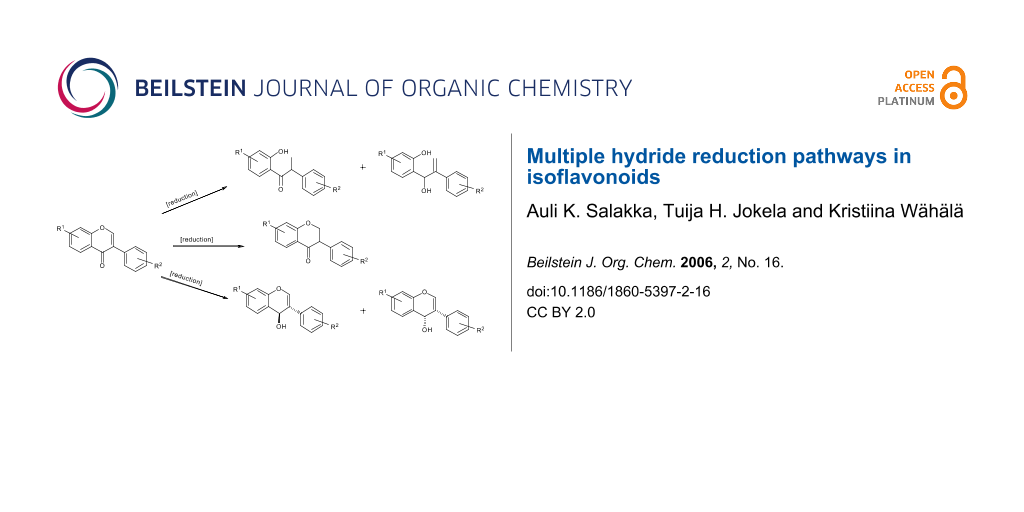
Abstract
Background
Isoflavonoids are of interest owing to their appearance in metabolic pathways of isoflavones, and their estrogenic and other physiological properties, making them promising lead compounds for drug design.
Results
The reduction of isoflavones by various hydride reagents occurs by a 1,4-pathway in contrast to ordinary β-alkoxy-α,β-unsaturated ketones. Isoflavan-4-ones, cis- and trans-isoflavan-4-ols, α-methyldeoxybenzoins or 1,2-diphenylprop-2-en-1-ols are obtained depending on the hydride reagent, mostly in good yields. The stereoselective reduction of isoflavan-4-ones is also discussed.
Conclusion
The work described in this paper shows that most structural types of reduced isoflavonoids are now reliably available in satisfactory or good yields by hydride reductions to be used as authentic reference compounds in analytical and biological studies.

Graphical Abstract
Background
The reduction of isoflavones 1 has been actively studied during the last twenty years, owing to the range of interesting biological effects [1-3] – estrogenic activity, promise in cancer, osteoporosis, and coronary heart disease prevention – shown by the isoflavones themselves and their reduced metabolites. There are some reports [4-8] of total syntheses of reduced isoflavonoid structures from commercially available starting materials but overall yields in these multistep procedures tend to be low, and free hydroxy groups are not compatible. Another strategy involves the hydrogenation of isoflavones using a palladium or platinum catalyst but mixtures of reduction products are often formed [9,10]. Certain hydride reagents have been tested, as discussed below, for the reduction of simple or protected isoflavones but many of the early results are contradictory or rely on indeterminate product characterization.
The reduction of simple (nonflavonoid) β-alkoxy-α,β-unsaturated ketones by hydride reagents (NaBH4, LiAlH4, DIBAH) occurs normally by 1,2-attack, this being a key step in the well known "carbonyl transposition" of 1,3-diketone enol ethers into enones (R3O-CR=CR1-COR2 → R-CO-CR1=CHR2). [11-15] Prior to our work there were no reports on the reduction of isoflavones containing free hydroxy groups which nevertheless are common, usually at one or several of the C-5, C-7 and C-4' sites, in the naturally occurring isoflavonoids. It is to be expected that the presence of the phenolic hydroxyls, or the derived phenolate anions, will alter the reactivity pattern of the parent isoflavone system, besides perhaps decreasing the overall reactivity due to solubility reasons. For example, any tendency of hydride attack at the C-2 will be opposed by electron feeding from the 4'-OH group while an OH group at C-5 or C-7 will discourage attack at C-2 and C-4. Thus there was ample room for the development of reliable methods for the synthesis of hydroxy-substituted isoflavone metabolites, and for clarification of the course of reduction of isoflavones with various hydride reducing agents. We present here experimental details of our own results in this field together with a thorough survey of the literature. The discussion is based on the types of reduced isoflavonoid structures formed (Figure 1), i.e., isoflavanones 2, cis-isoflavan-4-ols 3, trans-isoflavan-4-ols 4, the ring opened α-methyldeoxybenzoins 5 and 1,2-diaryl-2-propen-1-ols 6, and isoflavenes 7 and 8 (hydride reductions do not lead to isoflavans 9, which however are obtained by catalytic hydrogenation [10,16,20]). Incidentally, it is appropriate to point out that flavones do not undergo similar reductive metabolism in mammals as described above for isoflavones. We will nevertheless present at a later date certain findings on the hydride reduction pathways in flavones.
![[1860-5397-2-16-1]](/bjoc/content/figures/1860-5397-2-16-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Isoflavanones (2)
Isoflavanones result from the 1,4-reduction of isoflavones. The resonance contributor 10 will encourage this mode of attack. DIBAH, normally a preferential 1,2-reducer, reacts with methoxy-, benzyloxy-, MOMO- and MEMO-substituted isoflavones to give the isoflavanones in 40–93% yield [16,21-23] (Table 1). Our own work has shown that even unprotected hydroxy-substituted isoflavones are reduced by a large excess of DIBAH in 50–70% yield (Table 1). The simple borohydride reagents reduce isoflavones to the isoflavanols (see below) but the Selectrides® give 60–88% yields of isoflavanones in the absence of hydroxy substituents according to our results (Table 1). There is no significant difference between the K- and L-Selectride®. In the literature, there is an isolated report of the reduction of a MOM substituted isoflavone by L-Selectride® in 40% yield (Table 2) [16]. Methoxy substituted isoflavones have also been reduced by sodium hydrogen telluride to give isoflavan-4-ones in 61–71% yields [24].
