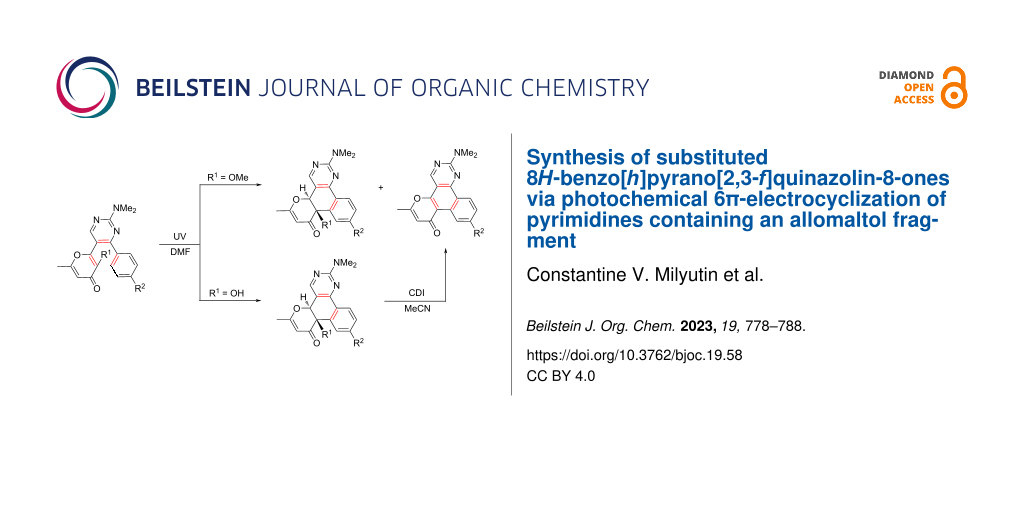
Abstract
For the first time, we elaborated a method for the synthesis of pyrimidines containing an allomaltol unit. The suggested approach is based on the reaction of 2-(1-(dimethylamino)-3-oxo-3-arylprop-1-en-2-yl)-3-hydroxy-6-methyl-4H-pyran-4-ones with cyanamide. The photochemical behavior of the obtained pyrimidines was investigated. It was shown that for the hydroxy derivatives the main pathway of phototransformation is a 6π-electrocyclization of the 1,3,5-hexatriene system and subsequent [1,9]-H sigmatropic shift leading to dihydrobenzo[h]pyrano[2,3-f]quinazolines. At the same time, for methylated analogues the photoreaction proceeds in two directions resulting in the formation of a mixture of the corresponding dihydrobenzo[h]pyrano[2,3-f]quinazolines and polyaromatic products. The obtained dihydro derivatives are stable compounds and do not undergo aromatization upon further UV irradiation. The structures of two of the dihydrobenzo[h]pyrano[2,3-f]quinazolines were confirmed by X-ray diffraction analysis. Based on the performed studies, a two-stage telescopic method for the synthesis of polyaromatic benzo[h]pyrano[2,3-f]quinazolines including the initial photocyclization of the starting pyrimidines and the final dehydration was proposed.

Graphical Abstract
Introduction
Photochemical processes involve absorption of UV light leading to the generation of molecules in the excited state and subsequent chemical transformations [1,2]. After excitation the electron distribution significantly changes leading to substantial alterations in the chemical behavior of the starting compound. Thereby, UV-induced reactions are a useful addition to common thermal processes. In this regard, photochemical transformations have found wide applications in various areas of science and technology [3-5]. For example, the use of photoreactions for the preparation of complex natural products allows reducing the number of synthetic steps [6-9]. Moreover, in some cases photochemical processes can open access to complicated multifunctional systems which are unavailable by other methods. Wherein, usually UV-promoted reactions are environmentally friendly due to the absence of toxic reagents, which is especially perspective in context of green chemistry [9,10]. Thus, studies of the photochemical behavior of organic compounds play an important role in modern chemistry allowing to create novel original synthetic methods.
