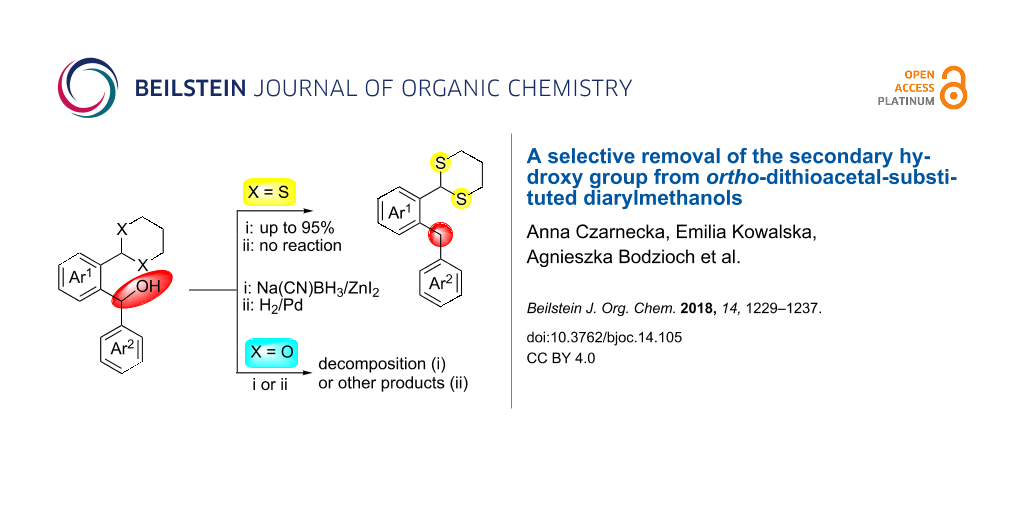
Abstract
We present a successful deoxygenation reaction of ortho-1,3-dithianylaryl(aryl)methanols which enables a selective removal of the secondary hydroxy group in presence of the 1,3-dithianyl moiety under reductive conditions. This reaction proceeds well with ZnI2/Na(CN)BH3 in dichloroethane or benzene for both unsubstituted and substituted aryls (by electron-rich groups). This is leading to formyl-protected diarylmethanes with potential application in the synthesis of new pharmaceuticals and optoelectronic materials. This synthetic approach gives an access to a wide variety of functionalized ortho-1,3-dithianylaryl(aryl)methanes in 26–95% yields and is recommended for the substrates containing sulfur atoms, for which transition metal-induced reactions fail.

Graphical Abstract
Introduction
In last decades, diarylmethanes, such as I–IV and cyclic compounds possessing a diarylmethyl motif, like podophyllotoxin (V) and lasofoxifene (VI), have been a subject of interest as bioactive molecules (Figure 1) [1]. The diarylmethane derivatives, isolated from natural sources, showed antibacterial [2], antiestrogenic and antitumorigenic (I) [3-5] or vasodilating activities (II) [6]. Other ones have been reported as inhibitors of HIV-1 integrase and viral replication in cells [7] or antitubercular agents (III) [8,9]. Various diarylmethane-based molecules, like tolterodine (IV) [10], podophyllotoxin (V) [11] and lasofoxifene (VI) [12] have been used in the treatment of overactive bladder, external genital warts and osteoporosis (Figure 1). The diarylmethyl motif also plays a pivotal role in supramolecular architectures of calixarenes [13] and orthocyclophanes [14].
![[1860-5397-14-105-1]](/bjoc/content/figures/1860-5397-14-105-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of biologically active diarylmethanes and commercially available pharmaceuticals based on the diarylmethyl moiety.
Figure 1: Structures of biologically active diarylmethanes and commercially available pharmaceuticals based o...
A special, ortho-carbonyl-substituted diarylmethyl scaffold is present in the key substrates for aromatic cyclodehydration, also known as the Bradsher reaction [15], which leads to fused, polycyclic aromatic hydrocarbons [16-19]. This type of reaction found applications in synthesis of organic, optoelectronic materials [20-22].
Metal-catalyzed cross-coupling reactions (e.g., Suzuki–Miyaura, Stille, Kumada, Hiyama or Negishi couplings) play a key role in the preparation of diarylmethanes. However, the reactions involving sulfur-containing moieties have not been reported yet (Scheme 1) [1]. It is noteworthy that under certain conditions, the Pd-catalyzed cross-coupling reactions of substrates containing an 1,3-dithiane moiety are feasible, like in the case of the 2-arylation of 2-aryl-substituted 1,3-dithianes. However, in the case of 2-benzyl-substituted 1,3-dithianes, a tandem elimination/1,3-dithiane ring opening followed by a Pd-catalyzed C−S bond formation was observed instead [23].
Diarylmethanes were also obtained in the Friedel–Crafts reactions of arenes with primary benzyl alcohols, aryl acetals, and benzyl esters [1]. Benzyl fluorides (in 1,1,1,3,3,3-hexafluoroisopropanol in the presence of a catalytic amount of trifluoroacetic acid [24]) as well as benzotrifluorides (employed in hydrodefluorinative Friedel–Crafts alkylations catalyzed by alumenium ions [25]) were interesting, alternative substrates for alcohols, acetals and esters.
A large group of synthetic procedures for the synthesis of diarylmethanes are deoxygenations of secondary diarylmethyl alcohols with hydride sources. These reactions require a preliminary C–OH bond activation by Brønsted or Lewis acids. Several reagent systems have recently been employed to achieve this goal, including: NaBH4–CF3COOH [26], ZnI2–NaBH3CN [27], HI–Pred [28], H3PO2–I2 [29,30], Mo(CO)6-Lawesson’s reagent [31], PBr3 [32], BF3·Et2O–dibutyl ether [33] and silanes (Si–H) in the presence of various Lewis acids: B(C6F5)3 [34-36], InCl3 [37,38], H3[PW12O40]×nH2O [39], Ca(NTf2)2 [40], Bi(OTf)3 [41], Sn(IV)-montmorillonite [42] and PdCl2 [43]. It is interesting that Seto et al. reported that Et3SiH–CF3COOH, TMSCl/NaI failed to reduce secondary OH groups in diheteroaromatic systems and the reduction with H2/Pd/C-ZnBr2 was very slow [32]. On the other hand, some rigid diarylmethanols were successfully reduced to benzo[b]indeno[2,1-d]thiophenes using the Et3SiH–CF3COOH system which was reported in the patent literature [44].
Benzophenones were also reduced to diarylmethanes using supercritical iPrOH at 350 °C [1], BF3·OEt2/H2O [33] and PhSiH3/MoCl2O2(H2O)2 [45].