Table 1: Reduction of isoflavones to isoflavanones.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-2-16-i1.svg?max-width=637&scale=1.0)
|
||||||||||
| a all R groups = H | ||||||||||
| b R7 = OMe, other R groups = H | ||||||||||
| c R4' = R7 = OMe, other R groups = H | ||||||||||
| d R7 = OH, other R groups = H | ||||||||||
| e R4' = OMe, R7 = OH, other R groups = H | ||||||||||
| f R4' = OMe, R5 = R7 = OH, other R groups = H | ||||||||||
| R2' | R3' | R4' | R5' | R6' | R5 | R6 | R7 | R8 | reductant, eqs | yield % |
|---|---|---|---|---|---|---|---|---|---|---|
| DIBb, 4 | 72 | |||||||||
| SELc, 4 | 96 | |||||||||
| OH | DIB, 10 | 68 | ||||||||
| SEL, 8 | 0 | |||||||||
| OMe | DIB, 2.5 | 90d,e | ||||||||
| DIB, 6 | 57 | |||||||||
| SEL, 4 | 84 | |||||||||
| Me | OMe | OMe | DIB, 2.5 | 75e | ||||||
| OMe | OMe | OMe | DIB, 2.5 | 89e | ||||||
| OMe | OMe | OMe | NaHTe, 2 | 68f | ||||||
| OH | OH | DIB, 25 | 70 | |||||||
| SEL, 8 | 0 | |||||||||
| OMe | OH | DIB, 25 | 60 | |||||||
| OMe | OMe | DIB, 2.5 | 87e | |||||||
| DIB, 7 | 54 | |||||||||
| NaHTe, 2 | 61f | |||||||||
| SEL, 4 | 82 | |||||||||
| OMOM | OMOM | DIB, 4.7 | 93g | |||||||
| OH | OH | OH | DIB, 25 | 50 | ||||||
| SEL, 9 | 0 | |||||||||
| OMe | OH | OH | DIB, 25 | 57 | ||||||
| OMe | OMe | OMe | OMe | NaHTe, 2 | 71f | |||||
| OMOM | OMOM | OMOM | DIB, 4.7 | 87g,h | ||||||
| SEL, 2 | 40g | |||||||||
| OMe | OMe | OBn | OBn | DIB, 2.5 | 40i | |||||
| OBn | OMe | OMe | OMe | DIB, 2.5 | 56e | |||||
| OBn | OBn | OMe | OMe | OMe | DIB, 1.9 | 52j | ||||
| OBn | OBn | OBn | OMe | OMe | DIB, 1.9 | 57j | ||||
| OBn | OBn | OMe | OMe | OMe | OBn | DIB, 1.9 | 67j | |||
| OBn | OMe | OMe | OMe | OBn | DIB, 1.9 | 61j | ||||
| OBn | OBn | OBn | OMe | OMe | OBn | DIB, 1.9 | 58j | |||
a Only substituents other than H are shown in the Table. Items without a reference are results reported here for the first time. b DIB = di-isobutylaluminiumhydride. c SEL = K- or L-Selectride®. d 2-Methyl-7-methoxyisoflavone reacted similarly in 63% yield.[21] e Ref. 21. f Ref. 24. g Ref. 16. h The corresponding tris(methoxyethoxymethoxy)flavone reacted similarly. i Ref. 23. j Ref. 22.
Table 2: Reduction of isoflavones to isoflavan-4-olsa
![[Graphic 2]](/bjoc/content/inline/1860-5397-2-16-i2.svg?max-width=637&scale=1.0)
|
||||||||||
| SM | R2 | R2' | R4' | R6 | R7 | R8 | reductant, eqs, solvent | yield 3+4 % | 3:4 ratio | Yld 6 % |
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | NaBH4,2.5,EtOH | 86 | 70:30 | 12 | ||||||
| LiBH4,2.5,THF | 56 | 80:20 | 40 | |||||||
| NaBH4,2.5,MeOH | 57b | 3 only | ||||||||
| NaBH4,2,EtOH | 75c,d | |||||||||
| NaBH4,2,diglyme | 88d,e | |||||||||
| NaBH4,3.5,PdCl2,aq. THF | 85 | 82:18 | ||||||||
| NaBH4,H3BO3,2.5, EtOH | 98 | 71:29 | ||||||||
| NaBH4,CeCl3,1.0, DMSO | 99 | 3 only | ||||||||
| NaBH4,AlCl3,excess., digl. | 81d | |||||||||
| Zn(BH4)2,4,Et2O | 69 | 62:38 | 19 | |||||||
| LiEt3BH,4,THF | 50 | 70:30 | ||||||||
| B2H6, excess,THF | 76c,d | |||||||||
| 1b | OMe | NaBH4,2.5,EtOH | 90 | 70:30 | 9 | |||||
| NaBH4,H3BO3,3.1, EtOH | 80f | 3 only | ||||||||
| NaBH4,H3BO3,2.5, EtOH | 97 | 70:30 | ||||||||
| NaBH4,CeCl3,1.5, DMSO | 88 | 3 only | 12 | |||||||
| LiBH4,10,THF | 54 | 70:30 | 37 | |||||||
| LiEt3BH,4,THF | 55 | 65:35 | ||||||||
| 1c | OMe | OMe | NaBH4,2.5,MeOH | 37b | 3 only | |||||
| NaBH4,2.5,EtOH | 88 | 70:30 | 10 | |||||||
| NaBH4,CeCl3,2.5, DMSO | 86 | 3 only | 10 | |||||||
| NaBH4,H3BO3,2.5, EtOH | 96 | 70:30 | ||||||||
| LiBH4,10,THF | 70 | 77:23 | 10 | |||||||
| Zn(BH4)2,2,Et2O | 74 | 62:38 | ||||||||
| LiEt3BH,4,THF | 36 | 78:22 | ||||||||
| 1 | OMOM | OMOM | OMOM |
NaBH4,15.9,EtOH, THF
LiBH4,10,THF |
87g
82g |
65:35
57:43 |
||||
| 1 | Me | Br | OMe | NaBH4,4.2,EtOH | 25e,h | |||||
| 1 | Me | Br | OBn | NaBH4,4.2,EtOH | 22e,h | |||||
| 1 | Me | OMe | Br | NaBH4,4.2,EtOH | 20e,h | |||||
| 1 | Me | Br | OMe | Br | NaBH4,4.2,EtOH | 20e,h | ||||
a Only substituents other than H are shown. According to our results, OH substituted isoflavones are not reduced, nor are such reactions reported in the literature. Items without a reference are results reported here for the first time. b Ref. 25. c Product was given as 2-isoflaven-4-ol. d Ref. 28. e Product ratio not given. f Ref. 26. g Ref. 16. h Ref. 27.