UV-initiated reactions of heterocyclic systems are a significant part of organic photochemistry and there are numerous such transformations for various classes of heterocycles known in the literature [11-15]. Among them, photoreactions of compounds containing the 3-hydroxypyran-4-one (allomaltol) fragment attract considerable attention due to the unique photochemical properties of compounds of this class. Previously it was shown that allomaltol derivatives under UV irradiation undergo contraction of the pyranone ring leading to the formation of α-hydroxy-1,2-diketones. This phototransformation is initiated by an excited state intramolecular proton transfer (ESIPT) process. In most cases the resulting α-hydroxy-1,2-diketones are unstable and their further reactions open access to various original products [16-20]. The photochemical behavior of terarylenes containing an allomaltol fragment deserves special attention. In this case, two types of photoprocesses are possible: a 6π-electrocyclization of the 1,3,5-hexatriene system and the aforementioned ESIPT-induced transformation to α-hydroxy-1,2-diketone [21-25]. However, the direction of the transformation in each case depends on the structure of the bridge fragment. Thus, for example, the 1,3,5-hexatriene cyclization does not occur for pyrazole 1 and imidazole 2 derivatives, and the obtained products are formed exclusively as a result of ESIPT-induced reactions (Scheme 1A and B). At the same time, both directions of phototransformation are realized for terarylenes with furan, pyrrole, and oxazolone-bridge fragments (Scheme 1C). However, the suppression of the ESIPT process in such systems makes it possible to perform the 6π-electrocyclization leading to polyaromatic products. Therefore, it is complicate to predict the photochemical behavior of terarylenes containing a 3-hydroxypyran-4-one unit.
![[1860-5397-19-58-i1]](/bjoc/content/inline/1860-5397-19-58-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photochemical behavior of terarylenes containing an allomaltol fragment.
Scheme 1: Photochemical behavior of terarylenes containing an allomaltol fragment.
It should be noted that all the products considered above were constructed on the basis of five-membered heterocyclic bridge fragments. At the same time, it is obvious that the ring size can significantly influence the photochemical properties. In this regard, it seemed interesting to obtain terarylenes with a six-membered bridge and to study their behavior under the action of UV light. Continuing our research in the field of terarylenes containing a 3-hydroxy-4-pyranone fragment, in the present communication we investigated the photochemical properties of 2-(4-(4-aryl)-2-(dimethylamino)pyrimidin-5-yl)-6-methyl-4H-pyran-4-ones 9 and 10. It is important to note that the 6π-electrocyclization of 1,3,5-hexatriene systems with a pyrimidine-bridge fragment has been described in the literature [26-28]. Herein, we show that for pyrimidines 9 and 10 containing an allomaltol fragment the main direction of a phototransformation is also the cyclization of the triene system. In this case, condensed dihydropyranone derivatives 11 were obtained along with the expected polyaromatic compounds 12 (Scheme 1D).
Results and Discussion
The starting terarylenes 9 were obtained based on the reaction of the corresponding enaminones 13 with cyanamide (14). It should be noted that a similar approach is described in the literature and involves the condensation of 3-(dimethylamino)-1-phenylprop-2-en-1-one with cyanamide (14) in NMP in the presence of N-methylmorpholine [29]. However, this reaction led to a mixture of products and the yield of the target pyrimidine did not exceed 10%. It was assumed that the reported conditions were not optimal and the process could be significantly improved. In order to employ the aforementioned approach for the preparation of pyrimidines containing an allomaltol moiety we initially selected the interaction of enaminone 13a with cyanamide as a model reaction. We varied time and temperature of the process, as well as basic reagents and solvents to identify the optimal conditions and the best results are summarized in Table 1.
Table 1: Optimization of the condensation conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-58-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Time, h | Solvent | Temperature, °C | Basic reagent | Yield, % |
| 1 | 16 | NMP | 100 | N-methylmorpholine | 15 |
| 2 | 16 | NMP | 100 | – | 44 |
| 3 | 16 | MeCN | reflux | – | 52 |
| 4 | 16 | toluene | reflux | – | 40 |
| 5 | 16 | EtOH | reflux | – | 79 |
| 6 | 16 | dioxane | reflux | – | 62 |
| 7 | 16 | MeOH | reflux | – | 73 |
| 8 | 2 | EtOH | reflux | – | 80 |
| 9 | 2 | EtOH | reflux | N-methylmorpholine | 22 |
| 10 | 2 | EtOH | reflux | Et3N | 25 |
| 11 | 16 | dioxane | rt | – | 32 |
| 12b | 16 | EtOH | rt | – | – |
aReaction conditions: 13a (1 mmol, 0.33 g), 14 (3 mmol, 0,13 g), solvent (5 mL), time, basic reagent (1 mmol), T in °C. b13a is not soluble in EtOH at rt.