The reductive deoxygenation reactions of diarylmethanols proceed via carbocationic species, which are formed by protonation or complexation of alcohols by Brønsted or Lewis acids. Therefore, diarylmethanols bearing electron-donating substituents (EDG = methyl, methoxy) are reduced much faster than diarylmethanols with electron-withdrawing groups (EWG = CF3, C(O)OR) on (hetero)aryl moieties. Consequently, dramatically decreased yields have been observed in these cases [32,33]. It has also been reported that ortho-substituents on aryl moieties give lower yields in the reduction processes [33]. Moreover, the OH reduction under acidic conditions cannot be carried out in the presence of other sensitive functional groups. For instance, ether cleavage and dehalogenation (I but not Cl and Br) are common, side reactions due to either the high acidity which is necessary to generate carbocationic species or due to the use of strongly reducing reaction conditions [28-30,46-52]. In this context, a serious challenge is still the selective and direct removal of the OH group from alcohols Ar2CH(OH) without excessive, side reactions. Especially in the presence of other functionalities such as, for instance, when a reductively sensitive ortho-1,3-dithianyl group is attached to one of the aryl moieties, side products are to be expected. This task is additionally difficult to accomplish due to a chemical similarity of oxygen and sulfur, two neighboring heteroatoms from the main group VI of the periodic table. A longer atomic radius of sulfur than oxygen should make the C–S bond weaker and more reactive than the C–O bond and consequently the reduction of the former should a priori occur preferentially [53-55].
According to the best of our knowledge, there are no literature reports concerning selective reductions of dibenzylic hydroxy groups in the presence of ortho-acetal or ortho-thioacetal functions. Such reductions may give an access to new series of ortho-formyl-protected diarylmethanes as well as their formyl-modified derivatives, obtained after deprotection of the 1,3-dithianyl group with one of the available methods [56].
Herein, we present the first example of a selective and efficient reduction of ortho-1,3-dithianylaryl(aryl)methanols leading to ortho-1,3-dithianylaryl(aryl)methanes using the ZnI2-Na(CN)BH3 reductive system (Scheme 1). The use of zinc iodide is critical in this system. It was used for the first time in dichloroethane by Lau et al. to reduce aryl ketones, aldehydes, benzylic, allylic and tertiary alcohols, including the first example of a diarylmethanol (benzhydrol) reduction to diarylmethane (diphenylmethane) [27].
![[1860-5397-14-105-i1]](/bjoc/content/inline/1860-5397-14-105-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Various synthetic approaches to diarylmethanols (literature review and this work).
Scheme 1: Various synthetic approaches to diarylmethanols (literature review and this work).
Results and Discussion
The synthesis of ortho-1,3-dithianylaryl(aryl)methanols 5 and 6, as key substrates for the OH reduction, has been realized according to the procedure shown in Scheme 2 including: 1) protection of the formyl group in ortho-bromoaldehydes 1 and 2 with 1,3-propanedithiol, 2) the Br/Li exchange reaction in the resulting ortho-bromo-1,3-dithianes 3 and 4 with n-BuLi followed by condensation with a second (hetero)aromatic aldehyde Ar2-CHO.
![[1860-5397-14-105-i2]](/bjoc/content/inline/1860-5397-14-105-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A general strategy for the synthesis of ortho-1,3-dithianylaryl(aryl)methanols 5 and 6, and their reduction to the corresponding ortho-1,3-dithianylaryl(aryl)methanes.
Scheme 2: A general strategy for the synthesis of ortho-1,3-dithianylaryl(aryl)methanols 5 and 6, and their r...
The reduction of ortho-1,3-dithianylaryl(aryl)methanols 5 and 6 to ortho-1,3-dithianylaryl(aryl)methanes 7 and 8 has been carried out using Lau’s procedure with ZnI2 (1.5 equiv) and Na(CN)BH3 (7 equiv) in dichloroethane (DCE) and a solvent-modified protocol employing the same reagents in benzene. In literature, only few applications of the ZnI2-Na(CN)BH3 system in DCE have been described for reduction of diarylmethanols that do not contain acid-sensitive, formyl protecting, acetal or thioacetal groups [20,27,57-60] and for the reduction of aromatic aldehydes and ketones [28]. The bromo function in such diarylmethanols [60], as well as Cl, Br, C(O)OMe, MeO, MeS and NO2 groups in aromatic aldehydes and ketones, being reduced to methyl and methylene groups, respectively [27], are tolerated by this reagent system.
The use of other procedures for deoxygenation of alcohols 5d, 6a and 6b with metal hydride donors, such as NaBH4 and LiAlH4 in various combinations with ZnI2 and AlCl3 failed [61-63]. For instance, the reaction with NaBH4 itself and NaBH4/ZnI2/THF at room temperature and reflux only recovered the substrates. The same result was obtained at room temperature with NaBH4/AlCl3/THF, while at reflux the whole substrate was consumed and three unidentified products were formed lacking the SCHS and ArCH2Ar characteristic signals in 1H NMR spectra. With NaBH4/CF3COOH/rt, the substrate underwent decomposition.
In our case, the use of the Lau’s procedure in DCE for the reductive deoxygenation of ortho-1,3-dithianyl-substituted alcohols 5a–h containing unsubstituted Ar1 phenyl ring and electron-donating Ar2 groups, gave diarylmethanes 7a–h in 26–95% yields (Table 1, entries 1–9).
Table 1: A selective reduction of ortho-1,3-dithianylaryl(aryl)methanols to ortho-1,3-dithianylaryl(aryl)methanes using the ZnI2/Na(CN)BH3 reducing system.