Isoflavanols (3, 4)
Full reduction at the heterocyclic ring of isoflavones by LiBH4 or NaBH4 leads to isoflavanols in 20–91% yield (Table 2), [16,25-28]except with hydroxy substituted substrates which do not react at all. Apparently the first step involves a 1,4-addition to give the isoflavanone enolate which picks up a proton from the solvent and is reduced further to the saturated alcohol. There are no reports of the intermediacy of the alternative 1,2-reduction products, the allylic alcohols 11 which in fact appear very incompletely known in the chemical literature (see below). Similarly, the reduction of isoflavanones 2 by LiBH4, NaBH4, L-Selectride® or Li(t-BuO)3AlH gives mixtures of cis-3 and trans-isoflavan-4-ols 4 (Table 3). [16,29,34] Isoflavanones are reduced by electrophilic hydrides (borane-tetrahydrofuran, bis-tert-butylthioethane borane) diastereoselectively to cis-isoflavan-4-ols, but a large excess of the reducing agent is usually needed [29].
Table 3: Reduction of isoflavanones to isoflavan-4-olsa
![[Graphic 3]](/bjoc/content/inline/1860-5397-2-16-i3.svg?max-width=637&scale=1.0)
|
|||||||
| SM | R2' | R3' | R4' | R7 | reductant, eqs, solvent | yield 3+4 % | 3:4 ratio |
|---|---|---|---|---|---|---|---|
| 2a |
NaBH4, ng.,EtOH
NaBH4,2,MeOH,THF Li(t-BuO)3AlH,10,THF B2H6,80,THF |
99b
99c 99c 98c |
70:30
42:58 34:66 3 only |
||||
| 2b | OMe |
NaBH4,2,MeOH,THF
NaBH4,1.2,EtOH Li(t-BuO)3AlH,10,THF L-Selectride, ng B2H6,80,THF BTEDg,0.7,THF |
99c
67d 99c 99c 99c 98c |
44:56
67:33 33:67 37:63 3 only 3 only |
|||
| 2 | OH | OH | LiBH4,6.9,THF | 94e | 70:30 | ||
| 2 | OTBDMS | OTBDMS | LiBH4,2,THF | 96f | 70:30 | ||
| 2c | OMe | OMe |
NaBH4,2,THF,MeOH
NaBH4,1.2,EtOH Li(t-BuO)3AlH,10,THF B2H6,80,THF |
99c
50d 99c 97c |
43:57
3 only 34:66 3 only |
||
| BTEDg,0.7,THF | 97c | 3 only | |||||
| 2 | OMe | OMe | OMe | NaBH4,2,THF,MeOH | 99c | 36:64 | |
| Li(t-BuO)3AlH,10,THF | 99c | 32:68 | |||||
| B2H6,80,THF | 98c | 3 only | |||||
| BTEDg,0.7,THF | 98c | 3 only | |||||
| 2 | OMOM | OMOM | OMOM | LiBH4,10,THF | 73h | 55:45 | |
| LiAlH4,17,THF | 78h | 45:55 | |||||
| NaBH4,15.9,THF,MeOH | 91h | 70:30 | |||||
a Only substituents other than H are shown. ng not given b Ref. 31. c Ref. 29. d Ref. 30. e Ref. 32. f Ref. 33. g bis-t-butylthioethane diborane. h Ref. 16.
Although it was realized by the early workers that diastereomeric mixtures of isoflavanols would presumably be formed, there were no reliable methods to determine their structures. In some papers, the products are summarily assigned the cis [25,26] or trans [35-38] structures. However, rigorous NMR analysis has recently made it possible to establish cis- and trans-structures for the isoflavanol products and to study their conformational equilibria [32,34]. Borohydride reductions generally give a small preference for the cis products as suggested by Cram's rule [39]. We are not aware of any examples in the literature of single enantiomers of isoflavanone or isoflavanol metabolites, nor have such compounds been reported as hydride reduction products of isoflavones. In view of the significant biological properties of the reduced metabolites it will be interesting to examine the behaviour of the pure enantiomers.
2-Isoflaven-4-ols (11)
There are very few reports of this class of compounds, either from reductive processes or otherwise. In 1965, the reduction of the parent isoflavone by NaBH4 in EtOH or diborane in THF was claimed [28] to furnish 2-isoflaven-4-ol in 75–76% yield, but unfortunately the characterization of this product relied on elemental analysis only. More recently, Japanese workers [40] reported that the reduction of 12 (Figure 2) by NaBH4 in the presence of PdCl2 in THF-H2O gave a 1:1 mixture of the ketone 13a and the diol 13b (Figure 2). A 1H NMR spectrum was given for the latter isoflavenol but there appear to be certain discrepancies, notably in the δ value (8.69) reported for the H-8 which is some 2 δ units in excess of what would be expected for such a vinyl ether proton. Thus more work is required to fully confirm the nature of this class of reduction products. In the event, in our studies the reduction of isoflavone 1a by NaBH4 in the presence of PdCl2 in THF-H2O gave a 82:18 mixture of cis- and trans-isoflavan-4-ol 3a, 4a while no 2-isoflaven-4-ols were observed. The absence of such 1,2-reduction products, or structures conceivably derivable thereof such as 2- or 3-isoflavenes (7, 8), even in the CeCl3-complexed NaBH4 reductions, must reflect the good stabilization obtainable via resonance heteroring stabilization (10) in the isoflavones. As already mentioned, this is in contrast to the behaviour of simple non-flavonoid β-alkoxy-α,β-unsaturated ketones which prefer 1,2-attack by hydride.
![[1860-5397-2-16-2]](/bjoc/content/figures/1860-5397-2-16-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 2-Isoflaven-4-ols and 1-(2-hydroxyphenyl)-2-phenyl-2-propen-1-one
Figure 2: 2-Isoflaven-4-ols and 1-(2-hydroxyphenyl)-2-phenyl-2-propen-1-one
Isoflavenes (7, 8) and isoflavans (9)
In the early work, [41] there is a mention of 7,4'-dimethoxy-2-methyl-3-isoflavene being obtained in 15% yield from the reduction of the corresponding isoflavone by LiAlH4 in Et2O-benzene but there is no structural data on the product other than elemental analysis, itself quite accurate. Similarly 2',4'-dimethoxy-3',6,7-trihydroxyisoflav-3-ene was reported from the reduction of 2',4'-dimethoxy-3',6,7-trihydroxyisoflavan-4-one with LiAlH4 in low yield [42]. More recent work by us [43] and others [16,44] would indicate that the normal course of LiAlH4 reduction of isoflavones leads to deoxybenzoins and propenols (see below and Table 4).