At first, based on literature data we carried out the reaction in NMP at 100 °C for 16 h in the presence of N-methylmorpholine. In this case the target pyrimidine 9a was obtained in 15% yield (Table 1, entry 1). Next, we repeated the above-mentioned reaction under similar conditions without a basic reagent and the exclusion of the base led to a significant increase in the yield of the target product 9a (Table 1, entry 2). Next, we tested the reaction in various solvents in the absence of a base (Table 1, entries 3–7) and the best results were obtained using alcoholic media (Table 1, entries 5 and 7). Probably, this is due to the protic nature of these solvents facilitating the studied condensation. Also, we have found that a decrease in the reaction time to 2 h did not affect the yield of pyrimidine 9a (Table 1, entry 8). It is important to emphasize that the use of bases essentially impaired the efficiency of the method (Table 1, entries 9 and 10). Apparently, the addition of a basic reagent does not influence the main process and the decrease in the yield is connected to side reactions. It is also of note that an elevated reaction temperature is beneficial for the protocol (Table 1, entries 11 and 12). Thus, the highest yield of the target pyrimidine 9a was obtained in refluxing EtOH for 2 h (Table 1, entry 8).
Hence, optimal conditions were developed for this reaction, allowing for the synthesis of pyrimidines 9 containing an allomaltol unit in good yields (Scheme 2). The process is of a general nature and is suitable for the synthesis of various target products 9 with electron-rich or deficient substituents.
![[1860-5397-19-58-i2]](/bjoc/content/inline/1860-5397-19-58-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of starting compounds 9. Reaction conditions: 13 (1 mmol), NH2CN (14, 3 mmol, 0.13 g), EtOH (5 mL), reflux, 2 h.
Scheme 2: Synthesis of starting compounds 9. Reaction conditions: 13 (1 mmol), NH2CN (14, 3 mmol, 0.13 g), Et...
The plausible reaction mechanism for the formation of pyrimidines 9 is presented in Scheme 3. At first, intermediate A is formed by Michael addition of cyanamide to enaminone 13. Further elimination of dimethylamine leads to cyanoenaminone B. Next, interaction of the cyano group with dimethylamine results in the formation of guanidine C. Finally, the intramolecular cyclization of the guanidine moiety and the carbonyl group leads to the target pyrimidine 9.
![[1860-5397-19-58-i3]](/bjoc/content/inline/1860-5397-19-58-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the formation of compounds 9.
Scheme 3: Proposed mechanism for the formation of compounds 9.
After the general synthetic method for pyrimidines containing the allomaltol fragment had been established, the photochemical behavior of the obtained compounds 9 was investigated.
It should be noted that we have previously studied the photochemical properties of various terarylenes with a 3-hydroxy-4-pyranone moiety [21-25]. It was shown that UV irradiation of such systems leads to a complex mixture of products, apparently due to the simultaneous occurrence of a 6π-electrocyclization of the hexatriene system and ESIPT-induced contraction of the pyranone ring. At the same time, the blocking of the ESIPT-promoted process via alkylation of the hydroxy group allows one to realize the regiospecific cyclization of the triene system. Based on these previous results, we started the photochemistry study of the considered pyrimidines from the corresponding methylated derivatives of 9. The products 10 were obtained by alkylation of compounds 9 with methyl iodide in DMF in the presence of K2CO3 (Scheme 4).
![[1860-5397-19-58-i4]](/bjoc/content/inline/1860-5397-19-58-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of methylated derivatives 10. Reaction conditions: 9 (1 mmol), MeI (3 mmol, 0.43 g), K2CO3 (3 mmol, 0.41 g), DMF (5 mL), stirring at rt for 4 h.
Scheme 4: Synthesis of methylated derivatives 10. Reaction conditions: 9 (1 mmol), MeI (3 mmol, 0.43 g), K2CO3...
At first, the photochemical behavior was investigated for the model compound 10a in DMSO-d6 solution using 1H NMR monitoring (Figure 1). Thus, the NMR spectrum recorded after UV irradiation (365 nm) for 24 h contained signals of protons of two products along with signals of the starting pyrimidine 10a (Figure 1B). The complete conversion of the starting compound 10a to the aforementioned mixture was observed after 48 h (Figure 1C). It should be noted that further irradiation of the test sample does not change the ratio of the formed products. In order to obtain the compounds in pure form we repeated the reaction in DMF irradiating at 365 nm for 48 h. As a result, both products could be isolated and characterized using 1H, 13C NMR spectroscopy and mass spectrometry. Moreover, the structure of product 11a was also confirmed by single-crystal X-ray diffraction analysis. It might be noted that precipitated crystals of 11a contained two polymorph modifications of triclinic (P1̄ and monoclinic (P21/c) crystal systems (see Supporting Information File 1 for details); the structure of one modification is shown in Figure 2. Thus, the first obtained compound is the expected polycycle 12a, while the second one is product 11a formed as a result of 6π-electrocyclization and subsequent [1,9]-H sigmatropic shift (Scheme 5). The considered photoprocess is of a general nature and the formation of a similar mixture of products 11 and 12 is observed upon UV irradiation of all starting compounds 10. Of note, the substituent in the aromatic ring influences only the ratio of the photoproducts (Table 2).