| entry | substrate | solvent | time [h] | temperature [°C] | product | yield |
|---|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-105-i4.svg?max-width=637&scale=1.0)
5a |
DCE | 24 | rt |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-105-i5.svg?max-width=637&scale=1.0)
7a |
95 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-105-i6.svg?max-width=637&scale=1.0)
5b |
DCE | 72 | rt |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-105-i7.svg?max-width=637&scale=1.0)
7b |
95a |
| 3 | DCE | 72 | reflux | 85 | ||
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-105-i8.svg?max-width=637&scale=1.0)
5c |
DCE | 72 | rt |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-105-i9.svg?max-width=637&scale=1.0)
7c |
70 |
| 5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-105-i10.svg?max-width=637&scale=1.0)
5d |
DCE | 2 | rt |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-105-i11.svg?max-width=637&scale=1.0)
7d |
95 |
| 6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-105-i12.svg?max-width=637&scale=1.0)
5e |
DCE | 2 | rt |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-105-i13.svg?max-width=637&scale=1.0)
7e |
26 |
| 7 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-105-i14.svg?max-width=637&scale=1.0)
5f |
DCE | 1 | rt |
![[Graphic 12]](/bjoc/content/inline/1860-5397-14-105-i15.svg?max-width=637&scale=1.0)
7f |
95 |
| 8 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-14-105-i16.svg?max-width=637&scale=1.0)
5g |
DCE | 3 | rt |
![[Graphic 14]](/bjoc/content/inline/1860-5397-14-105-i17.svg?max-width=637&scale=1.0)
7g |
59 |
| 9 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-14-105-i18.svg?max-width=637&scale=1.0)
5h |
DCE | 5 | rt |
![[Graphic 16]](/bjoc/content/inline/1860-5397-14-105-i19.svg?max-width=637&scale=1.0)
7h |
64 |
| 10 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-14-105-i20.svg?max-width=637&scale=1.0)
6a |
C6H6 | 24 | rt |
![[Graphic 18]](/bjoc/content/inline/1860-5397-14-105-i21.svg?max-width=637&scale=1.0)
8a |
95 |
| 11 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-14-105-i22.svg?max-width=637&scale=1.0)
6b |
C6H6 | 72 | rt |
![[Graphic 20]](/bjoc/content/inline/1860-5397-14-105-i23.svg?max-width=637&scale=1.0)
8b |
95a |
| 12 | C6H6 | 24 | reflux | 60 | ||
aYield calculated based on the consumed substrate.
Surprisingly, the reduction of the Ar1 piperonyl series 6a,b failed under the same reaction conditions and only the substrates could be isolated. In this case, a replacement of DCE by benzene resulted in a successful formation of the desired diarylmethanes 8a,b in 95% yield (Table 1, entries 10–12). In addition, diarylmethanols 5b and 6b with the bulky group Ar2 = 3,4,5-trimethoxyphenyl required refluxing in the relevant solvent (benzene or DCE) to complete the reduction (Table 1, entries 3 and 12). At room temperature, only 40% of the substrates underwent conversion to diarylmethanes 7b and 8b due to their low solubility at this temperature. Attempts to synthesize diarylmethanols 6 with electron-withdrawing groups (Ar2 = p-NO2, p-CF3) were also unsuccessful. They proved to be unstable: the reaction of lithiated 4 with p-nitro and p-trifluoromethylbenzaldehyde gave five products in the crude reaction mixture and even more complex mixtures of products after silica gel column chromatography. We also tried to remove the OH group using the Pd-catalytic hydrogenolysis (Scheme 3).
![[1860-5397-14-105-i3]](/bjoc/content/inline/1860-5397-14-105-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Attempts of the OH removal in ortho-1,3-dithianyl- 6b and ortho-1,3-dioxanylaryl(aryl)methanols 9 using the Pd-catalyzed hydrogenolysis reaction.
Scheme 3: Attempts of the OH removal in ortho-1,3-dithianyl- 6b and ortho-1,3-dioxanylaryl(aryl)methanols 9 u...
In case of 6b (X = S), no reaction occurred and only the substrate was recovered, most probably due to poisoning of the catalyst by the 1,3-dithianyl sulfur atoms. However, the successful catalytic hydrogenolysis (5% Pd/C) of diphenylmethanol to diphenylmethane, described in literature [64], encouraged us to try the Pd-catalyzed reduction of the oxygen analog 9 (X = O) as a representative of the ortho-1,3-dioxanyl series. In this case, instead of the expected ortho-1,3-dioxanyl diarylmethane analog of 8b, 1,3-dihydroisobenzofuran 10 was obtained in 90% yield, within 30 min, as a result of preferential deacetalization over the dibenzylic OH deoxygenation. This is followed by cyclization and final deoxygenation of the secondary monobenzylic OH group in the resulting lactol. This product is known in literature and was obtained by hydrogenolysis or pyrolysis of the corresponding ortho-1,3-dioxolanyl [65] and ortho-hydroxymethyl [66] substituted diarylmethanols, respectively. It is worth mentioning that the reduction of 9 with ZnI2–Na(CN)BH3 caused decomposition of the starting material. The described reduction process involving 1,3-dithianyl derivatives is facilitated by oxophilic zinc which preferentially binds to the OH oxygen atoms (bond dissociation energies for Zn–O and Zn–S are 284 and 205 kJ/mol, respectively) [67]. The weakened C–O bond is thus more susceptible to the borohydride attack to produce the corresponding diarylmethanes. Most probably, electron-donating groups on aryls facilitate both Zn complexation and stabilization of the intermediate carbocationic species formed.
Conclusion
In summary, a new example of the selective functional group transformation of diarylmethanols (Ar1Ar2CH(OH)) to diarylmethanes (Ar1Ar2CH2) has been performed. It is important for the sulfur-containing substrates for which metal-catalyzed cross-coupling reactions fail. The dibenzylic OH group has been preferentially reduced with the ZnI2/Na(CN)BH3 system in the presence of an ortho-dithioacetal moiety to give ortho-1,3-dithianylaryl(aryl)methanes in 26–95% yields under mild reaction conditions. Thus, the ortho-1,3-dithianyl moiety joins other functionalities tolerated by this reagent system [47]. Since analogous 1,3-dioxanyl derivatives, in our hands, decomposed or led to other products during the attempted catalytic hydrogenolysis, the elaborated protocol is the only one up to date that enables synthesis of ortho-formyl-protected diarylmethane derivatives under reductive conditions. The latter may be used as key substrates both for the synthesis of biologically active diarylmethanes or for the synthesis of (hetero)acenes via the Bradsher protocol.