Table 4: Reduction of isoflavones to α-methyldeoxybenzoins and 1,2-diaryl-2-propen-1-olsa
![[Graphic 4]](/bjoc/content/inline/1860-5397-2-16-i4.svg?max-width=637&scale=1.0)
|
||||||
| a all R groups = H | ||||||
| b R7 = Me, other R groups = H | ||||||
| c R4 = R7 = Me, other R groups = H | ||||||
| R2' | R4' | R5 | R7 | reductant, eqs, solvent | yield (%) | |
|---|---|---|---|---|---|---|
| of 5 | of 6 | |||||
| LiAlH4, 2.5,THF | 27b | 48 | ||||
| Li(t-BuO)3AlH, 5,THF | 74 | 6 | ||||
| Red-Al, 2.5,THF | 12 | 50 | ||||
| LiBH4, 10, THF | 9 | 68 | ||||
| OMe | LiAlH4, 2.5,THF | 29b | 50 | |||
| LiAlH4, 1, THF | 62c | - | ||||
| Li(t-BuO)3AlH,10,THF | 88 | 12 | ||||
| LiBH4, 10, THF | 7 | 37 | ||||
| OMe | OMe | LiAlH4,3.3,THF | 27b | 70 | ||
| Li(t-BuO)3AlH,10,THF | 88 | 12 | ||||
| LiBH4, 10, THF | 5 | 10 | ||||
| OH | LiAlH4,3.3,THF | 60b | - | |||
| Li(t-BuO)3AlH,8,THF | - | - | ||||
| OH | OH | LiAlH4,5.5,THF | 42b | - | ||
| Li(t-BuO)3AlH,10,THF | - | - | ||||
| OMe | OH | LiAlH4,3.3,THF | 42b | - | ||
| OH | OH | OH | LiAlH4,5.5,THF | 66b | - | |
| OMe | OH | OH | LiAlH4,4.3,THF | 70b | - | |
| OH | OMe | LiAlH4,3.3,THF | 17b | 34 | ||
| OMOM | OMOM | OMOM | LiAlH4,7.9,THF | 6d | - | |
a Only substituents other than H are shown in the Table. Items without a reference are results reported here for the first time. b Ref. 43. c Ref. 44. d Ref. 16.
Published syntheses of 2- and 3-isoflavenes involve the reduction of 3-arylcoumarins, [45-47] the corresponding aldehyde hemiacetals, [7] isoflavylium salts [48-50] or of isoflavones by the Clemmensen reaction [51]. Low-yielding non-reductive routes to 2- and/or 3-isoflavenes have also been reported [52,53]. 2-Isoflavenes 7 however remain mostly poorly characterized, and some of the NMR spectral details reported [7,51,52] appear inconsistent with the 2-isoflavenoid structure. As far as the NMR spectra of 2-isoflavenes are concerned, a recent study clears this issue by 2D NMR work on a natural 2-isoflavene, [54] establishing that the H-2 and H-4 protons appear at δ 6.87 and 3.61, respectively, much as expected by correlation data shift calculations. Isoflavans (9) have not been prepared by hydride reductions, but by catalytic hydrogenation of isoflavones [10,16-20].
Deoxybenzoins (5) and propenols (6)
Isoflavanones undergo a facile retro-Michael-type ring opening [21,55] under basic conditions to give the propenone intermediate 14 (Figure 2), sometimes considered [56] to be an independent isoflavone metabolite but presumably just an artefact in reality. As regards the synthesis of isoflavanones by DIBAH reduction of isoflavones (see above), we found that unless the workup is done with cold methanolic HCl, some amount of the propenone 14 will be formed and reduced further to the deoxybenzoin 5. If on the other hand the deoxybenzoins are the actual synthetic targets, the reducing agent of choice is LiAlH4 in THF. This works very well for isoflavones bearing a hydroxy group at C-7 such as genistein, but in isoflavones lacking a 7-OH group another reaction pathway competes leading to the propenols 6 [43] as byproducts (see below). We have discussed a possible mechanism to explain these hydroxyl-dependent divergent pathways [43].
To summarize, all hydride addition reactions with isoflavones appear to involve an initial 1,4-addition to give the isoflavanone enolate. In a hydroxylic solvent, or even on workup under basic conditions, the ketone is generated, and reduced further to the saturated alcohol (NaBH4). In a nonprotic solvent, the β-aryloxyenolate will undergo a retro-Michael addition, giving the phenolate anion of the ring opened 2-propen-1-one which may undergo a 1,2- or 1,4-addition of hydride (LiAlH4, LiBH4). If the O-metal bond in the initial enolate is very tight, the ring opening does not occur and allows the isolation of the isoflavanone (DIBAH, Selectrides®). The presence and number of hydroxy or alkoxy substituents in the substrates does not have a major effect in these reductions except in the case of NaBH4 reduction which fails completely presumably due to solubility reasons, and the LiAlH4 reduction where the outcome depends on the presence or absence of an OH group at C-7. Based on our and previous results by other workers, the reducing agents of choice for the synthesis of reduced isoflavonoids are as follows:
isoflavanones (2) from non-hydroxylated isoflavones DIBAH or Selectrides®
isoflavanones (2) from hydroxylated isoflavones DIBAH
cis-isoflavanols (3) from non-hydroxylated isoflavones NaBH4/CeCl3
cis-isoflavanols (3) from hydroxylated isoflavones no good methods
cis-isoflavanols (3) from non-hydroxylated isoflavanones B2H6 or BTED
trans-isoflavanols (4) from isoflavones no good methods
trans-isoflavanols (4) from isoflavanones Li(t-BuO)3AlH
2-isoflaven-4-ols (11) from isoflavones uncertain
isoflavenes (7, 8) from isoflavones Clemmensen
isoflavans (9) from isoflavones H2, Pd/BaSO4
α-methyldeoxybenzoins (5) from non-hydroxylated isoflavones Li(t-BuO)3AlH
α-methyldeoxybenzoins (5) from hydroxylated isoflavones LiAlH4
1,2-diaryl-2-propen-1-ols (6) from non-hydroxylated isoflavones LiBH4 or LiAlH4
Conclusion
Dietary isoflavonoids in vegetables, beans, peas and other legumes are possible cancer preventing agents, particularly in hormone based cancers such as breast and prostate cancer. [57-59] Epidemiological studies have shown that they decrease the risk of colon cancer, osteoporosis, and coronary heart disease. [1-3] Significantly, health claims of soy foods, rich in isoflavonoids, have recently received FDA authorization [60].
The dietary isoflavonoids are mainly metabolized in man via reductive pathways, leading to the reduced structural types discussed above (Figure 3). These compounds are often more estrogenic than the starting isoflavones. Research interest in many fields including medicine, nutrition and biosynthesis and metabolism thus converge on the reduced isoflavonoids. The work described in this paper shows that most structural types of reduced isoflavonoids are now reliably available in satisfactory or good yields by hydride reductions. Although not discussed here, it is clear that D atoms may be introduced in the same way which is very useful in the quantitation of the naturally occurring compounds by GC-MS selected ion monitoring techniques [61].