![[1860-5397-19-58-1]](/bjoc/content/figures/1860-5397-19-58-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR monitoring of the photoreaction of compound 10a under UV irradiation (365 nm) in DMSO-d6 solution.
Figure 1: 1H NMR monitoring of the photoreaction of compound 10a under UV irradiation (365 nm) in DMSO-d6 sol...
![[1860-5397-19-58-2]](/bjoc/content/figures/1860-5397-19-58-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The crystal structure of compound 11a (one of two polymorph modifications; p = 50%), CCDC 2248033.
Figure 2: The crystal structure of compound 11a (one of two polymorph modifications; p = 50%), CCDC 2248033.
![[1860-5397-19-58-i5]](/bjoc/content/inline/1860-5397-19-58-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Photochemical synthesis of compounds 11 and 12.
Scheme 5: Photochemical synthesis of compounds 11 and 12.
Table 2: Ratio of photoproducts 11 and 12.a
| Starting compound | Ratio of photoproducts 11 and 12 |
| 10a | 1:2 |
| 10b | 1:2 |
| 10c | 1:1.4 |
| 10d | 1:1 |
| 10e | 1:1 |
| 10f | 1:1 |
aRatio was calculated based on the 1H NMR spectrum of the crude reaction mixture.
Based on the structure of compound 11a we assumed that it could be converted into a polyaromatic product using conventional synthetic methods. However, the use of different systems (TsOH/toluene, HCl/EtOH, DBU/EtOH, MeONa/MeOH) resulted only in the decomposition of the derivative 11a. Thus, all our attempts towards the regiospecific photochemical conversion of pyrimidines 10 into polycyclic compounds 12 were unsuccessful.
A plausible mechanism for the considered phototransformation of compounds 9 and 10 is depicted in Scheme 6. At first, pyrimidines 10 undergo a 6π-electrocyclization under UV irradiation resulting in the formation of unstable intermediate A, starting from which two directions of further transformation are possible. The most obvious pathway is a simple elimination of a methanol molecule with aromatization of the central benzene ring leading to polycyclic product 12. In addition to the considered variant thermal suprafacial [1,9]-H sigmatropic shift resulting in compound 11 is implemented in this case. This transformation is energetically favorable due to the restoration of aromaticity of the benzene and pyrimidine rings. At the same time, the elimination of the methanol molecule occurs only for intermediate A and is not observed for product 11. It is important to emphasize that earlier, when discussing the mechanisms of the 6π-electrocyclization of various terarylenes accompanied by subsequent elimination of small molecules, we assumed a [1,9]-H sigmatropic shift step [23-25,30]. However, in all cases we failed to detect these intermediates even in the reaction mixture using NMR monitoring. However, in the present communication, for the first time, we managed to isolate and fully characterize a product of this class.
![[1860-5397-19-58-i6]](/bjoc/content/inline/1860-5397-19-58-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Proposed mechanism for the studied photoreaction.
Scheme 6: Proposed mechanism for the studied photoreaction.
Previously, we have shown that an alternative option for blocking ESIPT-induced processes for allomaltol derivatives is the use of DMF as a solvent [24]. Based on this fact, we assumed that the photoreaction in DMF would also allow the conversion of the starting pyrimidines 9 into the corresponding polycycles 12. However, the UV irradiation of compound 9a at 365 nm for 48 hours led only to product 11a resulting from the aforementioned [1,9]-H sigmatropic shift (Scheme 7). In addition, a significant amount of unreacted starting pyrimidine 9a was present in the reaction mixture. When increasing the process time to 96 h a complete conversion of substrate 9a could be achieved. Note, the phototransformation in this case is accompanied by side reactions, which are apparently associated with incomplete suppression of ESIPT-induced processes. At the same time, the prolonged irradiation of the reaction mixture did not lead to the conversion of the compound 11a into the polyaromatic product 12a and only a slight decrease in the yield as a result of photodecomposition was observed. Thus, the use of the above-mentioned conditions (365 nm, DMF, 96 h) allows one to synthesize products 11 (Scheme 7).
![[1860-5397-19-58-i7]](/bjoc/content/inline/1860-5397-19-58-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of compounds 11g–j starting from pyrimidines 9. Reaction conditions: 9 (0.5 mmol), DMF (15 mL), irradiation with a Vilber Lourmat VL-6.LM lamp (365 nm, 6 W) for 96 h at 27 °C in a commercial 25 mL-round-bottomed glass flask.
