.-B.; Zhang, X.-Z. J. Org. Chem. 2021, 86, 7490–7499. doi:10.1021/acs.joc.1c00484
Return to citation in text: [1] -
Zhan, G.; Shi, M.-L.; He, Q.; Du, W.; Chen, Y.-C. Org. Lett. 2015, 17, 4750–4753. doi:10.1021/acs.orglett.5b02279
Return to citation in text: [1] [2] -
Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021–15025. doi:10.1002/anie.201907797
Return to citation in text: [1] [2] -
Yan, R.-J.; Liu, B.-X.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2020, 22, 4240–4244. doi:10.1021/acs.orglett.0c01283
Return to citation in text: [1] [2] -
Du, J.-Y.; Ma, Y.-H.; Meng, F.-X.; Zhang, R.-R.; Wang, R.-N.; Shi, H.-L.; Wang, Q.; Fan, Y.-X.; Huang, H.-L.; Cui, J.-C.; Ma, C.-L. Org. Lett. 2019, 21, 465–468. doi:10.1021/acs.orglett.8b03709
Return to citation in text: [1] [2] -
Liu, D.; Sun, J.; Sun, Q.; Yan, C.-G. Org. Chem. Front. 2023, 10, 540–547. doi:10.1039/d2qo01771g
Return to citation in text: [1] [2] -
Pan, L.-N.; Sun, J.; Shi, R.-G.; Yan, C.-G. Org. Chem. Front. 2020, 7, 3202–3208. doi:10.1039/d0qo00845a
Return to citation in text: [1] [2] -
Pan, L.-N.; Sun, J.; Liu, X.-Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 7099–7104. doi:10.1039/d2ob01257j
Return to citation in text: [1] [2] -
Wang, D.; Sun, J.; Han, Y.; Sun, Q.; Yan, C.-G. Org. Lett. 2022, 24, 7790–7795. doi:10.1021/acs.orglett.2c03123
Return to citation in text: [1] [2] -
Cao, J.; Sun, J.; Yan, C.-G. Org. Biomol. Chem. 2019, 17, 9008–9013. doi:10.1039/c9ob01779h
Return to citation in text: [1] -
Sun, J.; Zhang, Y.; Shi, R.-G.; Yan, C.-G. Org. Biomol. Chem. 2019, 17, 3978–3983. doi:10.1039/c9ob00166b
Return to citation in text: [1] -
Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331
Return to citation in text: [1] -
Liu, D.; Liu, X.; Sun, J.; Yan, C.-G. J. Org. Chem. 2021, 86, 14705–14719. doi:10.1021/acs.joc.1c01513
Return to citation in text: [1] -
Liu, D.; Liu, X.; Sun, J.; Han, Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 4964–4969. doi:10.1039/d2ob00815g
Return to citation in text: [1] -
Xiao, Z.; Xu, F.; Sun, J.; Yan, C.-G. Beilstein J. Org. Chem. 2023, 19, 1234–1242. doi:10.3762/bjoc.19.91
Return to citation in text: [1]
| 1. | Ogawa, H.; Yamashita, H.; Kondo, K.; Yamamura, Y.; Miyamoto, H.; Kan, K.; Kitano, K.; Tanaka, M.; Nakaya, K.; Nakamura, S.; Mori, T.; Tominaga, M.; Yabuuchi, Y. J. Med. Chem. 1996, 39, 3547–3555. doi:10.1021/jm960133o |
| 2. | Tabata, H.; Nakagomi, J.; Morizono, D.; Oshitari, T.; Takahashi, H.; Natsugari, H. Angew. Chem., Int. Ed. 2011, 50, 3075–3079. doi:10.1002/anie.201007772 |
| 3. | Michelini, S.; Cassano, G. B.; Frare, F.; Perugi, G. Pharmacopsychiatry 2007, 29, 127–134. doi:10.1055/s-2007-979558 |
| 4. | Chakraborty, S.; Shah, N. H.; Fishbein, J. C.; Hosmane, R. S. Bioorg. Med. Chem. Lett. 2011, 21, 756–759. doi:10.1016/j.bmcl.2010.11.109 |
| 28. | Yu, B.; Yu, Z.; Qi, P.-P.; Yu, D.-Q.; Liu, H.-M. Eur. J. Med. Chem. 2015, 95, 35–40. doi:10.1016/j.ejmech.2015.03.020 |