![[1860-5397-2-16-3]](/bjoc/content/figures/1860-5397-2-16-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3:
Dietary isoflavones and their metabolites in humans.
e R4’=OMe, R7= OH, other R groups = H
f R4’= OMe, R5=R7= OH, other R groups = H
g R4’= R5=R7= OH, other R groups = H
h R4’=R7= OH, other R groups = H
i R4’=R7= OH, R6=OCH3, other R groups = H
j R4’= R6=R7= OH, other R groups = H
k R3’= R7= OH, other R groups = H
l R3’=R4’=R7= OH, other R groups = H
Figure 3:
Dietary isoflavones and their metabolites in humans.
e R4’=OMe, R7= OH, other R groups = H
f R4’= OMe...
Supporting Information
| Supporting Information File 1: Experimental details and characterisation data. | ||
| Format: DOC | Size: 48.5 KB | Download |
Acknowledgements
This work was partially funded within the Finnish Academy project no 178253. Funding to AKS and THJ from the Foundation of Emil Aaltonen and from the Jenny and Antti Wihuri Foundation to KW are gratefully acknowledged. We thank Dr Jorma Matikainen for running the mass spectra and Ms. Jenni Maria Petäjistö for the skilful laboratory assistance.
References
-
Cornwell, T.; Cohick, W.; Raskin, I. Phytochemistry 2004, 65, 995–1016. doi:10.1016/j.phytochem.2004.03.005
Return to citation in text: [1] [2] -
Cos, P.; De Bruyne, T.; Apers, S.; Van den Berghe, D.; Pieters, L.; Vlietinck, A. J. Planta Med. 2003, 69, 589–599. doi:10.1055/s-2003-41122
Return to citation in text: [1] [2] -
Duncan, A. M.; Phipps, W. R.; Kurzer, M. S. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 253–271. doi:10.1016/S1521-690X(02)00103-3
Return to citation in text: [1] [2] -
Bezuidenhoudt, B. C. B.; Brandt, E. V.; Roux, D. G. J. Chem. Soc., Perkin Trans. 1 1981, 263–269. doi:10.1039/p19810000263
Return to citation in text: [1] -
Jain, A. C.; Mehta, A. J. Chem. Soc., Perkin Trans. 1 1986, 215–220. doi:10.1039/p19860000215
Return to citation in text: [1] -
Shih, T. L.; Wyvratt, M. J.; Mrozik, H. J. Org. Chem. 1987, 52, 2029–2033. doi:10.1021/jo00386a024
Return to citation in text: [1] -
Liepa, A. J. Aust. J. Chem. 1984, 37, 2545–2558.
Return to citation in text: [1] [2] [3] -
Gopal, D.; Rajagopalan, K. Indian J. Chem. 1987, 26B, 401.
Return to citation in text: [1] -
Szabó, V.; Antal, E. Tetrahedron Lett. 1973, 19, 1659–1662. doi:10.1016/S0040-4039(01)96021-6
Return to citation in text: [1] -
Szabó, V.; Antal, E. Acta Chim. Acad. Sci. Hung. 1976, 90, 381–393.
Return to citation in text: [1] [2] [3] -
Jensen, N. P.; Brown, R. D.; Schmitt, S. M.; Windholz, T. B.; Patchett, A. A. J. Org. Chem. 1972, 37, 1639–1647. doi:10.1021/jo00975a040
Return to citation in text: [1] -
Gannon, W. F.; House, H. O. Org. Synth. 1960, 40, 14–15.
Return to citation in text: [1] -
Danishefsky, S.; Kerwin, J. F.; Kobayashi, S. J. Am. Chem. Soc. 1982, 104, 358–360. doi:10.1021/ja00365a095
Return to citation in text: [1] -
Kende, A. S.; Benechie, M.; Curran, D. P.; Fludzinski, P.; Swenson, W.; Clardy, J. Tetrahedron Lett. 1979, 20, 4513–4516. doi:10.1016/S0040-4039(01)86636-3
Return to citation in text: [1] -
Denmark, S. E.; Habermas, K. L.; Hite, G. A. Helv. Chim. Acta 1988, 71, 168–194. doi:10.1002/hlca.19880710120
Return to citation in text: [1] -
Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Adlercreutz, H.; Musey, P. I.; Fotsis, T.; Bannwart, C.; Wähälä, K.; Mäkelä, T.; Brunow, G.; Hase, T. Clin. Chim. Acta 1986, 158, 147–154. doi:10.1016/0009-8981(86)90230-5
Return to citation in text: [1] -
Luk, K.-C.; Stern, L.; Weigele, M. J. Nat. Prod. 1983, 46, 852–861. doi:10.1021/np50030a005
Return to citation in text: [1] -
Antus, S.; Gottsegen, Á.; Kolonits, P.; Nógrádi, M. Liebigs Ann. Chem. 1986, 2179–2181.
Return to citation in text: [1] -
Wähälä, K.; Valo, T.; Brunow, G.; Hase, T. Finn. Chem. Lett. 1989, 16, 79–83.
Return to citation in text: [1] [2] -
Antus, S.; Gottsegen, Á.; Nógrádi, M. Synthesis 1981, 574–576. doi:10.1055/s-1981-29535
Return to citation in text: [1] [2] -
Antus, S.; Gottsegen, Á.; Kolonits, P.; Nagy, Z.; Nógrádi, M.; Vermes, B. J. Chem. Soc., Perkin Trans. 1 1982, 1389–1394. doi:10.1039/p19820001389
Return to citation in text: [1] -
Májor, Á.; Nógrádi, M.; Vermes, B.; Kajtár-Peredy, M. Liebigs Ann. Chem. 1988, 555–558.
Return to citation in text: [1] -
Jain, A. C.; Kumar, A.; Sharma, N. K. Indian J. Chem. 1991, 30B, 290–291.
Return to citation in text: [1] -
Yamaguchi, S.; Ito, S.; Nakamura, A.; Inoue, N. Bull. Chem. Soc. Jpn. 1965, 38, 2187–2189. doi:10.1246/bcsj.38.2187
Return to citation in text: [1] [2] -
Anjaneylu, A. S. R.; Sri Krishna, C.; Ramachandra Row, L. Tetrahedron 1965, 21, 2677–2681. doi:10.1016/S0040-4020(01)93923-7
Return to citation in text: [1] [2] -
Badran, M. M.; El-Saba, H. M. Egypt. J. Pharm. Sci. 1991, 32, 149–155.
Return to citation in text: [1] -
Thakar, G. P.; Janaki, N.; Subba Rao, B. C. Indian J. Chem. 1965, 74–77.
Return to citation in text: [1] [2] -
Chidiak, H.; Kirkiacharian, S. Arm. Khim. Zh. 1996, 49, 94–104.