Scheme 7: Synthesis of compounds 11g–j starting from pyrimidines 9. Reaction conditions: 9 (0.5 mmol), DMF (1...
The structures of the obtained products 11g–j were confirmed by 1H, 13C NMR spectroscopy and high-resolution mass spectrometry. In the 1H NMR spectra of the products, characteristic singlets corresponding to the protons of the dihydropyranone fragment in the region δ 5.3–5.4 ppm and the protons of the hydroxy group in the region δ 5.4–5.5 ppm are present. In addition to the aforementioned characterization methods, the crystal structure of 11g·0.5EtOH was determined by single-crystal X-ray diffraction analysis. Its asymmetric unit contains 12 crystallographically unique molecules of 11g (Z' = 12, Z = 24) and 6 independent ethanol molecules. One molecule of 11g is shown in Figure 3. The other molecules have very similar conformations, but the methoxy group in some molecules exhibits an opposite orientation.
![[1860-5397-19-58-3]](/bjoc/content/figures/1860-5397-19-58-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: One of crystallographically unique molecules of 11g (p = 50%), CCDC 2248035.
Figure 3: One of crystallographically unique molecules of 11g (p = 50%), CCDC 2248035.
The mechanism of formation of products 11g–j is similar to the presented mechanism above for the methoxy derivatives 11a–f (Scheme 6). However, the key difference is the regioselectivity of the process and the absence of a pathway associated with the direct elimination of a water molecule and the formation of the polyaromatic compound 12. It can be assumed that for the considered system a [1,9]-H sigmatropic shift is a much more preferable process compared to aromatization.
The above-mentioned photochemical reaction of pyrimidines 10 allows to synthesize polycyclic products 12 (Scheme 8, method A). In this case, the yields of compounds 12 did not exceed 41%, which is associated with low regioselectivity of the process and the simultaneous formation of the products 11a–f. At the same time, we failed to increase the yield of the polycyclic products 12 by additional conversion of the byproducts 11a–f. At this point, we assumed that the conversion of the analogous hydroxy derivatives 11g–j into the polycyclic compounds 12 could be achieved by using suitable dehydrating agents (TsOH/toluene, Ac2O/MeCN, SOCl2/toluene, CDI/MeCN, POCl3/toluene). Gratifyingly, it was shown that the application of 1,1-carbonyldiimidazole (CDI) in acetonitrile allows to convert compound 11g into the polyaromatic product 12a in 94% yield (see Scheme S5 in Supporting Information File 1).
![[1860-5397-19-58-i8]](/bjoc/content/inline/1860-5397-19-58-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of photoproducts 12. Reaction conditions: method A) 10 (0.5 mmol), DMF (15 mL) irradiation for 48 h; method B) 1) 9 (0.5 mmol), DMF (15 mL), irradiation for 96 h; 2) CDI (1.75 mmol, 0.28 g), MeCN (5 mL), reflux, 2 h.
Scheme 8: Synthesis of photoproducts 12. Reaction conditions: method A) 10 (0.5 mmol), DMF (15 mL) irradiatio...
Based on the obtained results, we have developed a preparative telescopic method for the synthesis of polycycles 12. The presented two-step one-pot approach includes the preliminary photoconversion of the starting pyrimidines 9 in DMF to the corresponding hydroxy intermediates 11 and subsequent dehydration using the CDI/acetonitrile system (Scheme 8, method B). This approach allowed to increase the yield of products 12 in comparison with the phototransformation of methylated derivatives 10 (method A in Scheme 8).
It should be mentioned that the presented photoreaction can be carried out for starting compounds with both donor and acceptor substituents in the aryl fragment. However, it is interesting to note that the UV irradiation of thiophene containing pyrimidines 9g and 10g leads to a complicated mixture of unidentified products. Probably, this is due to photochemical side processes involving the thiophene ring.