Supporting Information
| Supporting Information File 1: General experimental information, characterization data and copies of 1H, 13C NMR spectra. | ||
| Format: PDF | Size: 3.9 MB | Download |
References
-
Mondal, S.; Panda, G. RSC Adv. 2014, 4, 28317–28358. doi:10.1039/C4RA01341G
Return to citation in text: [1] [2] [3] [4] -
Sun, H. H.; Paul, V. J.; Fencial, W. Phytochemistry 1983, 22, 743–745. doi:10.1016/S0031-9422(00)86974-5
Return to citation in text: [1] -
Chen, I.; McDougal, A.; Wang, F.; Safe, S. Carcinogenesis 1998, 19, 1631–1639. doi:10.1093/carcin/19.9.1631
Return to citation in text: [1] -
McDougal, A.; Gupta, M. S.; Ramamoorthy, K.; Sun, G.; Safe, S. H. Cancer Lett. 2000, 151, 169–179. doi:10.1016/S0304-3835(99)00406-1
Return to citation in text: [1] -
Abdelrahim, M.; Newman, K.; Vanderlaag, K.; Samudio, I.; Safe, S. Carcinogenesis 2006, 27, 717–728. doi:10.1093/carcin/bgi270
Return to citation in text: [1] -
Li, H.; Zhu, X.; Yao, G.; Wang, Z. J. Surg. Res. 2015, 195, 271–276. doi:10.1016/j.jss.2015.01.016
Return to citation in text: [1] -
Wai, J. S.; Egbertson, M. S.; Payne, L. S.; Fisher, T. E.; Embrey, M. W.; Tran, L. O.; Melamed, J. Y.; Langford, H. M.; Guare, J. P.; Zhuang, L.; Grey, V. E.; Vacca, J. P.; Holloway, M. K.; Naylor-Olsen, A. M.; Hazuda, D. J.; Felock, P. J.; Wolfe, A. L.; Stillmock, K. A.; Schleif, W. A.; Gabryelski, L. J.; Young, S. D. J. Med. Chem. 2000, 43, 4923–4926. doi:10.1021/jm000176b
Return to citation in text: [1] -
Panda, G.; Parai, M. K.; Das, S. K.; Shagufta; Sinha, M.; Chaturvedi, V.; Srivastava, A. K.; Manju, Y. S.; Gaikwad, A. N.; Sinha, S. Eur. J. Med. Chem. 2007, 42, 410–419. doi:10.1016/j.ejmech.2006.09.020
Return to citation in text: [1] -
Upadhayaya, R. S.; Vandavasi, J. K.; Vasireddy, N. R.; Sharma, V.; Dixit, S. S.; Chattopadhyaya, J. Bioorg. Med. Chem. 2009, 17, 2830–2841. doi:10.1016/j.bmc.2009.02.026
Return to citation in text: [1] -
Kerrebroeck, P. V.; Kreder, K.; Jonas, U.; Zinner, N.; Wein, A. Urology 2001, 57, 414–421. doi:10.1016/S0090-4295(00)01113-4
Return to citation in text: [1] -
Canel, C.; Moraes, R. M.; Dayan, F. E.; Ferreira, D. Phytochemistry 2000, 54, 115–120. doi:10.1016/S0031-9422(00)00094-7
Return to citation in text: [1] -
Gennari, L.; Merlotti, D.; Martini, G.; Nuti, R. Expert Opin. Invest. Drugs 2006, 15, 1091–1103. doi:10.1517/13543784.15.9.1091
Return to citation in text: [1] -
Conn, M. M.; Rebek, J., Jr. Chem. Rev. 1997, 97, 1647–1668. doi:10.1021/cr9603800
Return to citation in text: [1] -
Lee, W. Y.; Park, C. H.; Kim, H.-J.; Kim, S. J. Org. Chem. 1994, 59, 878–884. doi:10.1021/jo00083a033
Return to citation in text: [1] -
Bradsher, C. K. J. Am. Chem. Soc. 1940, 62, 486–488. doi:10.1021/ja01860a006
Return to citation in text: [1] -
Bradsher, C. K. Chem. Rev. 1987, 87, 1277–1297. doi:10.1021/cr00082a001
Return to citation in text: [1] -
Bodzioch, A.; Kowalska, E.; Skalik, J.; Bałczewski, P. Chem. Heterocycl. Compd. 2017, 53, 11–20. doi:10.1007/s10593-017-2015-y
Return to citation in text: [1] -
Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E.; Jeszka, J. K.; Uznański, P.; Kania, S.; Kuliński, J.; Bałczewski, P. Synfacts 2012, 8, 619. doi:10.1055/s-0031-1291065
Return to citation in text: [1] -
Bałczewski, P.; Kowalska, E.; Skalik, J. 10-Thiosubstituted pentahydroxyanthracene derivatives, a method of their preparation and intermediate compounds. Eur. Pat. Appl. EP 2886539 A1 20150624, June 24, 2015.
Return to citation in text: [1] -
Park, J.-I.; Chung, J. W.; Kim, J.-Y.; Lee, J.; Jung, J. Y.; Koo, B.; Lee, B.-L.; Lee, S. W.; Jin, Y. W.; Lee, S. Y. J. Am. Chem. Soc. 2015, 137, 12175–12178. doi:10.1021/jacs.5b01108
Return to citation in text: [1] [2] -
Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E.; Jeszka, J. K.; Uznański, P.; Kania, S.; Kuliński, J.; Bałczewski, P. Chem. – Eur. J. 2012, 18, 4866–4877. doi:10.1002/chem.201101909
Return to citation in text: [1] -
Bałczewski, P.; Skalik, J.; Uznański, P.; Guziejewski, D.; Ciesielski, W. RSC Adv. 2015, 5, 24700–24704. doi:10.1039/C4RA14071K
Return to citation in text: [1] -
Dockrey, S. A. B.; Makepeace, A. K.; Schmink, J. R. Org. Lett. 2014, 16, 4730–4733. doi:10.1021/ol502428h
Return to citation in text: [1] -
Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003
Return to citation in text: [1] -
Forster, F.; Metsänen, T. T.; Irran, E.; Hrobárik, P.; Oestreich, M. J. Am. Chem. Soc. 2017, 139, 16334–16342. doi:10.1021/jacs.7b09444
Return to citation in text: [1] -
Gribble, G. W.; Leese, R. M.; Evans, B. E. Synthesis 1977, 172–176. doi:10.1055/s-1977-24308
Return to citation in text: [1] -
Lau, C. K.; Dufresne, C.; Belanger, P. C.; Pietre, S.; Scheigetz, J. J. Org. Chem. 1986, 51, 3038–3043. doi:10.1021/jo00365a034
Return to citation in text: [1] [2] [3] [4] -
Dobmeier, M.; Herrmann, J. M.; Lenoir, D.; König, B. Beilstein J. Org. Chem. 2012, 8, 330–336. doi:10.3762/bjoc.8.36
Return to citation in text: [1] [2] [3] -
Gordon, P. E.; Fry, A. J. Tetrahedron Lett. 2001, 42, 831–833. doi:10.1016/S0040-4039(00)02159-6
Return to citation in text: [1] [2] -
Gordon, P. E.; Fry, A. J.; Hicks, L. D. ARKIVOC 2005, No. 6, 393–400. doi:10.3998/ark.5550190.0006.634
Return to citation in text: [1] [2] -
Wu, X.; Mahalingam, A. K.; Alterman, M. Tetrahedron Lett. 2005, 46, 1501–1504. doi:10.1016/j.tetlet.2005.01.012
Return to citation in text: [1] -
Nishigaya, Y.; Umei, K.; Watanabe, D.; Kohno, Y.; Seto, S. Tetrahedron 2016, 72, 1566–1572. doi:10.1016/j.tet.2016.02.005