Return to citation in text: [1] [2] -
Inoue, N. Bull. Chem. Soc. Jpn. 1964, 37, 601–606. doi:10.1246/bcsj.37.601
-
Szabó, V.; Borbély, J.; Antal, E. Acta Chim. Acad. Sci. Hung. 1979, 102, 51–57.
-
Wähälä, K.; Koskimies, J. K.; Mesilaakso, M.; Salakka, A. K.; Leino, T. K.; Adlercreutz, H. J. Org. Chem. 1997, 62, 7690–7693. doi:10.1021/jo970892u
Return to citation in text: [1] -
Wähälä, K.; Salakka, A.; Adlercreutz, H. Proc. Soc. Exp. Biol. Med. 1998, 217, 293–299.
-
Pihlaja, K.; Tähtinen, P.; Klika, K. D.; Jokela, T.; Salakka, A.; Wähälä, K. J. Org. Chem. 2003, 68, 6864–6869. doi:10.1021/jo0301200
Return to citation in text: [1] [2] -
Anjaneyulu, A. S. R.; Rao, M. G.; Row, L. R.; Krishna, C. S. Tetrahedron Lett. 1966, 7, 3199–3202. doi:10.1016/S0040-4039(01)99937-X
Return to citation in text: [1] -
Anjaneyulu, A. S. R.; Krishna, C. S.; Row, L. R. Bull. Natl. Inst. Sci. India 1965, 118.
Return to citation in text: [1] -
Inoue, N.; Yamaguchi, S.; Fujiwara, S. Bull. Chem. Soc. Jpn. 1964, 37, 588–600. doi:10.1246/bcsj.37.588
Return to citation in text: [1] -
Yamaguchi, S.; Ito, S.; Suzuki, I.; Inoue, N. Bull. Chem. Soc. Jpn. 1968, 41, 2073. doi:10.1246/bcsj.41.2073
Return to citation in text: [1] -
Gomis, M.; Kirkiacharian, B. S. Tetrahedron 1990, 46, 1849–1858. doi:10.1016/S0040-4020(01)89754-4
Return to citation in text: [1] -
Tsukayama, M.; Kawamura, Y.; Tamaki, H.; Kubo, T.; Horie, T. Bull. Chem. Soc. Jpn. 1989, 62, 826–832. doi:10.1246/bcsj.62.826
Return to citation in text: [1] -
Bradbury, R. B.; White, D. E. J. Chem. Soc. 1953, 871–876. doi:10.1039/jr9530000871
Return to citation in text: [1] -
Shoukry, M. M.; Darwish, N. A.; Morsi, M. A. Gazz. Chim. Ital. 1982, 112, 289–291.
Return to citation in text: [1] -
Salakka, A.; Wähälä, K. J. Chem. Soc., Perkin Trans. 1 1999, 2601–2604. doi:10.1039/a904946k
Return to citation in text: [1] [2] [3] -
Vermes, B.; Antus, S.; Gottsegen, Á.; Nógrádi, M. Liebigs Ann. Chem. 1983, 2034–2037.
Return to citation in text: [1] -
Bulut, M. Chim. Acta Turc. 1991, 19, 17–26.
Return to citation in text: [1] -
Grese, T. A.; Pennington, L. D. Tetrahedron Lett. 1995, 36, 8913–8916. doi:10.1016/0040-4039(95)01916-6
Return to citation in text: [1] -
Verma, P.; Singh, S.; Dikshit, D. K.; Ray, S. Synthesis 1988, 68–70. doi:10.1055/s-1988-27468
Return to citation in text: [1] -
Liepa, A. J. Aust. J. Chem. 1981, 34, 2647–2655.
Return to citation in text: [1] -
Bouvier, P.; Adrieux, J.; Cunha, H.; Molho, D. Bull. Soc. Chim. Fr. 1977, 1187–1194.
Return to citation in text: [1] -
Deschamps-Vallet, C.; Ilotse, J.-B.; Meyer-Dayan, M. Tetrahedron Lett. 1983, 24, 3993–3996. doi:10.1016/S0040-4039(00)88245-3
Return to citation in text: [1] -
Dudley, K. H.; Miller, H. W.; Corley, R. C.; Wall, M. E. J. Org. Chem. 1967, 32, 2317–2321. doi:10.1021/jo01282a049
Return to citation in text: [1] [2] -
Diaz, P.; Gendre, F.; Stella, L.; Charpentier, B. Tetrahedron 1998, 54, 4579–4590. doi:10.1016/S0040-4020(98)00169-0
Return to citation in text: [1] [2] -
Baranton, F.; Fontaine, G.; Maitte, P. Bull. Soc. Chim. Fr. 1968, 4203–4208.
Return to citation in text: [1] -
Miyase, T.; Sano, M.; Yoshino, K.; Nonaka, K. Phytochemistry 1999, 52, 311–319. doi:10.1016/S0031-9422(99)00194-6
Return to citation in text: [1] -
Szabó, S.; Antal, E. Magy. Kem. Foly. 1976, 10, 474–477.
Chem. Abstr. 1977, 86, 55118a.
Return to citation in text: [1] -
Kelly, G. E.; Nelson, C.; Waring, M. A.; Joannou, G. E.; Reeder, A. Y. Clin. Chim. Acta 1993, 223, 9–22. doi:10.1016/0009-8981(93)90058-C
Return to citation in text: [1] -
Pollard, M.; Wolter, W. Prostate 2000, 45, 101–105. doi:10.1002/1097-0045(20001001)45:2<101::AID-PROS3>3.0.CO;2-P
Return to citation in text: [1] -
Lamartiniere, C. A. Am. J. Clin. Nutr. 2000, 71, 1705S–1707S.
Return to citation in text: [1] -
Wiseman, H. Expert Opin. Invest. Drugs 2000, 9, 1829–1840. doi:10.1517/13543784.9.8.1829
Return to citation in text: [1] -
Federal register 64FR57699, U.S. Food and Drug Administration, Oct 26, 1999.
Return to citation in text: [1] -
Adlercreutz, H.; Fotsis, T.; Lampe, J.; Wähälä, K.; Mäkelä, T.; Brunow, G.; Hase, T. Scand. J. Clin. Lab. Invest. 1993, 215, 5S–18S.