Conclusion
In summary we have developed an approach for the preparation of pyrimidines with an allomaltol fragment. The suggested method started with the synthesis of pyrimidines from the reaction of 2-(1-(dimethylamino)-3-oxo-3-arylprop-1-en-2-yl)-3-hydroxy-6-methyl-4H-pyran-4-ones with cyanamide. Subsequently, the photochemical properties of synthesized pyrimidines were studied. We have demonstrated that a 6π-electrocyclization of the 1,3,5-hexatriene system and subsequent [1,9]-H sigmatropic shift leading to dihydrobenzo[h]pyrano[2,3-f]quinazolines are realized for hydroxy derivatives under UV irradiation. At the same time, for similar allomaltols with a methoxy group the photoprocess results in the formation of a mixture of the polyaromatic compounds and the corresponding dihydrobenzo[h]pyrano[2,3-f]quinazolines. The prepared dihydro derivatives are stable products and do not convert into polyaromatic benzo[h]pyrano[2,3-f]quinazolines upon further UV irradiation. The structures of two of the dihydrobenzo[h]pyrano[2,3-f]quinazolines were confirmed by X-ray diffraction analysis. Based on the obtained results, a two-step one-pot protocol for the preparation of polycyclic benzo[h]pyrano[2,3-f]quinazolines was elaborated. The method comprises the preliminary photoreaction of the starting compounds and the conclusive dehydration under the action of CDI.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data of all products, copies of 1H, 13C NMR, HRMS spectra of all new compounds, and X-ray crystallographic data. | ||
| Format: PDF | Size: 4.8 MB | Download |
References
-
Hoffmann, N. Chem. Rev. 2008, 108, 1052–1103. doi:10.1021/cr0680336
Return to citation in text: [1] -
Di Filippo, M.; Bracken, C.; Baumann, M. Molecules 2020, 25, 356. doi:10.3390/molecules25020356
Return to citation in text: [1] -
Kaur, N. Curr. Org. Synth. 2018, 15, 298–320. doi:10.2174/1570179414666171011160355
Return to citation in text: [1] -
Sarkar, D.; Bera, N.; Ghosh, S. Eur. J. Org. Chem. 2020, 1310–1326. doi:10.1002/ejoc.201901143
Return to citation in text: [1] -
De Keukeleire, D.; He, S.-L. Chem. Rev. 1993, 93, 359–380. doi:10.1021/cr00017a017
Return to citation in text: [1] -
Zhang, Z.; Zhou, Y.-j.; Liang, X.-W. Org. Biomol. Chem. 2020, 18, 5558–5566. doi:10.1039/d0ob01204a
Return to citation in text: [1] -
Bach, T.; Hehn, J. P. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. doi:10.1002/anie.201002845
Return to citation in text: [1] -
Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760
Return to citation in text: [1] -
Albini, A.; Fagnoni, M. Green Chem. 2004, 6, 1–6. doi:10.1039/b309592d
Return to citation in text: [1] [2] -
Protti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Green Chem. 2009, 11, 239–249. doi:10.1039/b810594d
Return to citation in text: [1] -
Hoffmann, N. Photochem. Photobiol. Sci. 2021, 20, 1657–1674. doi:10.1007/s43630-021-00135-6
Return to citation in text: [1] -
Lefebvre, C.; Fortier, L.; Hoffmann, N. Eur. J. Org. Chem. 2020, 1393–1404. doi:10.1002/ejoc.201901190
Return to citation in text: [1] -
Kaur, N. Synth. Commun. 2018, 48, 2935–2964. doi:10.1080/00397911.2018.1514051
Return to citation in text: [1] -
Coote, S. C. Eur. J. Org. Chem. 2020, 1405–1423. doi:10.1002/ejoc.201901230
Return to citation in text: [1] -
Münster, N.; Parker, N. A.; van Dijk, L.; Paton, R. S.; Smith, M. D. Angew. Chem., Int. Ed. 2017, 56, 9468–9472. doi:10.1002/anie.201705333
Return to citation in text: [1] -
Shiozaki, M.; Hiraoka, T. Tetrahedron Lett. 1972, 13, 4655–4658. doi:10.1016/s0040-4039(01)94390-4
Return to citation in text: [1] -
Barton, D. H. R.; Hulshof, L. A. J. Chem. Soc., Perkin Trans. 1 1977, 1103–1106. doi:10.1039/p19770001103
Return to citation in text: [1] -
Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Minyaev, M. E. Org. Lett. 2021, 23, 5266–5270. doi:10.1021/acs.orglett.1c01814
Return to citation in text: [1] -
Milyutin, C. V.; Galimova, R. D.; Komogortsev, A. N.; Lichitskii, B. V.; Melekhina, V. G.; Migulin, V. A.; Fakhrutdinov, A. N.; Minyaev, M. E. Org. Biomol. Chem. 2021, 19, 9975–9985. doi:10.1039/d1ob01871j
Return to citation in text: [1] -
Milyutin, C. V.; Galimova, R. G.; Komogortsev, A. N.; Lichitsky, B. V.; Migulin, V. A.; Melekhina, V. G. ChemistrySelect 2022, 7, e202204000. doi:10.1002/slct.202204000
Return to citation in text: [1] -
Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Beilstein J. Org. Chem. 2022, 18, 588–596. doi:10.3762/bjoc.18.61
Return to citation in text: [1] [2] -
Melekhina, V. G.; Komogortsev, A. N.; Lichitsky, B. V.; Mityanov, V. S.; Fakhrutdinov, A. N.; Dudinov, A. A.; Migulin, V. A.; Nelyubina, Y. V.; Melnikova, E. K.; Krayushkin, M. M. Tetrahedron Lett. 2019, 60, 151080. doi:10.1016/j.tetlet.2019.151080
Return to citation in text: [1] [2] -
Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 124, 133012. doi:10.1016/j.tet.2022.133012
Return to citation in text: [1] [2] [3] -
Komogortsev, A. N.; Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 114, 132780. doi:10.1016/j.tet.2022.132780
Return to citation in text: [1] [2] [3] [4] -
Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902
Return to citation in text: [1] [2] [3] -
Verbitskiy, E. V.; Cheprakova, E. M.; Makarova, N. I.; Dorogan, I. V.; Metelitsa, A. V.; Minkin, V. I.; Slepukhin, P. A.; Svalova, T. S.; Ivanova, A. V.; Kozitsina, A. N.; Rusinov, G. L.; Chupakhin, O. N.; Charushin, V. N. Eur. J. Org. Chem. 2016, 1420–1428. doi:10.1002/ejoc.201501450
Return to citation in text: [1] -
Wei, W.; Li, C.; Wang, T.; Liu, D.; Zhang, Z. Tetrahedron 2016, 72, 5037–5046. doi:10.1016/j.tet.2016.04.080
Return to citation in text: [1] -
Fan, J.; Zhang, W.; Gao, W.; Wang, T.; Duan, W.-L.; Liang, Y.; Zhang, Z. Org. Lett. 2019, 21, 9183–9187. doi:10.1021/acs.orglett.9b03556
Return to citation in text: [1] -
Fu, R.-G.; Wang, Y.; Xia, F.; Zhang, H.-L.; Sun, Y.; Yang, D.-W.; Wang, Y.-W.; Yin, P. J. Org. Chem. 2019, 84, 12237–12245. doi:10.1021/acs.joc.9b02032
Return to citation in text: [1] -
Karibov, T. T.; Lichitsky, B. V.; Melekhina, V. G.; Komogortsev, A. N. Polycyclic Aromat. Compd. 2023, in press. doi:10.1080/10406638.2022.2112706
Return to citation in text: [1]
| 1. | Hoffmann, N. Chem. Rev. 2008, 108, 1052–1103. doi:10.1021/cr0680336 |
| 2. | Di Filippo, M.; Bracken, C.; Baumann, M. Molecules 2020, 25, 356. doi:10.3390/molecules25020356 |
| 11. | Hoffmann, N. Photochem. Photobiol. Sci. 2021, 20, 1657–1674. doi:10.1007/s43630-021-00135-6 |
| 12. | Lefebvre, C.; Fortier, L.; Hoffmann, N. Eur. J. Org. Chem. 2020, 1393–1404. doi:10.1002/ejoc.201901190 |
| 13. | Kaur, N. Synth. Commun. 2018, 48, 2935–2964. doi:10.1080/00397911.2018.1514051 |
| 14. | Coote, S. C. Eur. J. Org. Chem. 2020, 1405–1423. doi:10.1002/ejoc.201901230 |
| 15. | Münster, N.; Parker, N. A.; van Dijk, L.; Paton, R. S.; Smith, M. D. Angew. Chem., Int. Ed. 2017, 56, 9468–9472. doi:10.1002/anie.201705333 |
| 9. | Albini, A.; Fagnoni, M. Green Chem. 2004, 6, 1–6. doi:10.1039/b309592d |
| 10. | Protti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Green Chem. 2009, 11, 239–249. doi:10.1039/b810594d |
| 6. | Zhang, Z.; Zhou, Y.-j.; Liang, X.-W. Org. Biomol. Chem. 2020, 18, 5558–5566. doi:10.1039/d0ob01204a |
| 7. | Bach, T.; Hehn, J. P. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. doi:10.1002/anie.201002845 |
| 8. | Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760 |
| 9. | Albini, A.; Fagnoni, M. Green Chem. 2004, 6, 1–6. doi:10.1039/b309592d |