Return to citation in text: [1] [2] [3] -
Li, J.; Liu, Q.; Shen, H.; Huang, R.; Zhang, X.; Xiong, Y.; Chen, C. RSC Adv. 2015, 5, 85291–85295. doi:10.1039/C5RA14775A
Return to citation in text: [1] [2] [3] [4] -
Gevorgyan, V.; Liu, J.-X.; Rubin, M.; Benson, S.; Yamamoto, Y. Tetrahedron Lett. 1999, 40, 8919–8922. doi:10.1016/S0040-4039(99)01757-8
Return to citation in text: [1] -
Gevorgyan, V.; Rubin, M.; Benson, S.; Liu, J.-X.; Yamamoto, Y. J. Org. Chem. 2000, 65, 6179–6186. doi:10.1021/jo000726d
Return to citation in text: [1] -
Denancé, M.; Guyot, M.; Samadi, M. Steroids 2006, 71, 599–602. doi:10.1016/j.steroids.2006.03.002
Return to citation in text: [1] -
Miyai, T.; Ueba, M.; Baba, A. Synlett 1999, 182–184. doi:10.1055/s-1999-2588
Return to citation in text: [1] -
Baba, A.; Yasuda, M.; Nishimoto, Y.; Saito, T.; Onishi, Y. Pure Appl. Chem. 2008, 80, 845–854. doi:10.1351/pac200880050845
Return to citation in text: [1] -
Egi, M.; Kawai, T.; Umemura, M.; Akai, S. J. Org. Chem. 2012, 77, 7092–7097. doi:10.1021/jo300889p
Return to citation in text: [1] -
Meyer, V. J.; Niggemann, M. Chem. – Eur. J. 2012, 18, 4687–4691. doi:10.1002/chem.201103691
Return to citation in text: [1] -
Narayana Kumar, G. G. K. S.; Laali, K. K. Org. Biomol. Chem. 2012, 10, 7347–7355. doi:10.1039/c2ob26046h
Return to citation in text: [1] -
Tandiary, M. A.; Masui, Y.; Onaka, M. Tetrahedron Lett. 2014, 55, 4160–4162. doi:10.1016/j.tetlet.2014.05.042
Return to citation in text: [1] -
Mirza-Aghayan, M.; Boukherroub, R.; Rahimifard, M. Tetrahedron Lett. 2009, 50, 5930–5932. doi:10.1016/j.tetlet.2009.08.043
Return to citation in text: [1] -
Bell, M. G.; Muehl, B. S.; Winter, M. A. Dihydrobenzo(B)indeno(2,1-D)thiophene compounds, intermediates, processes, compositions, and methods. Eur. Pat. EP 832891, April 1, 1998.
Return to citation in text: [1] -
Sousa, S. C. A.; Fernandes, T. A.; Fernandes, A. C. Eur. J. Org. Chem. 2016, 3109–3112. doi:10.1002/ejoc.201600441
Return to citation in text: [1] -
Barton, D. H. R.; Jacob, M. Tetrahedron Lett. 1998, 39, 1331–1334. doi:10.1016/S0040-4039(98)00005-7
Return to citation in text: [1] -
Shi, Y.; Dayoub, W.; Chen, G.-R.; Lemaire, M. Tetrahedron Lett. 2011, 52, 1281–1283. doi:10.1016/j.tetlet.2011.01.038
Return to citation in text: [1] [2] -
Mirza-Aghayan, M.; Boukherroub, R.; Rahimifard, M.; Zadmard, R. J. Iran. Chem. Soc. 2011, 8, 570–573. doi:10.1007/BF03249092
Return to citation in text: [1] -
Purushothama Chary, K.; Santosh Laxmi, Y. R.; Iyengar, D. S. Synth. Commun. 1999, 29, 1257–1261. doi:10.1080/00397919908086100
Return to citation in text: [1] -
Kotsuki, H.; Ushio, Y.; Yoshimura, N.; Ochi, M. J. Org. Chem. 1987, 52, 2594–2596. doi:10.1021/jo00388a049
Return to citation in text: [1] -
Fleming, B.; Bolker, H. I. Can. J. Chem. 1974, 52, 888–893. doi:10.1139/v74-143
Return to citation in text: [1] -
Ishihara, K.; Hanaki, N.; Yamomoto, H. Synlett 1993, 127–129. doi:10.1055/s-1993-22373
Return to citation in text: [1] -
Luh, T.-Y.; Ni, Z.-J. Synthesis 1990, 89–103. doi:10.1055/s-1990-26798
Return to citation in text: [1] -
Rentner, J.; Kljajic, M.; Offner, L.; Breinbauer, R. Tetrahedron 2014, 70, 8983–9027. doi:10.1016/j.tet.2014.06.104
Return to citation in text: [1] -
Saito, K.; Kondo, K.; Akiyama, T. Org. Lett. 2015, 17, 3366–3369. doi:10.1021/acs.orglett.5b01651
Return to citation in text: [1] -
Burghardt, T. E. J. Sulfur Chem. 2005, 26, 411–427. doi:10.1080/17415990500195198
Return to citation in text: [1] -
Alesso, E. N.; Bianchi, D. E.; Finkielsztein, L. M.; Lantaño, B.; Moltrasio, G. Y.; Aguirre, J. M. Tetrahedron Lett. 1995, 36, 3299–3302. doi:10.1016/0040-4039(95)00525-H
Return to citation in text: [1] -
Wex, B.; Kaafarani, B. R.; Neckers, D. C. J. Org. Chem. 2004, 69, 2197–2199. doi:10.1021/jo035769j
Return to citation in text: [1] -
Wex, B.; Kaafarani, B. R.; Kirschbaum, K.; Neckers, D. C. J. Org. Chem. 2005, 70, 4502–4505. doi:10.1021/jo048010w
Return to citation in text: [1] -
Tylleman, B.; Vande Velde, C. M. L.; Balandier, J.-Y.; Stas, S.; Sergeyev, S.; Geerts, Y. H. Org. Lett. 2011, 13, 5208–5211. doi:10.1021/ol202089t
Return to citation in text: [1] [2] -
Pan, X.; Wang, K.; Yu, W.; Zhang, R.; Xu, L.; Liu, F. Chem. Lett. 2015, 44, 1170–1172. doi:10.1246/cl.150390
Return to citation in text: [1] -
Bhattacharyya, S.; Chatterjee, A.; Williamson, J. S. Synth. Commun. 1997, 27, 4265–4274. doi:10.1080/00397919708005050
Return to citation in text: [1] -
Ley, S. V.; Tackett, M. N.; Maddess, M. L.; Anderson, J. C.; Brennan, P. E.; Cappi, M. W.; Heer, J. P.; Helgen, C.; Kori, M.; Kouklovsky, C.; Marsden, S. P.; Norman, J.; Osborn, D. P.; Palomero, M. Á.; Pavey, J. B. J.; Pinel, C.; Robinson, L. A.; Schnaubelt, J.; Scott, J. S.; Spilling, C. D.; Watanabe, H.; Wesson, K. E.; Willis, M. C. Chem. – Eur. J. 2009, 15, 2874–2914. doi:10.1002/chem.200801656
Return to citation in text: [1] -
Marques, C. A.; Selva, M.; Tundo, P. J. Org. Chem. 1995, 60, 2430–2435. doi:10.1021/jo00113a024
Return to citation in text: [1] -
Arnold, B. J.; Mellows, S. M.; Sammes, P. G. J. Chem. Soc., Perkin Trans. 1 1973, 1266–1270. doi:10.1039/p19730001266
Return to citation in text: [1] -
Galletti, G. C.; Ward, R. S.; Pelter, A.; Goubet, D. J. Anal. Appl. Pyrolysis 1992, 24, 139–146. doi:10.1016/0165-2370(92)85025-G
Return to citation in text: [1] -
Dean, J. A. Lange’s Handbook of Chemistry, 15th ed.; Section 4; McGraw-Hill: New York, Montreal, 1998; 4.52.