Return to citation in text: [1] -
Gensler, W. J.; Johnson, F.; Sloan, A. D. B. J. Am. Chem. Soc. 1960, 82, 6074–6081. doi:10.1021/ja01508a026
-
Fisher, G. B.; Harrison, J.; Fuller, J. C.; Goralski, C. T.; Singaram, B. Tetrahedron Lett. 1992, 33, 4533–4536. doi:10.1016/S0040-4039(00)61305-9
-
Olah, G. A.; Wang, Q.; Prakash, G. K. S. Synlett 1992, 647–650. doi:10.1055/s-1992-21443
-
Ibrahim, A.-R.; Abul-Hajj, Y. J. J. Nat. Prod. 1990, 53, 644–656. doi:10.1021/np50072a011
-
Osawa, K.; Yasuda, H.; Maruyama, T.; Morita, H.; Takeya, K.; Itokawa, H. Chem. Pharm. Bull. 1992, 40, 2970–2974.
| 54. | Miyase, T.; Sano, M.; Yoshino, K.; Nonaka, K. Phytochemistry 1999, 52, 311–319. doi:10.1016/S0031-9422(99)00194-6 |
| 10. | Szabó, V.; Antal, E. Acta Chim. Acad. Sci. Hung. 1976, 90, 381–393. |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 17. | Adlercreutz, H.; Musey, P. I.; Fotsis, T.; Bannwart, C.; Wähälä, K.; Mäkelä, T.; Brunow, G.; Hase, T. Clin. Chim. Acta 1986, 158, 147–154. doi:10.1016/0009-8981(86)90230-5 |
| 18. | Luk, K.-C.; Stern, L.; Weigele, M. J. Nat. Prod. 1983, 46, 852–861. doi:10.1021/np50030a005 |
| 19. | Antus, S.; Gottsegen, Á.; Kolonits, P.; Nógrádi, M. Liebigs Ann. Chem. 1986, 2179–2181. |
| 20. | Wähälä, K.; Valo, T.; Brunow, G.; Hase, T. Finn. Chem. Lett. 1989, 16, 79–83. |
| 21. | Antus, S.; Gottsegen, Á.; Nógrádi, M. Synthesis 1981, 574–576. doi:10.1055/s-1981-29535 |
| 55. |
Szabó, S.; Antal, E. Magy. Kem. Foly. 1976, 10, 474–477.
Chem. Abstr. 1977, 86, 55118a. |
| 1. | Cornwell, T.; Cohick, W.; Raskin, I. Phytochemistry 2004, 65, 995–1016. doi:10.1016/j.phytochem.2004.03.005 |
| 2. | Cos, P.; De Bruyne, T.; Apers, S.; Van den Berghe, D.; Pieters, L.; Vlietinck, A. J. Planta Med. 2003, 69, 589–599. doi:10.1055/s-2003-41122 |
| 3. | Duncan, A. M.; Phipps, W. R.; Kurzer, M. S. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 253–271. doi:10.1016/S1521-690X(02)00103-3 |
| 10. | Szabó, V.; Antal, E. Acta Chim. Acad. Sci. Hung. 1976, 90, 381–393. |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 20. | Wähälä, K.; Valo, T.; Brunow, G.; Hase, T. Finn. Chem. Lett. 1989, 16, 79–83. |
| 39. | Gomis, M.; Kirkiacharian, B. S. Tetrahedron 1990, 46, 1849–1858. doi:10.1016/S0040-4020(01)89754-4 |
| 61. | Adlercreutz, H.; Fotsis, T.; Lampe, J.; Wähälä, K.; Mäkelä, T.; Brunow, G.; Hase, T. Scand. J. Clin. Lab. Invest. 1993, 215, 5S–18S. |
| 11. | Jensen, N. P.; Brown, R. D.; Schmitt, S. M.; Windholz, T. B.; Patchett, A. A. J. Org. Chem. 1972, 37, 1639–1647. doi:10.1021/jo00975a040 |
| 12. | Gannon, W. F.; House, H. O. Org. Synth. 1960, 40, 14–15. |
| 13. | Danishefsky, S.; Kerwin, J. F.; Kobayashi, S. J. Am. Chem. Soc. 1982, 104, 358–360. doi:10.1021/ja00365a095 |
| 14. | Kende, A. S.; Benechie, M.; Curran, D. P.; Fludzinski, P.; Swenson, W.; Clardy, J. Tetrahedron Lett. 1979, 20, 4513–4516. doi:10.1016/S0040-4039(01)86636-3 |
| 15. | Denmark, S. E.; Habermas, K. L.; Hite, G. A. Helv. Chim. Acta 1988, 71, 168–194. doi:10.1002/hlca.19880710120 |
| 9. | Szabó, V.; Antal, E. Tetrahedron Lett. 1973, 19, 1659–1662. doi:10.1016/S0040-4039(01)96021-6 |
| 10. | Szabó, V.; Antal, E. Acta Chim. Acad. Sci. Hung. 1976, 90, 381–393. |
| 35. | Anjaneyulu, A. S. R.; Rao, M. G.; Row, L. R.; Krishna, C. S. Tetrahedron Lett. 1966, 7, 3199–3202. doi:10.1016/S0040-4039(01)99937-X |
| 36. | Anjaneyulu, A. S. R.; Krishna, C. S.; Row, L. R. Bull. Natl. Inst. Sci. India 1965, 118. |
| 37. | Inoue, N.; Yamaguchi, S.; Fujiwara, S. Bull. Chem. Soc. Jpn. 1964, 37, 588–600. doi:10.1246/bcsj.37.588 |
| 38. | Yamaguchi, S.; Ito, S.; Suzuki, I.; Inoue, N. Bull. Chem. Soc. Jpn. 1968, 41, 2073. doi:10.1246/bcsj.41.2073 |
| 1. | Cornwell, T.; Cohick, W.; Raskin, I. Phytochemistry 2004, 65, 995–1016. doi:10.1016/j.phytochem.2004.03.005 |
| 2. | Cos, P.; De Bruyne, T.; Apers, S.; Van den Berghe, D.; Pieters, L.; Vlietinck, A. J. Planta Med. 2003, 69, 589–599. doi:10.1055/s-2003-41122 |
| 3. | Duncan, A. M.; Phipps, W. R.; Kurzer, M. S. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 253–271. doi:10.1016/S1521-690X(02)00103-3 |
| 4. | Bezuidenhoudt, B. C. B.; Brandt, E. V.; Roux, D. G. J. Chem. Soc., Perkin Trans. 1 1981, 263–269. doi:10.1039/p19810000263 |
| 5. | Jain, A. C.; Mehta, A. J. Chem. Soc., Perkin Trans. 1 1986, 215–220. doi:10.1039/p19860000215 |
| 6. | Shih, T. L.; Wyvratt, M. J.; Mrozik, H. J. Org. Chem. 1987, 52, 2029–2033. doi:10.1021/jo00386a024 |