| 29. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 30. | Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. J. Med. Chem. 2019, 62, 448–466. doi:10.1021/acs.jmedchem.8b00909 |
| 54. | Zhan, G.; Shi, M.-L.; He, Q.; Du, W.; Chen, Y.-C. Org. Lett. 2015, 17, 4750–4753. doi:10.1021/acs.orglett.5b02279 |
| 55. | Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021–15025. doi:10.1002/anie.201907797 |
| 56. | Yan, R.-J.; Liu, B.-X.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2020, 22, 4240–4244. doi:10.1021/acs.orglett.0c01283 |
| 57. | Du, J.-Y.; Ma, Y.-H.; Meng, F.-X.; Zhang, R.-R.; Wang, R.-N.; Shi, H.-L.; Wang, Q.; Fan, Y.-X.; Huang, H.-L.; Cui, J.-C.; Ma, C.-L. Org. Lett. 2019, 21, 465–468. doi:10.1021/acs.orglett.8b03709 |
| 58. | Liu, D.; Sun, J.; Sun, Q.; Yan, C.-G. Org. Chem. Front. 2023, 10, 540–547. doi:10.1039/d2qo01771g |
| 59. | Pan, L.-N.; Sun, J.; Shi, R.-G.; Yan, C.-G. Org. Chem. Front. 2020, 7, 3202–3208. doi:10.1039/d0qo00845a |
| 60. | Pan, L.-N.; Sun, J.; Liu, X.-Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 7099–7104. doi:10.1039/d2ob01257j |
| 61. | Wang, D.; Sun, J.; Han, Y.; Sun, Q.; Yan, C.-G. Org. Lett. 2022, 24, 7790–7795. doi:10.1021/acs.orglett.2c03123 |
| 22. | Matsuya, Y.; Ohsawa, N.; Nemoto, H. J. Am. Chem. Soc. 2006, 128, 13072–13073. doi:10.1021/ja065277z |
| 23. | Zhu, S.-Y.; Zhang, Y.; Wang, W.; Hui, X.-P. Org. Lett. 2017, 19, 5380–5383. doi:10.1021/acs.orglett.7b02657 |
| 24. | Deng, Y.; Pei, C.; Arman, H.; Dong, K.; Xu, X.; Doyle, M. P. Org. Lett. 2016, 18, 5884–5887. doi:10.1021/acs.orglett.6b02965 |
| 25. | Yuan, C.; Zhou, L.; Xia, M.; Sun, Z.; Wang, D.; Guo, H. Org. Lett. 2016, 18, 5644–5647. doi:10.1021/acs.orglett.6b02885 |
| 26. | Li, T.; Yang, Z.; Song, Z.; Chauvin, R.; Cui, X. Org. Lett. 2020, 22, 4078–4082. doi:10.1021/acs.orglett.0c01139 |
| 27. | Ding, Y.-L.; Zhao, Y.-L.; Niu, S.-S.; Wu, P.; Cheng, Y. J. Org. Chem. 2020, 85, 612–621. doi:10.1021/acs.joc.9b02693 |
| 14. | Guo, C.; Sahoo, B.; Daniliuc, C. G.; Glorius, F. J. Am. Chem. Soc. 2014, 136, 17402–17405. doi:10.1021/ja510737n |
| 15. | Wang, L.; Li, S.; Blümel, M.; Philipps, A. R.; Wang, A.; Puttreddy, R.; Rissanen, K.; Enders, D. Angew. Chem., Int. Ed. 2016, 55, 11110–11114. doi:10.1002/anie.201604819 |
| 16. | Mojikhalifeh, S.; Hasaninejad, A. Org. Chem. Front. 2018, 5, 1516–1521. doi:10.1039/c8qo00210j |
| 17. | Belyy, A. Y.; Levina, A. A.; Platonov, D. N.; Salikov, R. F.; Medvedev, M. G.; Tomilov, Y. V. Eur. J. Org. Chem. 2019, 4133–4138. doi:10.1002/ejoc.201801861 |
| 18. | Mohammed, K. S.; Elbeily, E. E.; El‐Taweel, F. M.; Fadda, A. A. J. Heterocycl. Chem. 2019, 56, 493–500. doi:10.1002/jhet.3425 |
| 19. | Asamdi, M.; Shaikh, M. M.; Chauhan, P. M.; Chikhalia, K. H. Tetrahedron 2018, 74, 3719–3727. doi:10.1016/j.tet.2018.05.051 |
| 20. | Attanasi, O. A.; De Crescentini, L.; Favi, G.; Filippone, P.; Mantellini, F.; Perrulli, F. R.; Santeusanio, S. Eur. J. Org. Chem. 2009, 3109–3127. doi:10.1002/ejoc.200900243 |
| 21. | Zhang, L.; Xu, Y.; Zhang, X.; Zhang, X.; Fan, X. Org. Chem. Front. 2020, 7, 2284–2290. doi:10.1039/d0qo00657b |