| 3. | Kaur, N. Curr. Org. Synth. 2018, 15, 298–320. doi:10.2174/1570179414666171011160355 |
| 4. | Sarkar, D.; Bera, N.; Ghosh, S. Eur. J. Org. Chem. 2020, 1310–1326. doi:10.1002/ejoc.201901143 |
| 5. | De Keukeleire, D.; He, S.-L. Chem. Rev. 1993, 93, 359–380. doi:10.1021/cr00017a017 |
| 29. | Fu, R.-G.; Wang, Y.; Xia, F.; Zhang, H.-L.; Sun, Y.; Yang, D.-W.; Wang, Y.-W.; Yin, P. J. Org. Chem. 2019, 84, 12237–12245. doi:10.1021/acs.joc.9b02032 |
| 23. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 124, 133012. doi:10.1016/j.tet.2022.133012 |
| 24. | Komogortsev, A. N.; Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 114, 132780. doi:10.1016/j.tet.2022.132780 |
| 25. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
| 30. | Karibov, T. T.; Lichitsky, B. V.; Melekhina, V. G.; Komogortsev, A. N. Polycyclic Aromat. Compd. 2023, in press. doi:10.1080/10406638.2022.2112706 |
| 26. | Verbitskiy, E. V.; Cheprakova, E. M.; Makarova, N. I.; Dorogan, I. V.; Metelitsa, A. V.; Minkin, V. I.; Slepukhin, P. A.; Svalova, T. S.; Ivanova, A. V.; Kozitsina, A. N.; Rusinov, G. L.; Chupakhin, O. N.; Charushin, V. N. Eur. J. Org. Chem. 2016, 1420–1428. doi:10.1002/ejoc.201501450 |
| 27. | Wei, W.; Li, C.; Wang, T.; Liu, D.; Zhang, Z. Tetrahedron 2016, 72, 5037–5046. doi:10.1016/j.tet.2016.04.080 |
| 28. | Fan, J.; Zhang, W.; Gao, W.; Wang, T.; Duan, W.-L.; Liang, Y.; Zhang, Z. Org. Lett. 2019, 21, 9183–9187. doi:10.1021/acs.orglett.9b03556 |
| 24. | Komogortsev, A. N.; Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 114, 132780. doi:10.1016/j.tet.2022.132780 |
| 21. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Beilstein J. Org. Chem. 2022, 18, 588–596. doi:10.3762/bjoc.18.61 |
| 22. | Melekhina, V. G.; Komogortsev, A. N.; Lichitsky, B. V.; Mityanov, V. S.; Fakhrutdinov, A. N.; Dudinov, A. A.; Migulin, V. A.; Nelyubina, Y. V.; Melnikova, E. K.; Krayushkin, M. M. Tetrahedron Lett. 2019, 60, 151080. doi:10.1016/j.tetlet.2019.151080 |
| 23. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 124, 133012. doi:10.1016/j.tet.2022.133012 |
| 24. | Komogortsev, A. N.; Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 114, 132780. doi:10.1016/j.tet.2022.132780 |
| 25. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
| 16. | Shiozaki, M.; Hiraoka, T. Tetrahedron Lett. 1972, 13, 4655–4658. doi:10.1016/s0040-4039(01)94390-4 |
| 17. | Barton, D. H. R.; Hulshof, L. A. J. Chem. Soc., Perkin Trans. 1 1977, 1103–1106. doi:10.1039/p19770001103 |
| 18. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Minyaev, M. E. Org. Lett. 2021, 23, 5266–5270. doi:10.1021/acs.orglett.1c01814 |
| 19. | Milyutin, C. V.; Galimova, R. D.; Komogortsev, A. N.; Lichitskii, B. V.; Melekhina, V. G.; Migulin, V. A.; Fakhrutdinov, A. N.; Minyaev, M. E. Org. Biomol. Chem. 2021, 19, 9975–9985. doi:10.1039/d1ob01871j |
| 20. | Milyutin, C. V.; Galimova, R. G.; Komogortsev, A. N.; Lichitsky, B. V.; Migulin, V. A.; Melekhina, V. G. ChemistrySelect 2022, 7, e202204000. doi:10.1002/slct.202204000 |
| 21. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Beilstein J. Org. Chem. 2022, 18, 588–596. doi:10.3762/bjoc.18.61 |
| 22. | Melekhina, V. G.; Komogortsev, A. N.; Lichitsky, B. V.; Mityanov, V. S.; Fakhrutdinov, A. N.; Dudinov, A. A.; Migulin, V. A.; Nelyubina, Y. V.; Melnikova, E. K.; Krayushkin, M. M. Tetrahedron Lett. 2019, 60, 151080. doi:10.1016/j.tetlet.2019.151080 |
| 23. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 124, 133012. doi:10.1016/j.tet.2022.133012 |
| 24. | Komogortsev, A. N.; Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G. Tetrahedron 2022, 114, 132780. doi:10.1016/j.tet.2022.132780 |
| 25. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
© 2023 Milyutin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.