| 7. | Liepa, A. J. Aust. J. Chem. 1984, 37, 2545–2558. |
| 8. | Gopal, D.; Rajagopalan, K. Indian J. Chem. 1987, 26B, 401. |
| 32. | Wähälä, K.; Koskimies, J. K.; Mesilaakso, M.; Salakka, A. K.; Leino, T. K.; Adlercreutz, H. J. Org. Chem. 1997, 62, 7690–7693. doi:10.1021/jo970892u |
| 34. | Pihlaja, K.; Tähtinen, P.; Klika, K. D.; Jokela, T.; Salakka, A.; Wähälä, K. J. Org. Chem. 2003, 68, 6864–6869. doi:10.1021/jo0301200 |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 25. | Yamaguchi, S.; Ito, S.; Nakamura, A.; Inoue, N. Bull. Chem. Soc. Jpn. 1965, 38, 2187–2189. doi:10.1246/bcsj.38.2187 |
| 26. | Anjaneylu, A. S. R.; Sri Krishna, C.; Ramachandra Row, L. Tetrahedron 1965, 21, 2677–2681. doi:10.1016/S0040-4020(01)93923-7 |
| 27. | Badran, M. M.; El-Saba, H. M. Egypt. J. Pharm. Sci. 1991, 32, 149–155. |
| 28. | Thakar, G. P.; Janaki, N.; Subba Rao, B. C. Indian J. Chem. 1965, 74–77. |
| 43. | Salakka, A.; Wähälä, K. J. Chem. Soc., Perkin Trans. 1 1999, 2601–2604. doi:10.1039/a904946k |
| 25. | Yamaguchi, S.; Ito, S.; Nakamura, A.; Inoue, N. Bull. Chem. Soc. Jpn. 1965, 38, 2187–2189. doi:10.1246/bcsj.38.2187 |
| 26. | Anjaneylu, A. S. R.; Sri Krishna, C.; Ramachandra Row, L. Tetrahedron 1965, 21, 2677–2681. doi:10.1016/S0040-4020(01)93923-7 |
| 57. | Pollard, M.; Wolter, W. Prostate 2000, 45, 101–105. doi:10.1002/1097-0045(20001001)45:2<101::AID-PROS3>3.0.CO;2-P |
| 58. | Lamartiniere, C. A. Am. J. Clin. Nutr. 2000, 71, 1705S–1707S. |
| 59. | Wiseman, H. Expert Opin. Invest. Drugs 2000, 9, 1829–1840. doi:10.1517/13543784.9.8.1829 |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 56. | Kelly, G. E.; Nelson, C.; Waring, M. A.; Joannou, G. E.; Reeder, A. Y. Clin. Chim. Acta 1993, 223, 9–22. doi:10.1016/0009-8981(93)90058-C |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 21. | Antus, S.; Gottsegen, Á.; Nógrádi, M. Synthesis 1981, 574–576. doi:10.1055/s-1981-29535 |
| 22. | Antus, S.; Gottsegen, Á.; Kolonits, P.; Nagy, Z.; Nógrádi, M.; Vermes, B. J. Chem. Soc., Perkin Trans. 1 1982, 1389–1394. doi:10.1039/p19820001389 |
| 23. | Májor, Á.; Nógrádi, M.; Vermes, B.; Kajtár-Peredy, M. Liebigs Ann. Chem. 1988, 555–558. |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 29. | Chidiak, H.; Kirkiacharian, S. Arm. Khim. Zh. 1996, 49, 94–104. |
| 34. | Pihlaja, K.; Tähtinen, P.; Klika, K. D.; Jokela, T.; Salakka, A.; Wähälä, K. J. Org. Chem. 2003, 68, 6864–6869. doi:10.1021/jo0301200 |
| 43. | Salakka, A.; Wähälä, K. J. Chem. Soc., Perkin Trans. 1 1999, 2601–2604. doi:10.1039/a904946k |
| 42. | Shoukry, M. M.; Darwish, N. A.; Morsi, M. A. Gazz. Chim. Ital. 1982, 112, 289–291. |
| 40. | Tsukayama, M.; Kawamura, Y.; Tamaki, H.; Kubo, T.; Horie, T. Bull. Chem. Soc. Jpn. 1989, 62, 826–832. doi:10.1246/bcsj.62.826 |
| 41. | Bradbury, R. B.; White, D. E. J. Chem. Soc. 1953, 871–876. doi:10.1039/jr9530000871 |
| 52. | Diaz, P.; Gendre, F.; Stella, L.; Charpentier, B. Tetrahedron 1998, 54, 4579–4590. doi:10.1016/S0040-4020(98)00169-0 |
| 53. | Baranton, F.; Fontaine, G.; Maitte, P. Bull. Soc. Chim. Fr. 1968, 4203–4208. |
| 7. | Liepa, A. J. Aust. J. Chem. 1984, 37, 2545–2558. |
| 51. | Dudley, K. H.; Miller, H. W.; Corley, R. C.; Wall, M. E. J. Org. Chem. 1967, 32, 2317–2321. doi:10.1021/jo01282a049 |
| 52. | Diaz, P.; Gendre, F.; Stella, L.; Charpentier, B. Tetrahedron 1998, 54, 4579–4590. doi:10.1016/S0040-4020(98)00169-0 |
| 48. | Liepa, A. J. Aust. J. Chem. 1981, 34, 2647–2655. |
| 49. | Bouvier, P.; Adrieux, J.; Cunha, H.; Molho, D. Bull. Soc. Chim. Fr. 1977, 1187–1194. |
| 50. | Deschamps-Vallet, C.; Ilotse, J.-B.; Meyer-Dayan, M. Tetrahedron Lett. 1983, 24, 3993–3996. doi:10.1016/S0040-4039(00)88245-3 |
| 51. | Dudley, K. H.; Miller, H. W.; Corley, R. C.; Wall, M. E. J. Org. Chem. 1967, 32, 2317–2321. doi:10.1021/jo01282a049 |
| 45. | Bulut, M. Chim. Acta Turc. 1991, 19, 17–26. |
| 46. | Grese, T. A.; Pennington, L. D. Tetrahedron Lett. 1995, 36, 8913–8916. doi:10.1016/0040-4039(95)01916-6 |
| 47. | Verma, P.; Singh, S.; Dikshit, D. K.; Ray, S. Synthesis 1988, 68–70. doi:10.1055/s-1988-27468 |
| 43. | Salakka, A.; Wähälä, K. J. Chem. Soc., Perkin Trans. 1 1999, 2601–2604. doi:10.1039/a904946k |
| 16. | Süsse, M.; Johne, S.; Hesse, M. Helv. Chim. Acta 1992, 75, 457–470. doi:10.1002/hlca.19920750205 |
| 44. | Vermes, B.; Antus, S.; Gottsegen, Á.; Nógrádi, M. Liebigs Ann. Chem. 1983, 2034–2037. |
© 2006 Salakka et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)