| 58. | Liu, D.; Sun, J.; Sun, Q.; Yan, C.-G. Org. Chem. Front. 2023, 10, 540–547. doi:10.1039/d2qo01771g |
| 5. | Wiethe, R. W.; Stewart, E. L.; Drewry, D. H.; Gray, D. W.; Mehbob, A.; Hoekstra, W. J. Bioorg. Med. Chem. Lett. 2006, 16, 3777–3779. doi:10.1016/j.bmcl.2006.04.055 |
| 6. | Łuszczki, J. J. Pharmacol. Rep. 2009, 61, 197–216. doi:10.1016/s1734-1140(09)70024-6 |
| 7. | Iwamoto, F. M.; Kreisl, T. N.; Kim, L.; Duic, J. P.; Butman, J. A.; Albert, P. S.; Fine, H. A. Cancer 2010, 116, 1776–1782. doi:10.1002/cncr.24957 |
| 8. | Murthy, K. S. K.; Knaus, E. E. Drug Dev. Res. 1999, 46, 155–162. doi:10.1002/(sici)1098-2299(199902)46:2<155::aid-ddr9>3.0.co;2-w |
| 9. | Isono, K. J. Antibiot. 1988, 41, 1711–1739. doi:10.7164/antibiotics.41.1711 |
| 10. | Kim, H.; Kim, M.; Lee, J.; Yu, H.; Hah, J.-M. Bioorg. Med. Chem. 2011, 19, 6760–6767. doi:10.1016/j.bmc.2011.09.042 |
| 11. | Assimon, V. A.; Tang, Y.; Vargas, J. D.; Lee, G. J.; Wu, Z. Y.; Lou, K.; Yao, B.; Menon, M.-K.; Pios, A.; Perez, K. C.; Madriaga, A.; Buchowiecki, P. K.; Rolfe, M.; Shawver, L.; Jiao, X.; Le Moigne, R.; Zhou, H.-J.; Anderson, D. J. ACS Chem. Biol. 2019, 14, 236–244. doi:10.1021/acschembio.8b00904 |
| 12. | Snieckus, V.; Streith, J. Acc. Chem. Res. 1981, 14, 348–355. doi:10.1021/ar00071a004 |
| 13. | Wang, M.; Huang, Z.; Xu, J.; Chi, Y. R. J. Am. Chem. Soc. 2014, 136, 1214–1217. doi:10.1021/ja411110f |
| 59. | Pan, L.-N.; Sun, J.; Shi, R.-G.; Yan, C.-G. Org. Chem. Front. 2020, 7, 3202–3208. doi:10.1039/d0qo00845a |
| 60. | Pan, L.-N.; Sun, J.; Liu, X.-Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 7099–7104. doi:10.1039/d2ob01257j |
| 61. | Wang, D.; Sun, J.; Han, Y.; Sun, Q.; Yan, C.-G. Org. Lett. 2022, 24, 7790–7795. doi:10.1021/acs.orglett.2c03123 |
| 62. | Cao, J.; Sun, J.; Yan, C.-G. Org. Biomol. Chem. 2019, 17, 9008–9013. doi:10.1039/c9ob01779h |
| 63. | Sun, J.; Zhang, Y.; Shi, R.-G.; Yan, C.-G. Org. Biomol. Chem. 2019, 17, 3978–3983. doi:10.1039/c9ob00166b |
| 64. | Liu, D.; Cao, J.; Sun, J.; Liu, R.-Z.; Yan, C.-G. Org. Lett. 2020, 22, 8931–8936. doi:10.1021/acs.orglett.0c03331 |
| 65. | Liu, D.; Liu, X.; Sun, J.; Yan, C.-G. J. Org. Chem. 2021, 86, 14705–14719. doi:10.1021/acs.joc.1c01513 |
| 66. | Liu, D.; Liu, X.; Sun, J.; Han, Y.; Yan, C.-G. Org. Biomol. Chem. 2022, 20, 4964–4969. doi:10.1039/d2ob00815g |
| 67. | Xiao, Z.; Xu, F.; Sun, J.; Yan, C.-G. Beilstein J. Org. Chem. 2023, 19, 1234–1242. doi:10.3762/bjoc.19.91 |
| 49. | Wei, F.; Huang, H.-Y.; Zhong, N.-J.; Gu, C.-L.; Wang, D.; Liu, L. Org. Lett. 2015, 17, 1688–1691. doi:10.1021/acs.orglett.5b00456 |
| 50. | Liu, J.; Wang, L.; Wang, X.; Xu, L.; Hao, Z.; Xiao, J. Org. Biomol. Chem. 2016, 14, 11510–11517. doi:10.1039/c6ob01953f |
| 51. | Wani, I. A.; Bhattacharyya, A.; Sayyad, M.; Ghorai, M. K. Org. Biomol. Chem. 2018, 16, 2910–2922. doi:10.1039/c8ob00228b |
| 52. | Gelis, C.; Levitre, G.; Merad, J.; Retailleau, P.; Neuville, L.; Masson, G. Angew. Chem., Int. Ed. 2018, 57, 12121–12125. doi:10.1002/anie.201807069 |