Return to citation in text: [1]
| 37. | Miyai, T.; Ueba, M.; Baba, A. Synlett 1999, 182–184. doi:10.1055/s-1999-2588 |
| 38. | Baba, A.; Yasuda, M.; Nishimoto, Y.; Saito, T.; Onishi, Y. Pure Appl. Chem. 2008, 80, 845–854. doi:10.1351/pac200880050845 |
| 39. | Egi, M.; Kawai, T.; Umemura, M.; Akai, S. J. Org. Chem. 2012, 77, 7092–7097. doi:10.1021/jo300889p |
| 40. | Meyer, V. J.; Niggemann, M. Chem. – Eur. J. 2012, 18, 4687–4691. doi:10.1002/chem.201103691 |
| 33. | Li, J.; Liu, Q.; Shen, H.; Huang, R.; Zhang, X.; Xiong, Y.; Chen, C. RSC Adv. 2015, 5, 85291–85295. doi:10.1039/C5RA14775A |
| 45. | Sousa, S. C. A.; Fernandes, T. A.; Fernandes, A. C. Eur. J. Org. Chem. 2016, 3109–3112. doi:10.1002/ejoc.201600441 |
| 44. | Bell, M. G.; Muehl, B. S.; Winter, M. A. Dihydrobenzo(B)indeno(2,1-D)thiophene compounds, intermediates, processes, compositions, and methods. Eur. Pat. EP 832891, April 1, 1998. |
| 43. | Mirza-Aghayan, M.; Boukherroub, R.; Rahimifard, M. Tetrahedron Lett. 2009, 50, 5930–5932. doi:10.1016/j.tetlet.2009.08.043 |
| 32. | Nishigaya, Y.; Umei, K.; Watanabe, D.; Kohno, Y.; Seto, S. Tetrahedron 2016, 72, 1566–1572. doi:10.1016/j.tet.2016.02.005 |
| 41. | Narayana Kumar, G. G. K. S.; Laali, K. K. Org. Biomol. Chem. 2012, 10, 7347–7355. doi:10.1039/c2ob26046h |
| 42. | Tandiary, M. A.; Masui, Y.; Onaka, M. Tetrahedron Lett. 2014, 55, 4160–4162. doi:10.1016/j.tetlet.2014.05.042 |
| 32. | Nishigaya, Y.; Umei, K.; Watanabe, D.; Kohno, Y.; Seto, S. Tetrahedron 2016, 72, 1566–1572. doi:10.1016/j.tet.2016.02.005 |
| 33. | Li, J.; Liu, Q.; Shen, H.; Huang, R.; Zhang, X.; Xiong, Y.; Chen, C. RSC Adv. 2015, 5, 85291–85295. doi:10.1039/C5RA14775A |
| 33. | Li, J.; Liu, Q.; Shen, H.; Huang, R.; Zhang, X.; Xiong, Y.; Chen, C. RSC Adv. 2015, 5, 85291–85295. doi:10.1039/C5RA14775A |
| 28. | Dobmeier, M.; Herrmann, J. M.; Lenoir, D.; König, B. Beilstein J. Org. Chem. 2012, 8, 330–336. doi:10.3762/bjoc.8.36 |
| 29. | Gordon, P. E.; Fry, A. J. Tetrahedron Lett. 2001, 42, 831–833. doi:10.1016/S0040-4039(00)02159-6 |
| 30. | Gordon, P. E.; Fry, A. J.; Hicks, L. D. ARKIVOC 2005, No. 6, 393–400. doi:10.3998/ark.5550190.0006.634 |
| 46. | Barton, D. H. R.; Jacob, M. Tetrahedron Lett. 1998, 39, 1331–1334. doi:10.1016/S0040-4039(98)00005-7 |
| 47. | Shi, Y.; Dayoub, W.; Chen, G.-R.; Lemaire, M. Tetrahedron Lett. 2011, 52, 1281–1283. doi:10.1016/j.tetlet.2011.01.038 |
| 48. | Mirza-Aghayan, M.; Boukherroub, R.; Rahimifard, M.; Zadmard, R. J. Iran. Chem. Soc. 2011, 8, 570–573. doi:10.1007/BF03249092 |
| 49. | Purushothama Chary, K.; Santosh Laxmi, Y. R.; Iyengar, D. S. Synth. Commun. 1999, 29, 1257–1261. doi:10.1080/00397919908086100 |
| 50. | Kotsuki, H.; Ushio, Y.; Yoshimura, N.; Ochi, M. J. Org. Chem. 1987, 52, 2594–2596. doi:10.1021/jo00388a049 |
| 51. | Fleming, B.; Bolker, H. I. Can. J. Chem. 1974, 52, 888–893. doi:10.1139/v74-143 |
| 52. | Ishihara, K.; Hanaki, N.; Yamomoto, H. Synlett 1993, 127–129. doi:10.1055/s-1993-22373 |
| 27. | Lau, C. K.; Dufresne, C.; Belanger, P. C.; Pietre, S.; Scheigetz, J. J. Org. Chem. 1986, 51, 3038–3043. doi:10.1021/jo00365a034 |
| 61. | Pan, X.; Wang, K.; Yu, W.; Zhang, R.; Xu, L.; Liu, F. Chem. Lett. 2015, 44, 1170–1172. doi:10.1246/cl.150390 |
| 62. | Bhattacharyya, S.; Chatterjee, A.; Williamson, J. S. Synth. Commun. 1997, 27, 4265–4274. doi:10.1080/00397919708005050 |
| 63. | Ley, S. V.; Tackett, M. N.; Maddess, M. L.; Anderson, J. C.; Brennan, P. E.; Cappi, M. W.; Heer, J. P.; Helgen, C.; Kori, M.; Kouklovsky, C.; Marsden, S. P.; Norman, J.; Osborn, D. P.; Palomero, M. Á.; Pavey, J. B. J.; Pinel, C.; Robinson, L. A.; Schnaubelt, J.; Scott, J. S.; Spilling, C. D.; Watanabe, H.; Wesson, K. E.; Willis, M. C. Chem. – Eur. J. 2009, 15, 2874–2914. doi:10.1002/chem.200801656 |