| 53. | Qiu, Z.-W.; Li, B. Q.; Liu, H.-F.; Zhu, Z.-Q.; Pan, H.-P.; Feng, N.; Ma, A.-J.; Peng, J.-B.; Zhang, X.-Z. J. Org. Chem. 2021, 86, 7490–7499. doi:10.1021/acs.joc.1c00484 |
| 55. | Chen, Z.-C.; Chen, Z.; Yang, Z.-H.; Guo, L.; Du, W.; Chen, Y.-C. Angew. Chem., Int. Ed. 2019, 58, 15021–15025. doi:10.1002/anie.201907797 |
| 56. | Yan, R.-J.; Liu, B.-X.; Xiao, B.-X.; Du, W.; Chen, Y.-C. Org. Lett. 2020, 22, 4240–4244. doi:10.1021/acs.orglett.0c01283 |
| 44. | Zhong, F.; Chen, G.-Y.; Han, X.; Yao, W.; Lu, Y. Org. Lett. 2012, 14, 3764–3767. doi:10.1021/ol301647g |
| 45. | Deng, H.-P.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2012, 354, 783–789. doi:10.1002/adsc.201101012 |
| 46. | Wang, Y.; Liu, L.; Zhang, T.; Zhong, N.-J.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2012, 77, 4143–4147. doi:10.1021/jo3002535 |
| 47. | Hu, F.-L.; Wei, Y.; Shi, M. Chem. Commun. 2014, 50, 8912–8914. doi:10.1039/c4cc03479a |
| 48. | Liu, Y.-L.; Wang, X.; Zhao, Y.-L.; Zhu, F.; Zeng, X.-P.; Chen, L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Angew. Chem., Int. Ed. 2013, 52, 13735–13739. doi:10.1002/anie.201307250 |
| 57. | Du, J.-Y.; Ma, Y.-H.; Meng, F.-X.; Zhang, R.-R.; Wang, R.-N.; Shi, H.-L.; Wang, Q.; Fan, Y.-X.; Huang, H.-L.; Cui, J.-C.; Ma, C.-L. Org. Lett. 2019, 21, 465–468. doi:10.1021/acs.orglett.8b03709 |
| 36. | Chen, Z.-C.; Chen, Z.; Du, W.; Chen, Y.-C. Chem. Rec. 2020, 20, 541–555. doi:10.1002/tcr.201900058 |
| 37. | Shanmugam, P.; Viswambharan, B.; Madhavan, S. Org. Lett. 2007, 9, 4095–4098. doi:10.1021/ol701533d |
| 38. | Gomes, J. C.; Sirvent, J.; Moyano, A.; Rodrigues, M. T., Jr.; Coelho, F. Org. Lett. 2013, 15, 5838–5841. doi:10.1021/ol4029034 |
| 39. | Warghude, P. K.; Sabale, A. S.; Bhat, R. G. Org. Biomol. Chem. 2020, 18, 1794–1799. doi:10.1039/d0ob00007h |
| 40. | Wang, Z.-H.; Lei, C.-W.; Zhang, X.-Y.; You, Y.; Zhao, J.-Q.; Yuan, W.-C. Org. Chem. Front. 2019, 6, 3342–3347. doi:10.1039/c9qo00890j |
| 41. | Zhou, T.; Xia, T.; Liu, Z.; Liu, L.; Zhang, J. Adv. Synth. Catal. 2018, 360, 4475–4479. doi:10.1002/adsc.201801152 |
| 42. | He, X.-H.; Fu, X.-J.; Zhan, G.; Zhang, N.; Li, X.; Zhu, H.-P.; Peng, C.; He, G.; Han, B. Org. Chem. Front. 2022, 9, 1048–1055. doi:10.1039/d1qo01785c |
| 43. | Zhang, L.; Liu, H.; Qiao, G.; Hou, Z.; Liu, Y.; Xiao, Y.; Guo, H. J. Am. Chem. Soc. 2015, 137, 4316–4319. doi:10.1021/jacs.5b01138 |
| 31. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 32. | Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808 |
| 33. | Liu, Y.; Wang, H.; Wan, J. Asian J. Org. Chem. 2013, 2, 374–386. doi:10.1002/ajoc.201200180 |
| 34. | Wang, Y.; Cobo, A. A.; Franz, A. K. Org. Chem. Front. 2021, 8, 4315–4348. doi:10.1039/d1qo00220a |
| 35. | Saeed, R.; Sakla, A. P.; Shankaraiah, N. Org. Biomol. Chem. 2021, 19, 7768–7791. doi:10.1039/d1ob01176f |
| 54. | Zhan, G.; Shi, M.-L.; He, Q.; Du, W.; Chen, Y.-C. Org. Lett. 2015, 17, 4750–4753. doi:10.1021/acs.orglett.5b02279 |
© 2023 Liu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.