| 28. | Dobmeier, M.; Herrmann, J. M.; Lenoir, D.; König, B. Beilstein J. Org. Chem. 2012, 8, 330–336. doi:10.3762/bjoc.8.36 |
| 60. | Tylleman, B.; Vande Velde, C. M. L.; Balandier, J.-Y.; Stas, S.; Sergeyev, S.; Geerts, Y. H. Org. Lett. 2011, 13, 5208–5211. doi:10.1021/ol202089t |
| 27. | Lau, C. K.; Dufresne, C.; Belanger, P. C.; Pietre, S.; Scheigetz, J. J. Org. Chem. 1986, 51, 3038–3043. doi:10.1021/jo00365a034 |
| 20. | Park, J.-I.; Chung, J. W.; Kim, J.-Y.; Lee, J.; Jung, J. Y.; Koo, B.; Lee, B.-L.; Lee, S. W.; Jin, Y. W.; Lee, S. Y. J. Am. Chem. Soc. 2015, 137, 12175–12178. doi:10.1021/jacs.5b01108 |
| 27. | Lau, C. K.; Dufresne, C.; Belanger, P. C.; Pietre, S.; Scheigetz, J. J. Org. Chem. 1986, 51, 3038–3043. doi:10.1021/jo00365a034 |
| 57. | Alesso, E. N.; Bianchi, D. E.; Finkielsztein, L. M.; Lantaño, B.; Moltrasio, G. Y.; Aguirre, J. M. Tetrahedron Lett. 1995, 36, 3299–3302. doi:10.1016/0040-4039(95)00525-H |
| 58. | Wex, B.; Kaafarani, B. R.; Neckers, D. C. J. Org. Chem. 2004, 69, 2197–2199. doi:10.1021/jo035769j |
| 59. | Wex, B.; Kaafarani, B. R.; Kirschbaum, K.; Neckers, D. C. J. Org. Chem. 2005, 70, 4502–4505. doi:10.1021/jo048010w |
| 60. | Tylleman, B.; Vande Velde, C. M. L.; Balandier, J.-Y.; Stas, S.; Sergeyev, S.; Geerts, Y. H. Org. Lett. 2011, 13, 5208–5211. doi:10.1021/ol202089t |
| 53. | Luh, T.-Y.; Ni, Z.-J. Synthesis 1990, 89–103. doi:10.1055/s-1990-26798 |
| 54. | Rentner, J.; Kljajic, M.; Offner, L.; Breinbauer, R. Tetrahedron 2014, 70, 8983–9027. doi:10.1016/j.tet.2014.06.104 |
| 55. | Saito, K.; Kondo, K.; Akiyama, T. Org. Lett. 2015, 17, 3366–3369. doi:10.1021/acs.orglett.5b01651 |
| 56. | Burghardt, T. E. J. Sulfur Chem. 2005, 26, 411–427. doi:10.1080/17415990500195198 |
| 65. | Arnold, B. J.; Mellows, S. M.; Sammes, P. G. J. Chem. Soc., Perkin Trans. 1 1973, 1266–1270. doi:10.1039/p19730001266 |
| 66. | Galletti, G. C.; Ward, R. S.; Pelter, A.; Goubet, D. J. Anal. Appl. Pyrolysis 1992, 24, 139–146. doi:10.1016/0165-2370(92)85025-G |
| 64. | Marques, C. A.; Selva, M.; Tundo, P. J. Org. Chem. 1995, 60, 2430–2435. doi:10.1021/jo00113a024 |
| 7. | Wai, J. S.; Egbertson, M. S.; Payne, L. S.; Fisher, T. E.; Embrey, M. W.; Tran, L. O.; Melamed, J. Y.; Langford, H. M.; Guare, J. P.; Zhuang, L.; Grey, V. E.; Vacca, J. P.; Holloway, M. K.; Naylor-Olsen, A. M.; Hazuda, D. J.; Felock, P. J.; Wolfe, A. L.; Stillmock, K. A.; Schleif, W. A.; Gabryelski, L. J.; Young, S. D. J. Med. Chem. 2000, 43, 4923–4926. doi:10.1021/jm000176b |
| 6. | Li, H.; Zhu, X.; Yao, G.; Wang, Z. J. Surg. Res. 2015, 195, 271–276. doi:10.1016/j.jss.2015.01.016 |
| 23. | Dockrey, S. A. B.; Makepeace, A. K.; Schmink, J. R. Org. Lett. 2014, 16, 4730–4733. doi:10.1021/ol502428h |
| 3. | Chen, I.; McDougal, A.; Wang, F.; Safe, S. Carcinogenesis 1998, 19, 1631–1639. doi:10.1093/carcin/19.9.1631 |
| 4. | McDougal, A.; Gupta, M. S.; Ramamoorthy, K.; Sun, G.; Safe, S. H. Cancer Lett. 2000, 151, 169–179. doi:10.1016/S0304-3835(99)00406-1 |
| 5. | Abdelrahim, M.; Newman, K.; Vanderlaag, K.; Samudio, I.; Safe, S. Carcinogenesis 2006, 27, 717–728. doi:10.1093/carcin/bgi270 |
| 16. | Bradsher, C. K. Chem. Rev. 1987, 87, 1277–1297. doi:10.1021/cr00082a001 |
| 17. | Bodzioch, A.; Kowalska, E.; Skalik, J.; Bałczewski, P. Chem. Heterocycl. Compd. 2017, 53, 11–20. doi:10.1007/s10593-017-2015-y |
| 18. | Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E.; Jeszka, J. K.; Uznański, P.; Kania, S.; Kuliński, J.; Bałczewski, P. Synfacts 2012, 8, 619. doi:10.1055/s-0031-1291065 |
| 19. | Bałczewski, P.; Kowalska, E.; Skalik, J. 10-Thiosubstituted pentahydroxyanthracene derivatives, a method of their preparation and intermediate compounds. Eur. Pat. Appl. EP 2886539 A1 20150624, June 24, 2015. |
| 2. | Sun, H. H.; Paul, V. J.; Fencial, W. Phytochemistry 1983, 22, 743–745. doi:10.1016/S0031-9422(00)86974-5 |
| 20. | Park, J.-I.; Chung, J. W.; Kim, J.-Y.; Lee, J.; Jung, J. Y.; Koo, B.; Lee, B.-L.; Lee, S. W.; Jin, Y. W.; Lee, S. Y. J. Am. Chem. Soc. 2015, 137, 12175–12178. doi:10.1021/jacs.5b01108 |
| 21. | Bodzioch, A.; Marciniak, B.; Różycka-Sokołowska, E.; Jeszka, J. K.; Uznański, P.; Kania, S.; Kuliński, J.; Bałczewski, P. Chem. – Eur. J. 2012, 18, 4866–4877. doi:10.1002/chem.201101909 |
| 22. | Bałczewski, P.; Skalik, J.; Uznański, P.; Guziejewski, D.; Ciesielski, W. RSC Adv. 2015, 5, 24700–24704. doi:10.1039/C4RA14071K |
| 12. | Gennari, L.; Merlotti, D.; Martini, G.; Nuti, R. Expert Opin. Invest. Drugs 2006, 15, 1091–1103. doi:10.1517/13543784.15.9.1091 |
| 14. | Lee, W. Y.; Park, C. H.; Kim, H.-J.; Kim, S. J. Org. Chem. 1994, 59, 878–884. doi:10.1021/jo00083a033 |
| 11. | Canel, C.; Moraes, R. M.; Dayan, F. E.; Ferreira, D. Phytochemistry 2000, 54, 115–120. doi:10.1016/S0031-9422(00)00094-7 |
| 10. | Kerrebroeck, P. V.; Kreder, K.; Jonas, U.; Zinner, N.; Wein, A. Urology 2001, 57, 414–421. doi:10.1016/S0090-4295(00)01113-4 |
| 67. | Dean, J. A. Lange’s Handbook of Chemistry, 15th ed.; Section 4; McGraw-Hill: New York, Montreal, 1998; 4.52. |
| 8. | Panda, G.; Parai, M. K.; Das, S. K.; Shagufta; Sinha, M.; Chaturvedi, V.; Srivastava, A. K.; Manju, Y. S.; Gaikwad, A. N.; Sinha, S. Eur. J. Med. Chem. 2007, 42, 410–419. doi:10.1016/j.ejmech.2006.09.020 |
| 9. | Upadhayaya, R. S.; Vandavasi, J. K.; Vasireddy, N. R.; Sharma, V.; Dixit, S. S.; Chattopadhyaya, J. Bioorg. Med. Chem. 2009, 17, 2830–2841. doi:10.1016/j.bmc.2009.02.026 |
| 13. | Conn, M. M.; Rebek, J., Jr. Chem. Rev. 1997, 97, 1647–1668. doi:10.1021/cr9603800 |
| 47. | Shi, Y.; Dayoub, W.; Chen, G.-R.; Lemaire, M. Tetrahedron Lett. 2011, 52, 1281–1283. doi:10.1016/j.tetlet.2011.01.038 |
| 25. | Forster, F.; Metsänen, T. T.; Irran, E.; Hrobárik, P.; Oestreich, M. J. Am. Chem. Soc. 2017, 139, 16334–16342. doi:10.1021/jacs.7b09444 |
| 24. | Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1–6. doi:10.1016/j.jfluchem.2016.08.003 |
| 33. | Li, J.; Liu, Q.; Shen, H.; Huang, R.; Zhang, X.; Xiong, Y.; Chen, C. RSC Adv. 2015, 5, 85291–85295. doi:10.1039/C5RA14775A |
| 34. | Gevorgyan, V.; Liu, J.-X.; Rubin, M.; Benson, S.; Yamamoto, Y. Tetrahedron Lett. 1999, 40, 8919–8922. doi:10.1016/S0040-4039(99)01757-8 |
| 35. | Gevorgyan, V.; Rubin, M.; Benson, S.; Liu, J.-X.; Yamamoto, Y. J. Org. Chem. 2000, 65, 6179–6186. doi:10.1021/jo000726d |
| 36. | Denancé, M.; Guyot, M.; Samadi, M. Steroids 2006, 71, 599–602. doi:10.1016/j.steroids.2006.03.002 |
| 31. | Wu, X.; Mahalingam, A. K.; Alterman, M. Tetrahedron Lett. 2005, 46, 1501–1504. doi:10.1016/j.tetlet.2005.01.012 |
| 32. | Nishigaya, Y.; Umei, K.; Watanabe, D.; Kohno, Y.; Seto, S. Tetrahedron 2016, 72, 1566–1572. doi:10.1016/j.tet.2016.02.005 |
| 28. | Dobmeier, M.; Herrmann, J. M.; Lenoir, D.; König, B. Beilstein J. Org. Chem. 2012, 8, 330–336. doi:10.3762/bjoc.8.36 |
| 29. | Gordon, P. E.; Fry, A. J. Tetrahedron Lett. 2001, 42, 831–833. doi:10.1016/S0040-4039(00)02159-6 |
| 30. | Gordon, P. E.; Fry, A. J.; Hicks, L. D. ARKIVOC 2005, No. 6, 393–400. doi:10.3998/ark.5550190.0006.634 |
| 26. | Gribble, G. W.; Leese, R. M.; Evans, B. E. Synthesis 1977, 172–176. doi:10.1055/s-1977-24308 |
| 27. | Lau, C. K.; Dufresne, C.; Belanger, P. C.; Pietre, S.; Scheigetz, J. J. Org. Chem. 1986, 51, 3038–3043. doi:10.1021/jo00365a034 |
© 2018 Czarnecka et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)